
Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities †


Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Plant Material
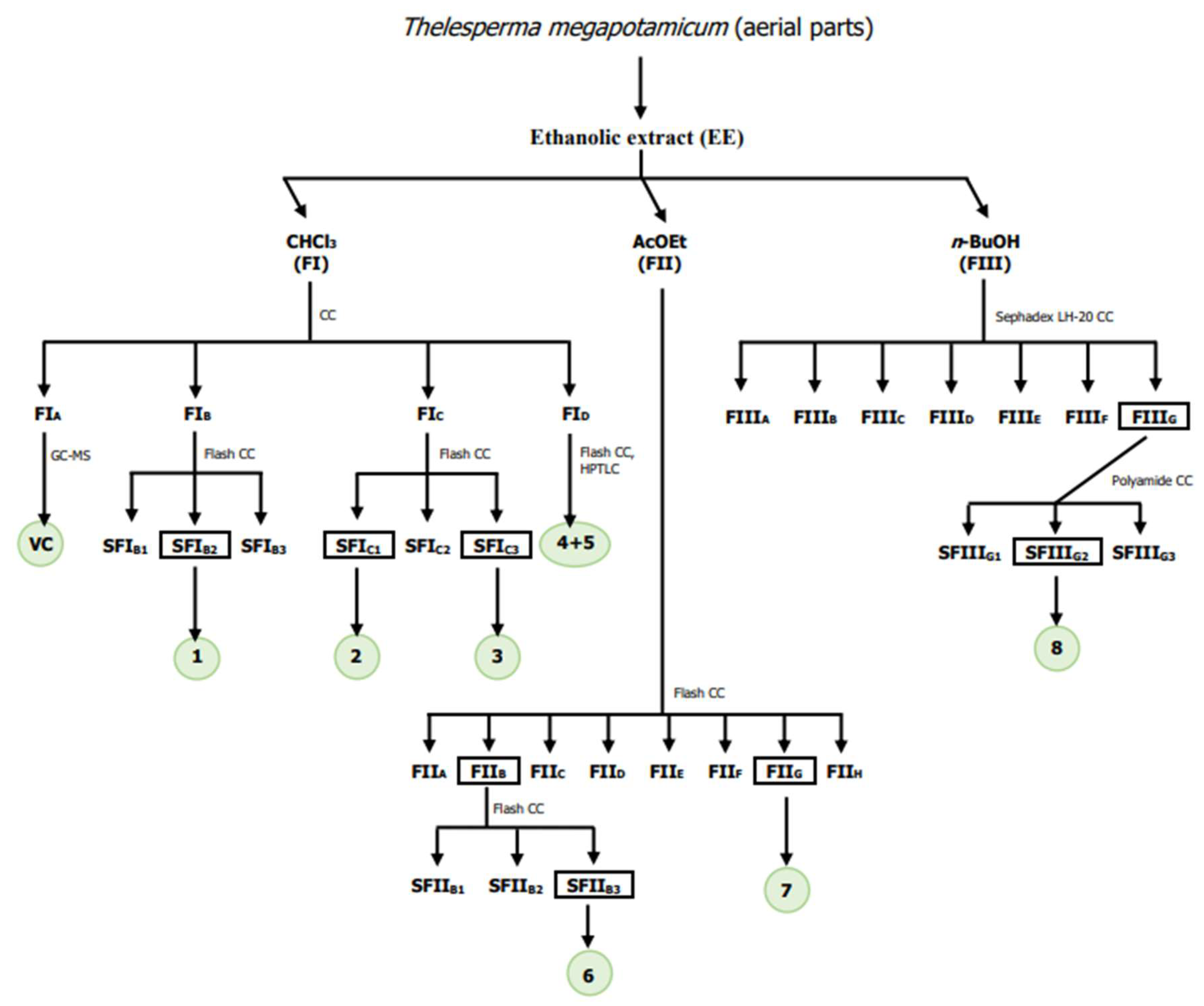
2.3. Extraction and Isolation
2.4. Inhibition Assay on AChE and BChE In Vitro
2.5. Antioxidant Activity
3. Results and Discussion
3.1. Phytochemical Investigation of T. megapotamicum
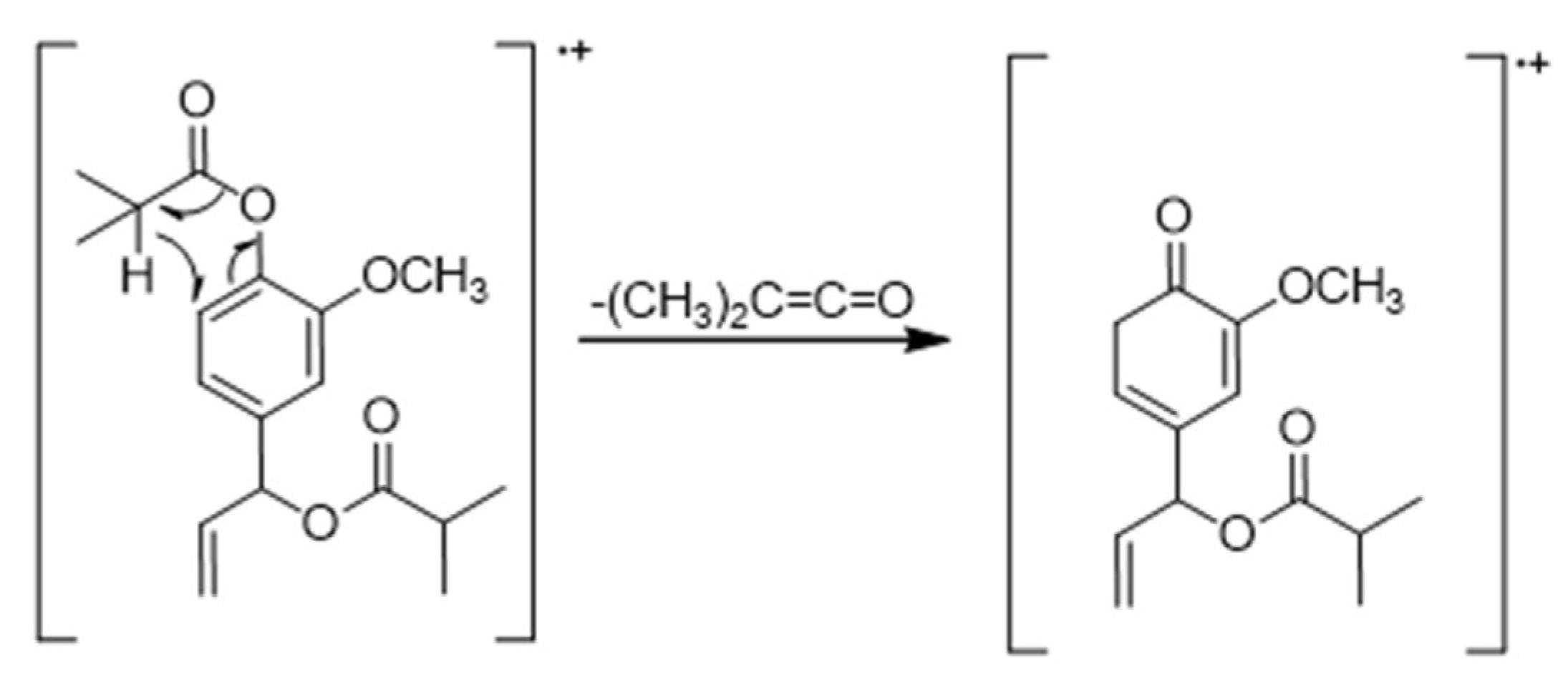
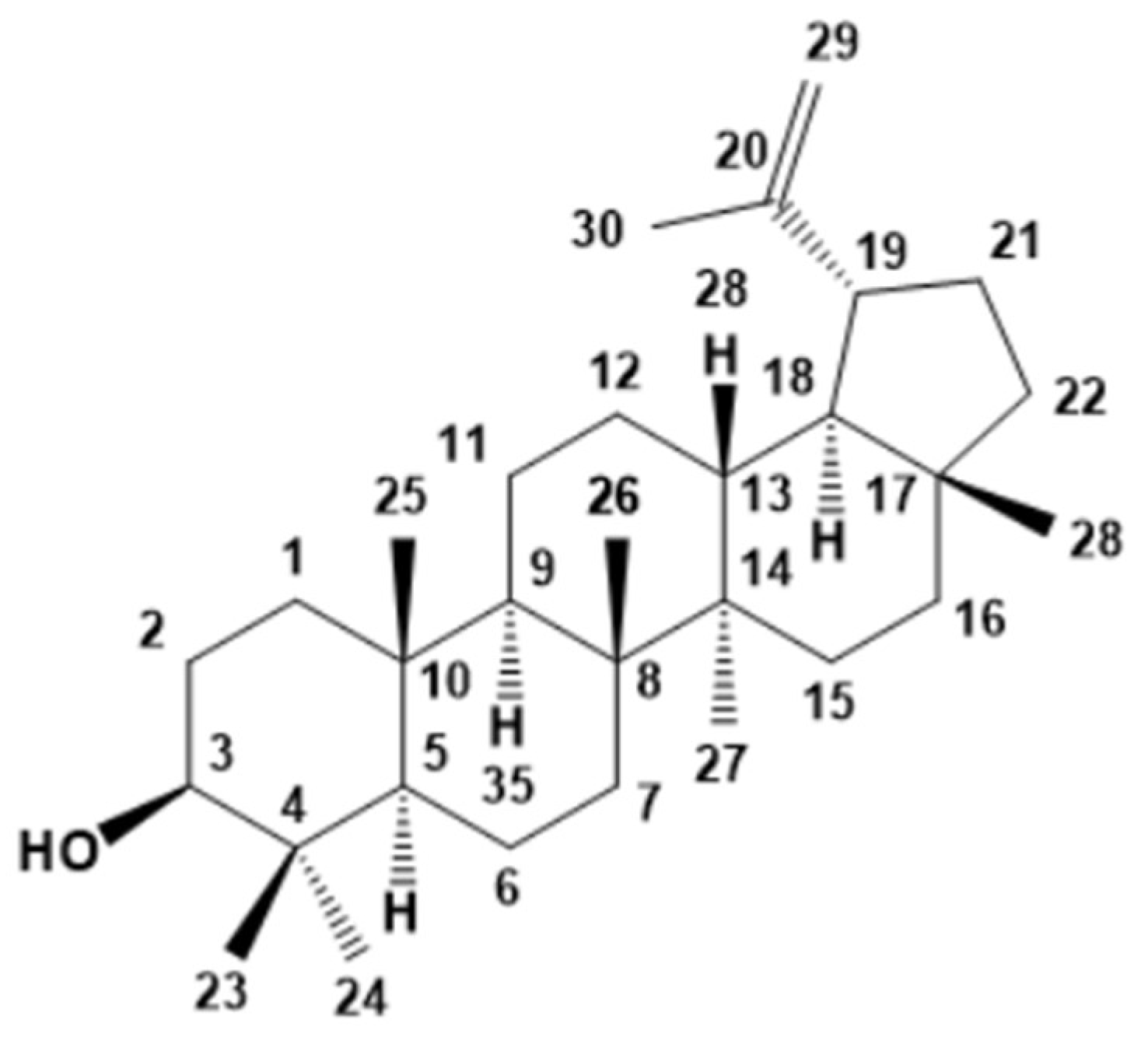
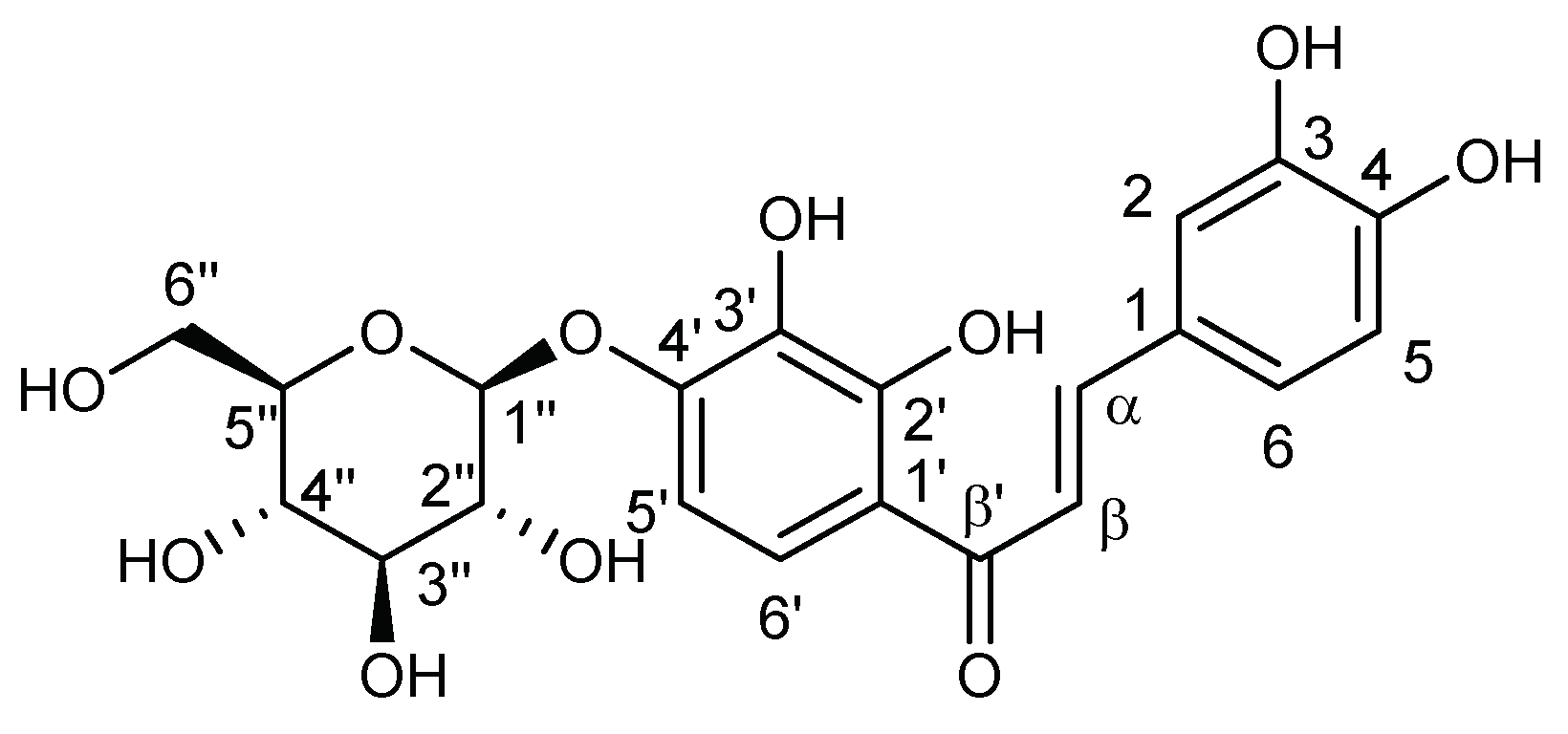
3.1.1. Chloroform Sub-Extract Composition
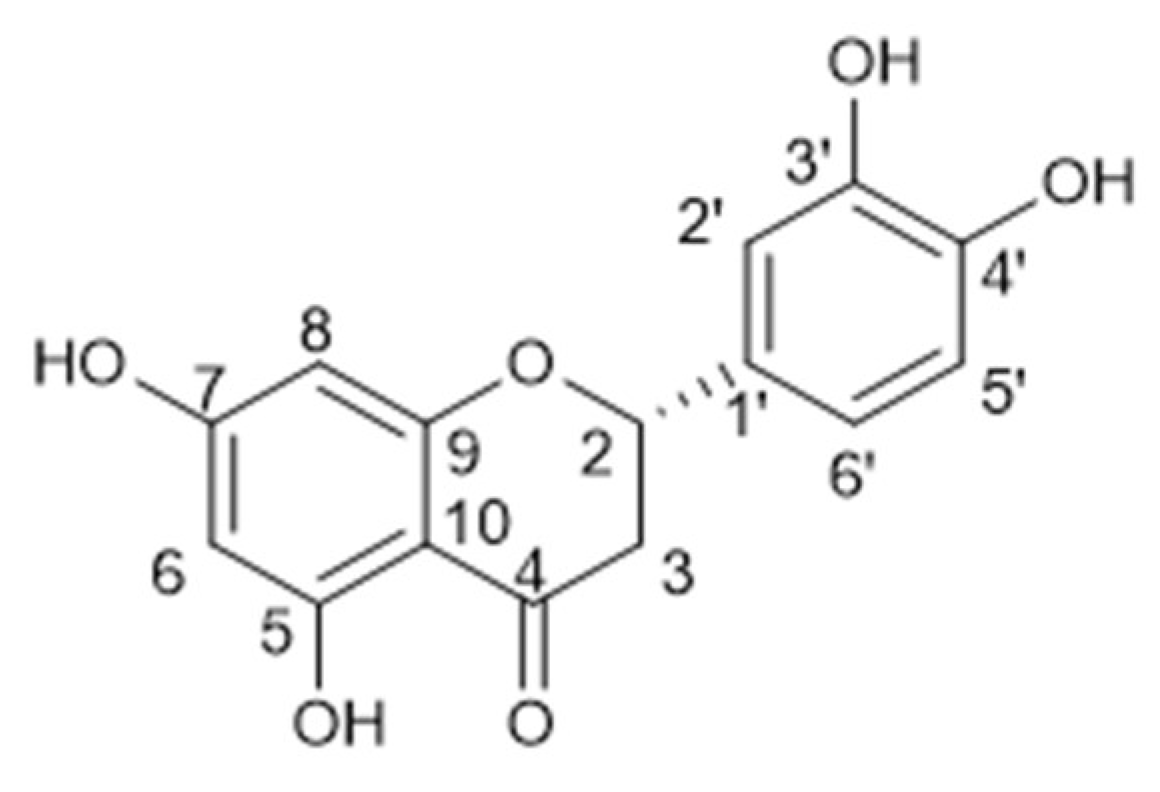
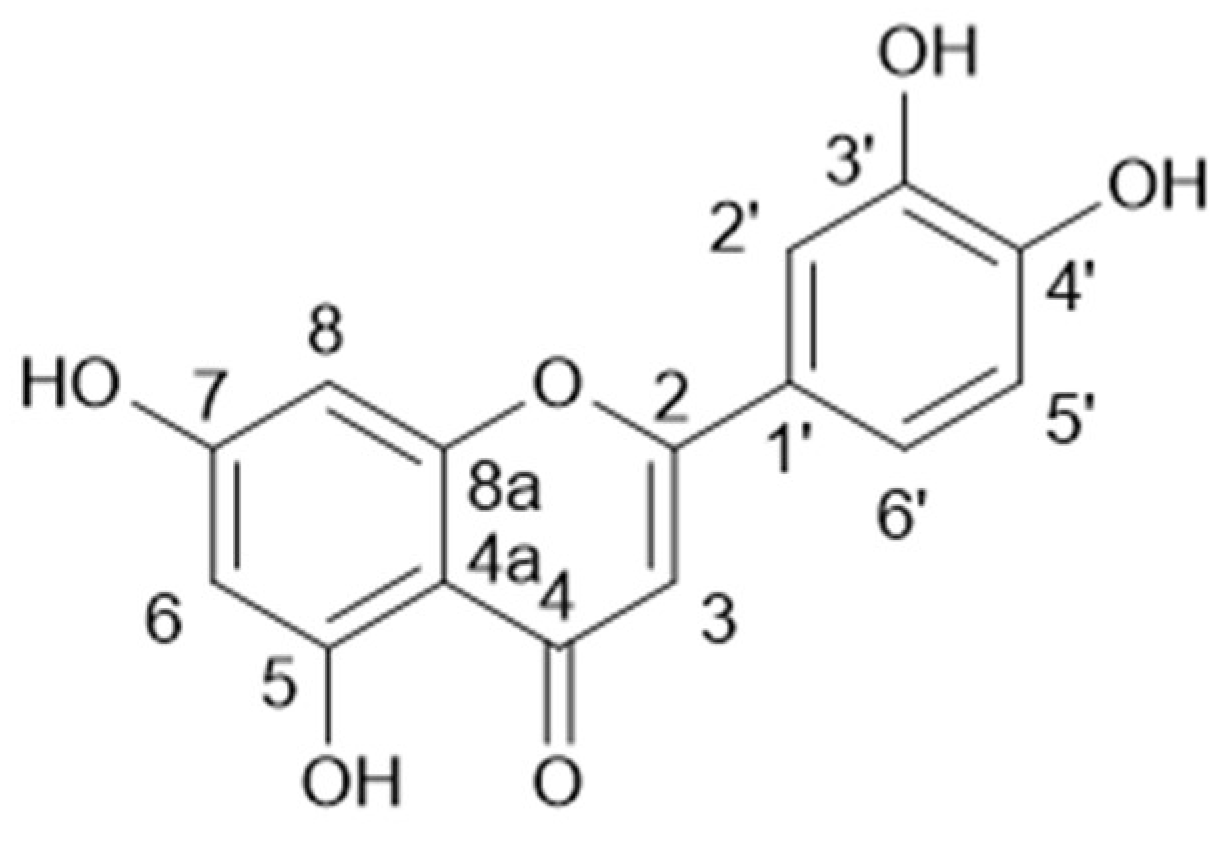
3.1.2. Ethyl Acetate Sub-Extract Composition
3.1.3. N-Butanol Sub-Extract Composition
3.2. Inhibition Assay on AChE and BChE In Vitro and Antioxidant Capacity of the Sub-Extracts and of the Major Metabolites
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Rao, A.V.; Balachandran, B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr. Neurosci. 