
Green Synthesis of Symmetric Dimaleamic Acids from Dianilines and Maleic Anhydride: Behind New Bidentate Ligands for MOFs †

 , and
, and 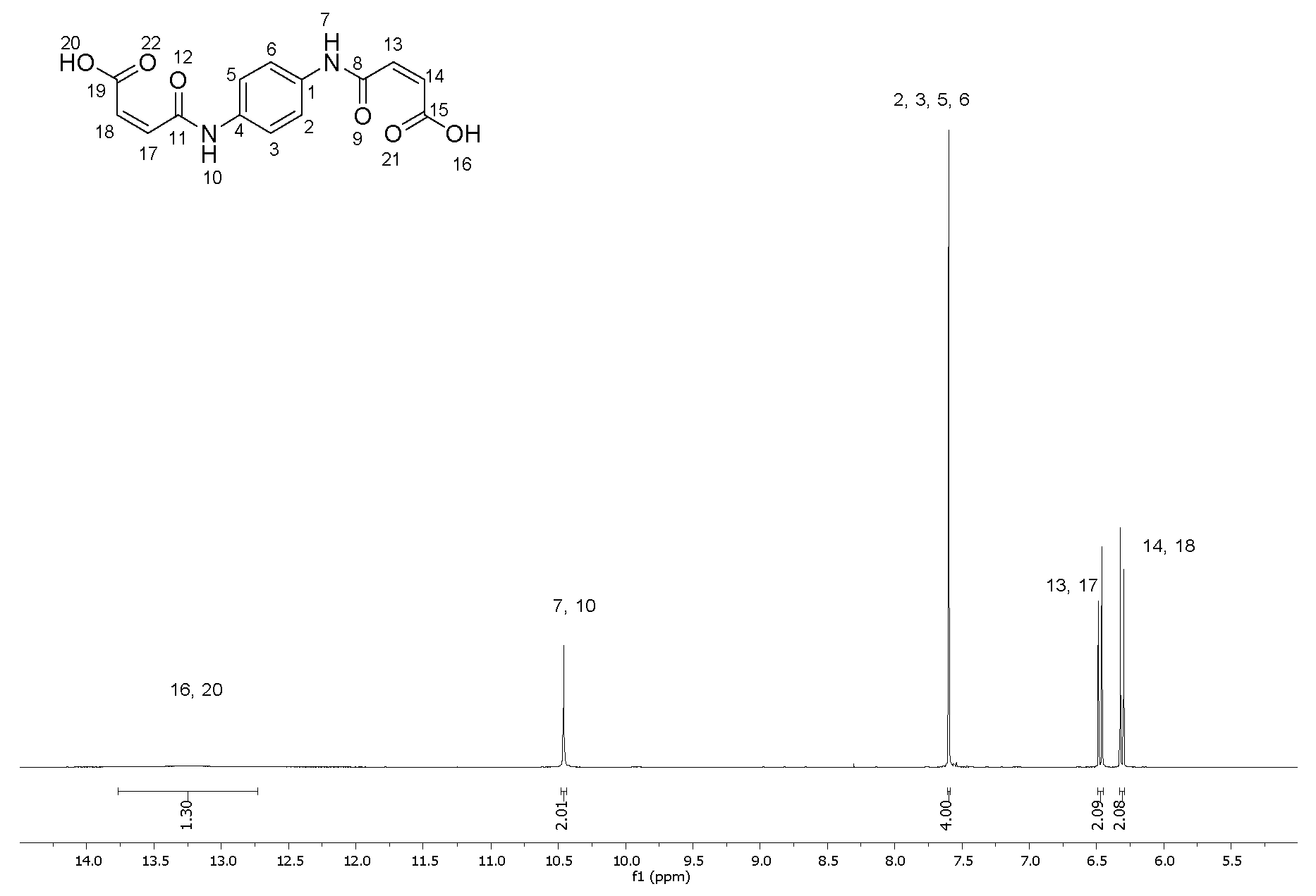