
Direct Arylation-Based Synthesis of Carbazoles Using an Efficient Palladium Nanocatalyst under Microwave Irradiation †


Abstract
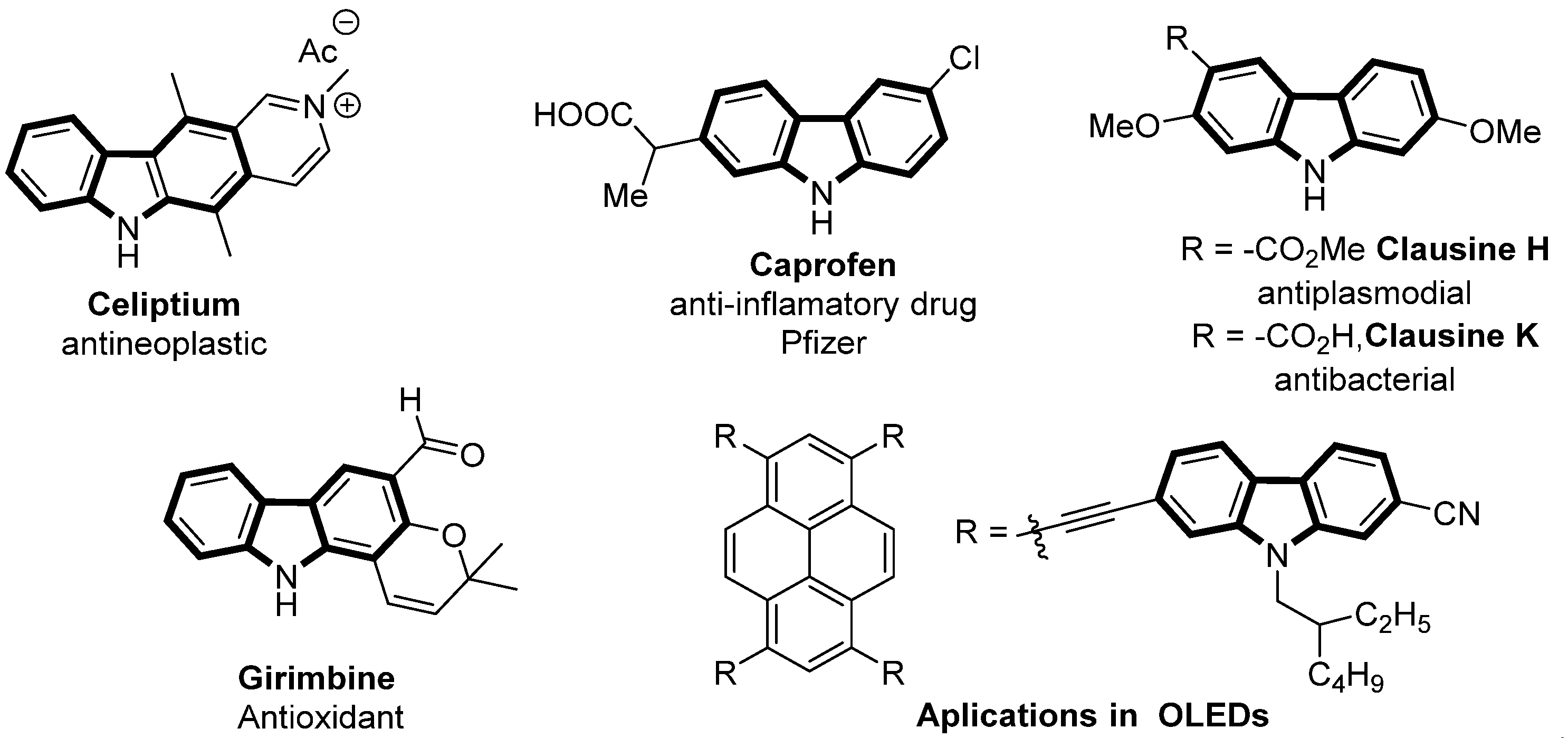
:1. Introduction
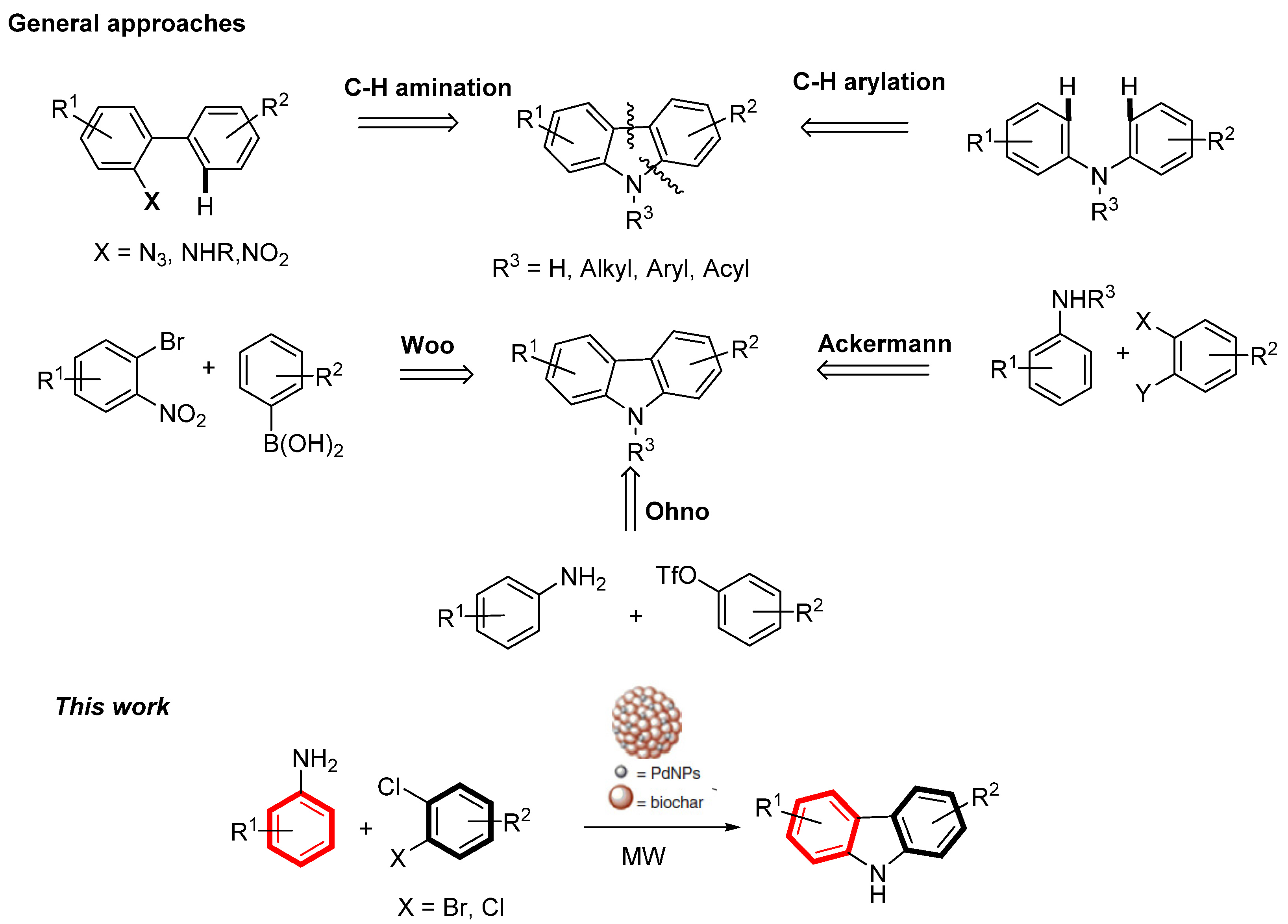
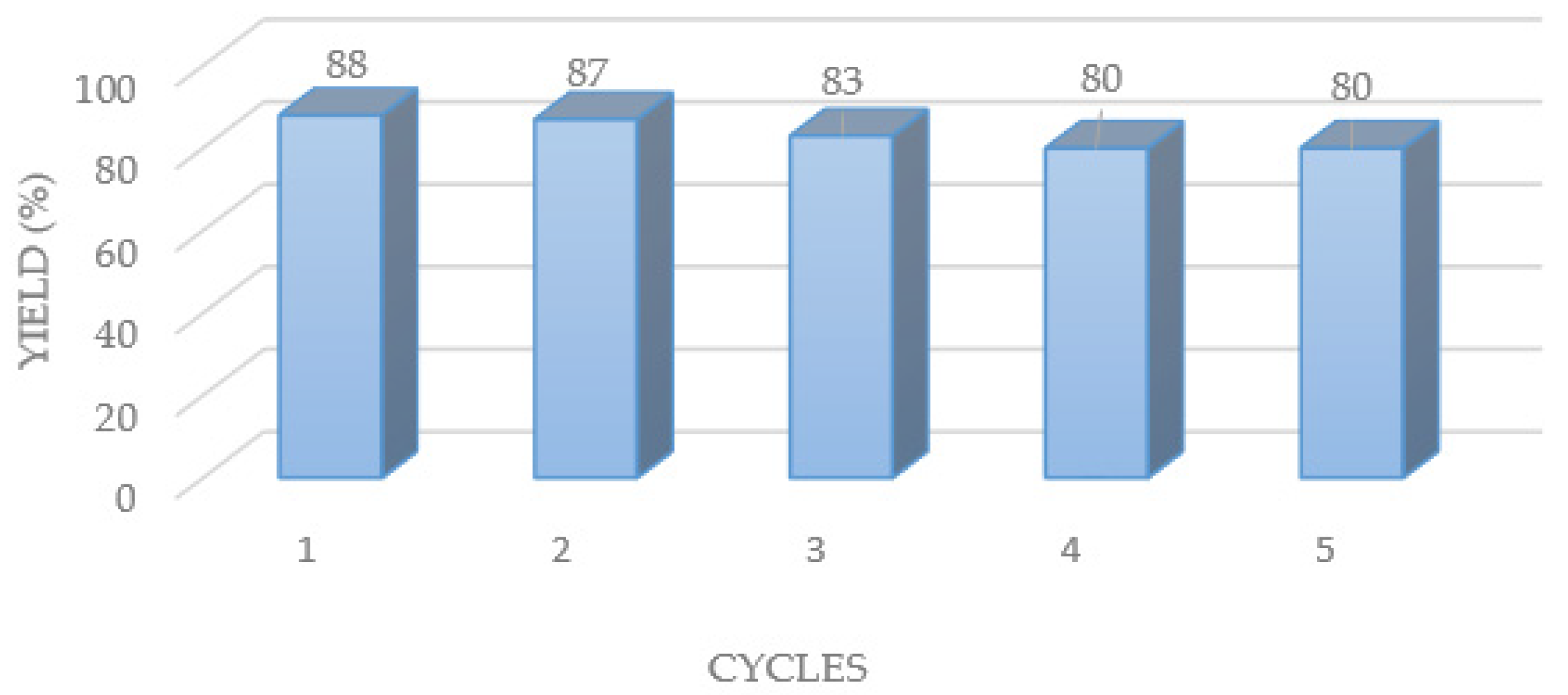
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Microwave Reactions
4.3. Synthesis and Characterization of the Catalyst

Funding
Acknowledgments
Conflicts of Interest
References
- Głuszynska, A. Biological potential of carbazole derivatives. Eur. J. Med. Chem. 2015, 94, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Oishi, S.; Watanabe, T.; Sawada, J.-I.; Asai, A.; Ohno, H.; Fujii, N. Kinesin Spindle Protein (KSP) Inhibitors with 2,3-Fused Indole Scaffolds. J. Med. Chem. 2010, 53, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Ledwon, P. Recent advances of donor-acceptor type carbazole-based molecules for light emitting applications. Org. Electron. 2019, 5, 1–15. [Google Scholar] [CrossRef]
- Campeau, C.L.; Fagnou, K. Palladium-catalyzed direct arylation of simple arenes in synthesis of biaryl molecules. Chem. Commun. 2006, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, S.N.; Nicolau, P.J. Recent advances in carbazole Syntheses. Adv. Heterocycl. Chem. 2019, 129, 1–88. [Google Scholar]
- Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Palladium-Catalyzed Direct Synthesis of Carbazoles via One-PotN-Arylation and Oxidative Biaryl Coupling: Synthesis and Mechanistic Study. J. Org. Chem. 2009, 74, 4720–4726. [Google Scholar] [CrossRef]
- Goo, D.-Y.; Woo, S.K. One-pot synthesis of carbazoles via tandem C–C cross-coupling and reductive amination. Org. Biomol. Chem. 2016, 14, 122–130. [Google Scholar] [CrossRef]
- Mpungose, P.P.; Vundla, Z.P.; Glenn, E.M.; Maguire, G.E.M.; Holger, B.; Friedrich, H.B. The Current Status of Heterogeneous Palladium Catalyzed Heck and Suzuki Cross-Coupling Reactions. Molecules 2018, 23, 1676–2000. [Google Scholar] [CrossRef]
- Kokel, A.; Schäfer, C.; Béla Török, B. Application of microwave-assisted heterogeneous catalysis in sustainable synthesis design. Green Chem. 2017, 19, 3729–3751. [Google Scholar] [CrossRef]
- Ackermann, L.; Althammer, A. Domino N-H/C-H bond activation: Palladium-catalyzed synthesis of annulated heterocycles using dichloro(hetero)arenes. Angew. Chem. 2007, 46, 1627. [Google Scholar] [CrossRef]
- Schölten, J.F.; Pijpers, A.P.; Hustings, M.L. Surface Characterization of Supported and Non-supported Hydrogenation Catalysts. Cold Reg. Sci. Eng. 1985, 27, 151–276. [Google Scholar]
- Takamatsu, K.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of Carbazoles by Copper-Catalyzed Intramolecular C–H/N–H Coupling. Org. Lett. 2014, 16, 2892–2895. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Mu, D.; Zhong, W.; Chen, H.; Yan, H. Amino-Directed Rh (III)-Catalyzed C-H Activation Leading to One-Pot Synthesis of N-H Carbazoles. Eur. J. 2013, 19, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Guerra, W.A.; Rossi, R.A.; Pierini, A.B.; Barolo, S.M. Transition-Metal-Free Synthesis of Carbazoles by Photostimulated Reactions of 2′-Halo[1,1′-biphenyl]-2-amines. J. Org. Chem. 2015, 80, 928–941. [Google Scholar] [CrossRef]
- Alcaide, P.; Almendros, J.M.; Alonso, M.T.; Quirós, G.P. Gold- or palladium-catalyzed allene carbocyclization/functionalization: Simple and efficient synthesis of carbazoles. Adv. Synth. Catal. 2011, 353, 1871–1876. [Google Scholar] [CrossRef]
- Stokes, B.J.; Jovanović, B.; Dong, H.; Richert, K.J.; Riell, R.D.; Driver, T.G. Rh2(II)-Catalyzed Synthesis of Carbazoles from Biaryl Azides. J. Org. Chem. 2009, 74, 3225–3228. [Google Scholar] [CrossRef]
- Laha, J.K.; Petrou, P.; Cuny, G.D. One-Pot Synthesis of α-Carbolines via Sequential Palladium-Catalyzed Aryl Amination and Intramolecular Arylation. J. Org. Chem. 2009, 74, 3152–3155. [Google Scholar] [CrossRef]
- Rasheed, S.; Rao, D.N.; Reddy, K.R.; Aravinda, S.; Vishwakarma, R.A.; Das, P. C–N bond formation via Cu-catalyzed cross-coupling with boronic acids leading to methyl carbazole-3-carboxylate: Synthesis of carbazole alkaloids. RSC Adv. 2014, 4, 4960–4969. [Google Scholar] [CrossRef]