2020, 5, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izaokovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Wen, L.L.; Huang, Y.N.; Chen, Y.T.; Ku, M.C. Dual effects of antioxidants in neurodegeneration: Direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar] [CrossRef]
- Linseman, D.A. Targeting oxidative stress for neuroprotection. Antioxid Redox Signal. 2009, 11, 421–423. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar] [CrossRef]
- Maestri, D.M.; Nepote, V.; Lamarque, A.L.; Zygadlo, J.A. Natural products as antioxidants. In Phytochemistry: Advances in Research, 1st ed.; Imperato, F., Ed.; Research Signpost: Kerala, India, 2006; pp. 105–135. [Google Scholar]
- Mandel, S.A.; Amit, T.; Weinreb, O.; Reznichenko, L.; Youdim, M.B. Simultaneous manipulation of multiple brain targets by green tea catechins: A potential neuroprotective strategy for Alzheimer and Parkinson diseases. CNS Neurosci. Ther. 2008, 14, 352–365. [Google Scholar] [CrossRef]
- Mandel, S.A.; Amit, T.; Kalfon, L.; Reznichenko, L.; Weinreb, O.; Youdim, M.B. Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: Special reference to epigallocatechin gallate (EGCG). J. Alzheimers Dis. 2008, 15, 211–222. [Google Scholar] [CrossRef]