{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
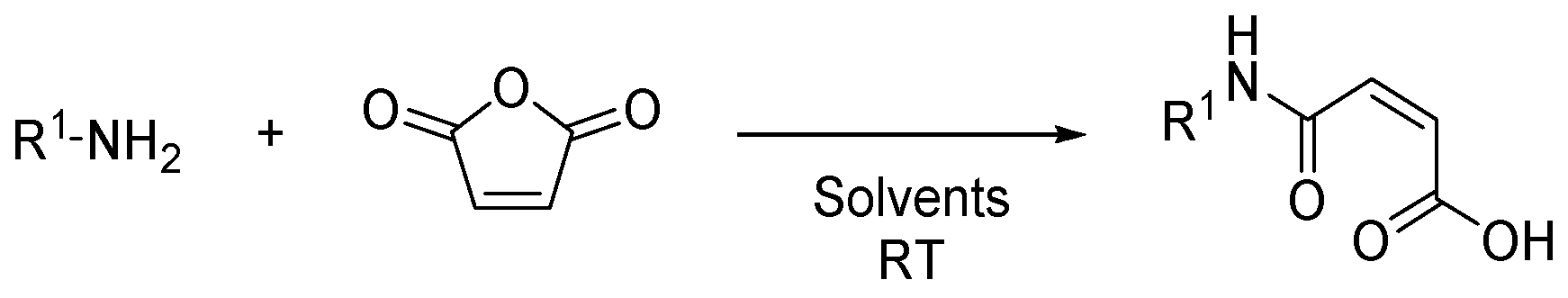
:1. Introduction
2. Results and Discussion
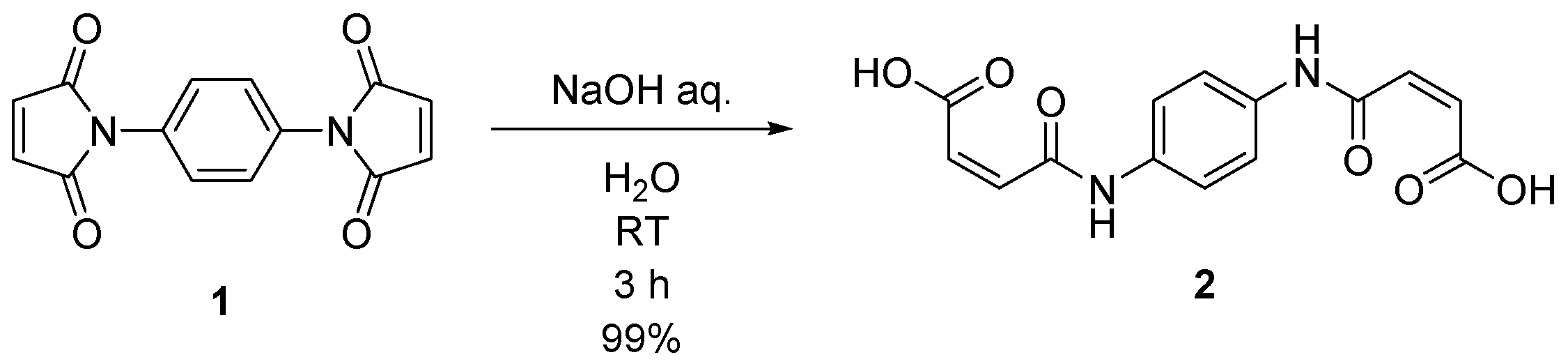