- Khan, A.; Karim, R.; Dhimane, H.; Alam, S. Mild and Efficient Synthesis of Functionalized Carbazoles via a DBU-Assisted Sequence Involving Cu- and Pd-Catalyzed Coupling Reactions. Chem. Sel. 2019, 4, 6598–6605. [Google Scholar] [CrossRef]
- Mozingo, R. Palladium Catalysts. Org. Synth. 1955, 3, 685–686. [Google Scholar]
- Casoni, A.I.; Hoch, P.; Volpe, M.; Gutierrez, V.J. Catalytic conversion of furfural from pyrolysis of sunflower seed hulls for producing bio-based furfuryl alcohol. J. Clean. Prod. 2018, 178, 237–246. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | [Pd] | Temperature (°C) | Yield (%) Thermal a,c | Yield (%) MW b,c |
|---|---|---|---|---|
| 1 | --- | 130 | --- | --- |
| 2 | PdCl2(PPh3)2 | 130 | 6 | trace |
| 3 | Pd2(dba)3 | 130 | --- | trace |
| 4 | PdCl2(MeCN)2 | 130 | 8 | -- |
| 5 | Pd(PPh3)4 | 130 | --- | --- |
| 6 | PdCl2(MeCN)2 | 160 | --- | --- |
| 7 | PdNPs/BC | 130 | 11 | 19 |
| 8 | PdNPs/PbO | 130 | -- | trace |
| 9 | PdNPs/Al2O3 | 130 | trace | 9 |
| 10 | PdNPs/BC | 180 | 18 | 43 |
| 11 | PdNPs/Al2O3 | 180 | 10 | 12 |

| Entry | PdNPs/BC (mol%) | Solvent | Base | Yield (%) b |
|---|---|---|---|---|
| 1 | 2.5 | NMP | K3PO4 | 16 |
| 2 | 10 | NMP | K3PO4 | 57 |
| 3 | 15 | NMP | K3PO4 | 76 |
| 4 | 15 | DMF | K3PO4 | 71 |
| 5 | 15 | DMAc | K3PO4 | 73 |
| 6 | 15 | DMSO | K3PO4 | 85 |
| 7 | 15 | DMSO | KOAc | 88 |
| 8 | 15 | DMSO | K2CO3 | 84 |