- Avramovich-Tirosh, Y.; Reznichenko, L.; Amit, T.; Zheng, H.; Fridkin, M.; Weinreb, O.; Mandel, S.; Youdim, M.B. Neurorescue activity, APP regulation and amyloid-b peptide reduction by novel multi-functional brain permeable iron- chelating- antioxidants, M-30 and green tea polyphenol, EGCG. Curr. Alzheimer Res. 2007, 4, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective–neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avramovich-Tirosh, Y.; Youdim, M.B.H.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and b-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem. 2006, 97, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Mandel, S.; Amit, T.; Youdim, M.B. Neurological mechanisms of green tea polyphenols in Alzheimer‘s and Parkinson’s diseases. J. Nutr. Biochem. 2004, 15, 506–516. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotection and neurorescue against Ab toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 952–954. [Google Scholar] [CrossRef]
- Mancuso, C.; Bates, T.E.; Butterfield, D.A.; Calafato, S.; Cornelius, C.; Lorenzo, A.D.; Dinkova Kostova, A.T.; Calabrese, V. Natural antioxidants in Alzheimer’s disease. Expert Opin. Investig. Drugs 2007, 16, 1921–1931. [Google Scholar] [CrossRef]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Botanical phenolics and brain health. Neuromol. Med. 2008, 10, 259–274. [Google Scholar] [CrossRef]
- Schroeter, H.; Spencer, J.P.E.; Rice-Evans, C.; Williams, R.J. Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem. J. 2001, 358, 547. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef]
- Uriarte-Pueyo, I.; Calvo, M. Flavonoids as acetylcholinesterase inhibitors. Curr. Med. Chem. 2001, 18, 5289. [Google Scholar] [CrossRef]
- Qingbo, L.; Jie, W.; Bin, L.; Zhuo-Yang, C.; Ming, B.; Shaochun, S.; Xiao-Xiao, H.; Shao-Jiang, S. Phenylpropanoids and lignans from Prunus tomentosa seeds as efficient β-amyloid (Aβ) aggregation inhibitors. Bioorg. Chem. 2019, 84, 269–275. [Google Scholar] [CrossRef]