2.1. Synthesis of (2E,2′E)-4,4′-(1,4-phenylenebis(azanediyl))bis(4-oxobut-2-enoic acid) (2)
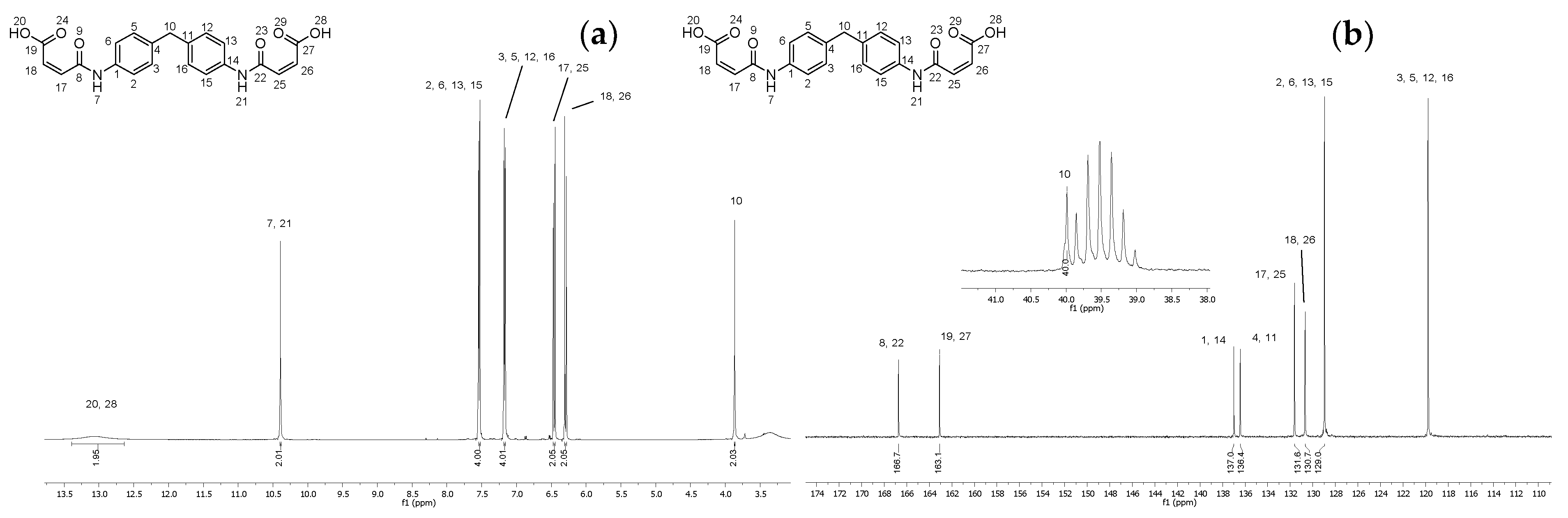
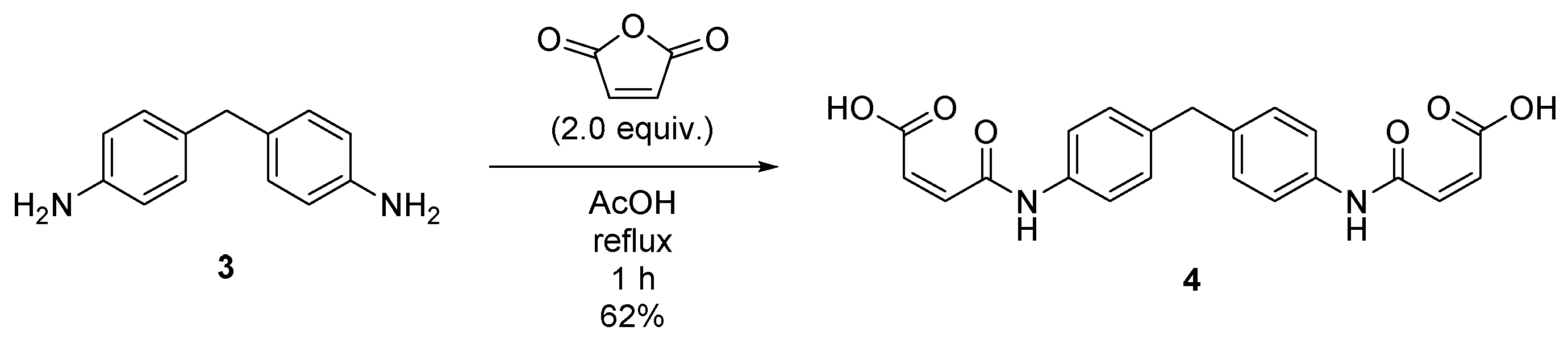