| 9 | 15 | DMSO | Cs2CO3 | 82 |
| 10 | 15 | DMSO | Na2CO3 | 81 |
| 11 | 15 | DMSO | NaOt-Bu | 61 |

| Entry | Aniline | 1,2-Dihalobenzene | 9H-Carbazole | Yield (%) b |
|---|---|---|---|---|
| 1 |  |  |  | 88 |
| 2 | 1a |  | 3 | 81 |
| 3 | 1a |  | 3 | 90 [c] |

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steingruber, H.S.; Mendioroz, P.; Volpe, M.A.; Gerbino, D.C. Direct Arylation-Based Synthesis of Carbazoles Using an Efficient Palladium Nanocatalyst under Microwave Irradiation. Chem. Proc. 2021, 3, 70. https://doi.org/10.3390/ecsoc-24-08314
Steingruber HS, Mendioroz P, Volpe MA, Gerbino DC. Direct Arylation-Based Synthesis of Carbazoles Using an Efficient Palladium Nanocatalyst under Microwave Irradiation. Chemistry Proceedings. 2021; 3(1):70. https://doi.org/10.3390/ecsoc-24-08314
Chicago/Turabian StyleSteingruber, H. Sebastián, Pamela Mendioroz, María A. Volpe, and Darío C. Gerbino. 2021. "Direct Arylation-Based Synthesis of Carbazoles Using an Efficient Palladium Nanocatalyst under Microwave Irradiation" Chemistry Proceedings 3, no. 1: 70. https://doi.org/10.3390/ecsoc-24-08314
APA StyleSteingruber, H. S., Mendioroz, P., Volpe, M. A., & Gerbino, D. C. (2021). Direct Arylation-Based Synthesis of Carbazoles Using an Efficient Palladium Nanocatalyst under Microwave Irradiation. Chemistry Proceedings, 3(1), 70. https://doi.org/10.3390/ecsoc-24-08314