- Cantero, J.J.; Nuñez, C.O. Las Plantas Medicinales del sur de la Provincia de Córdoba, 1st ed.; Fundación Universidad Nacional de Río Cuarto: Río Cuarto, Argentina, 2000. [Google Scholar]
- Borneo, R.; Leon, A.E.; Aguirre, A.; Ribotta, P.; Cantero, J.J. Antioxidant capacity of medicinal plants from the Province of Cordoba (Argentina) and their in vitro testing in a model food system. Food Chem. 2009, 112, 664–670. [Google Scholar] [CrossRef]
- Ateya, A.M.; Okarter, T.U.; Knapp, J.E.; Schiff, P.L.; Slatkin, D.J. Flavonoids of Thelesperma megapotanicum. Planta Med. 1982, 45, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Thron, U.; Martin, R.; Reichling, J. Rareeugenol and Z-coniferyl alcohol derivatives in roots of three Coreopsis species. Z. Nat. Schung C 1989, 44, 7–11. [Google Scholar]
- Vela Gurovic, M.S.; Castro, M.J.; Richmond, V.; Faraoni, M.B.; Maier, M.S.; Murray, A.P. Triterpenoids with Acetylcholinesterase Inhibition from Chuquiraga erinacea D. Don. subsp. erinacea (Asteraceae). Planta Med. 2010, 76, 607–610. [Google Scholar] [CrossRef]