2.2. Synthesis of (2Z,2′Z)-4,4′-((methylenebis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (4)
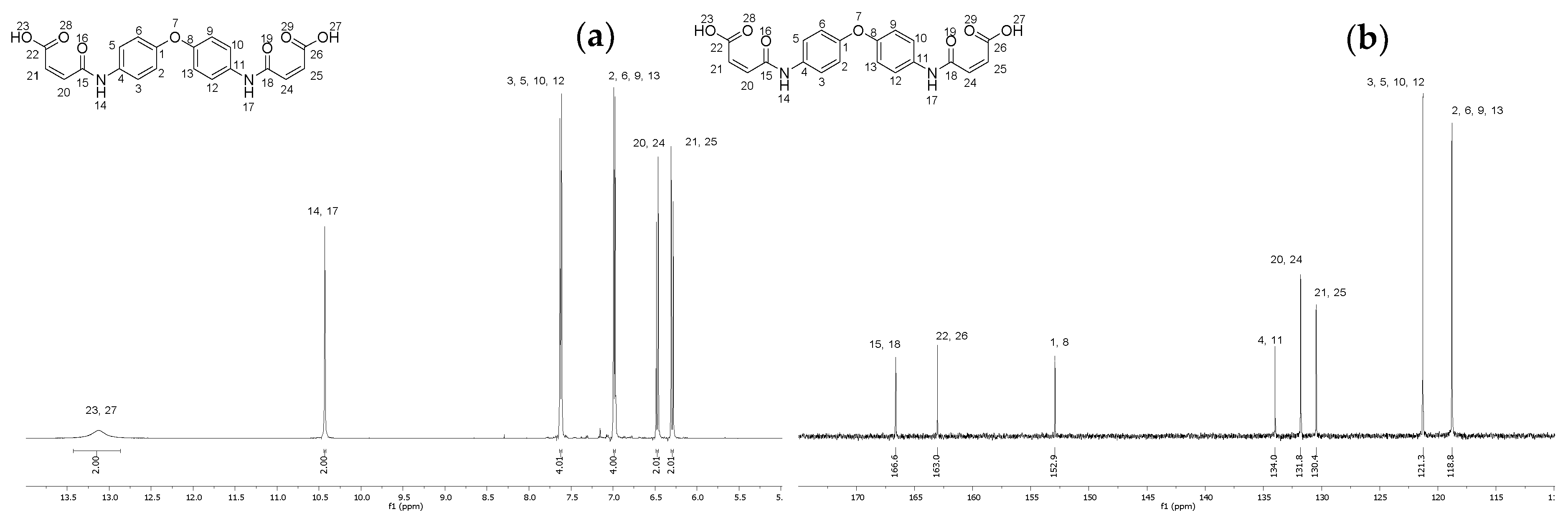