- Da Silva, L.A.L.; Faqueti, L.G.; Reginatto, F.H.; dos Santos, A.D.C.; Barison, A.; Biavatti, M.W. Phytochemical analysisof Vernonanthura tweedieana and a validated UPLC-PDA method for the quantification of eriodictyol. Rev. Bras. Farmacogn. 2015, 25, 375–381. [Google Scholar] [CrossRef]
- Nissler, L.; Gebhardt, R.; Berger, S. Flavonoid binding to a multi-drug-resistance transporter protein: An STD-NMR study. Anal. Bioanal. Chem. 2004, 379, 1045. [Google Scholar] [CrossRef]
- Redl, K.; Davis, B.; Bauer, R. Chalcone glycosides from Bidens campylotheca. Phytochemistry 1992, 32, 218–220. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Lebenson Wiss Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Erdogan Orhan, I.; Kartal, M.; Kan, Y.; Sener, B. Activity of essential oils and individual components against acetyl and butyrylcholinesterase. Z. Nat. 2008, 63, 547–553. [Google Scholar] [CrossRef]
- Mitsui, S.; Kobayashi, S.; Nagahori, N.; Ogiso, A. Constituents from seeds of Alpinia galanga WILD and their anti-ulcer activities. Chem. Pharm. Bull. 1976, 24, 2377–2382. [Google Scholar] [CrossRef] [PubMed]
- Lajter, I. Biologically Active Secondary Metabolites from Asteraceae and Polygonaceae Species. Ph.D. Thesis, University of Szeged, Szeged, Hungary, 2015. [CrossRef]
- Gade, S.; Rajamanikyam, M.; Vadlapudi, V.; Madhav Nukala, K.; Aluvala, R.; Giddigari, C.; Jyothi Karanam, N.C.; Barua, N.; Pandey, R.; Saradhi, V.; et al. Acetylcholinesterase inhibitory activity of stigmasterol & hexacosanol is responsible for larvicidal and repellent properties of Chromolaena odorata. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1861, 541–550. [Google Scholar] [CrossRef]
- Podoly, E.Y.; Bruck, T.; Diamant, S.; Melamed-Book, N.; Weiss, A.; Huang, Y.; Livnah, O.; Langermann, S.; Wilgus, H.; Soreq, H. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. Neurodegener. Dis. 2008, 5, 232. [Google Scholar] [CrossRef]
- Diamant, S.Z.; Podoly, E.; Friedler, A.; Ligumsky, H.; Livnah, O.; Soreq, H. Alzheimer’s disease and type 2 diabetes mellitus: The cholinesterase connection? Proc. Natl. Acad. Sci. USA 2006, 103, 8628. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Extracts | IC50 AChE (μg/mL) | IC50 BChE (μg/mL) | IC50 DPPH (μg/mL) |
|---|---|---|---|
| Chloroform (FI) | 562.7 ± 8.0 | 30.7 ± 1.5 | >200 |
| Ethyl acetate (FII) | 168.0 ± 6.0 | 505.8 ± 9.4 | 19.6 ± 0.7 |
| n-butanol (FIII) | 337.1 ± 7.2 | 60.3 ± 2.3 | 49.5 ± 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siben, B.A.; Castro, M.J.; Faraoni, M.B. Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities. Chem. Proc. 2021, 3, 122. https://doi.org/10.3390/ecsoc-24-08393
Siben BA, Castro MJ, Faraoni MB. Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities. Chemistry Proceedings. 2021; 3(1):122. https://doi.org/10.3390/ecsoc-24-08393
Chicago/Turabian StyleSiben, Braian Alberto, María Julia Castro, and María Belén Faraoni. 2021. "Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities" Chemistry Proceedings 3, no. 1: 122. https://doi.org/10.3390/ecsoc-24-08393
APA StyleSiben, B. A., Castro, M. J., & Faraoni, M. B. (2021). Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities. Chemistry Proceedings, 3(1), 122. https://doi.org/10.3390/ecsoc-24-08393