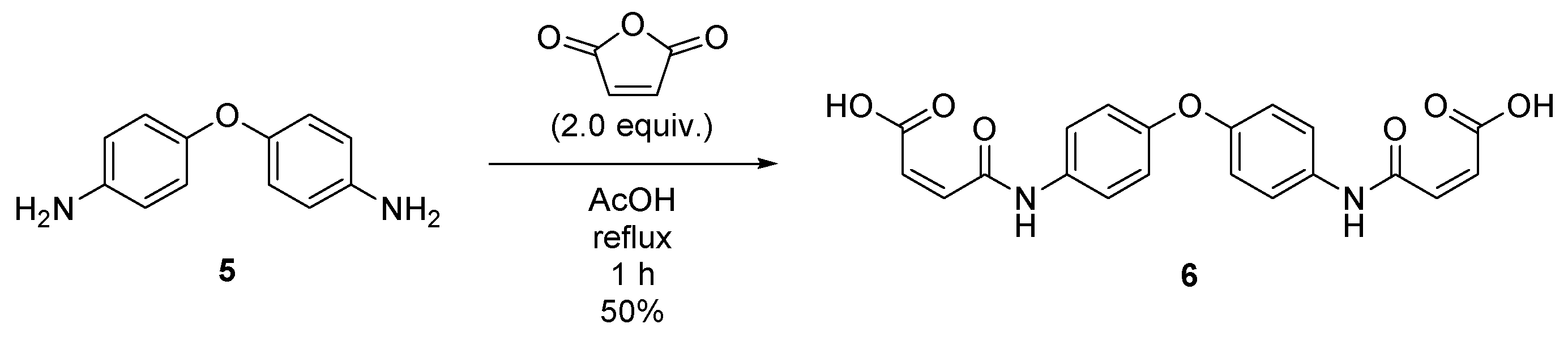
2.3. Synthesis of (2Z,2′Z)-4,4′-((oxybis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (6)
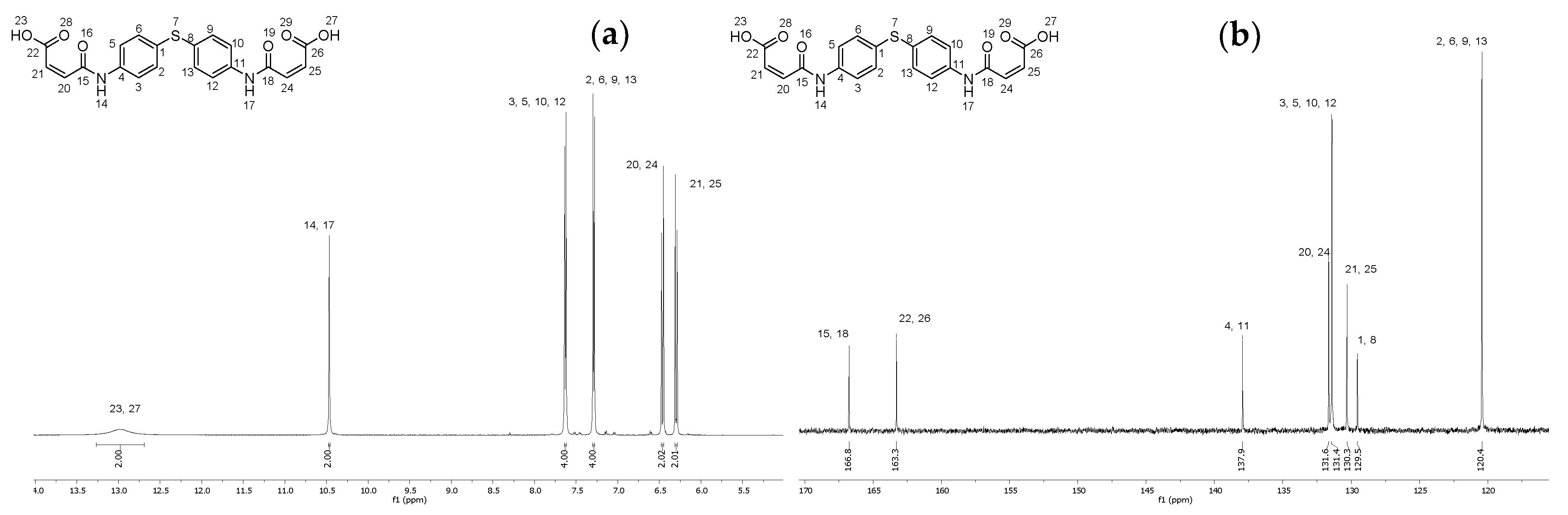
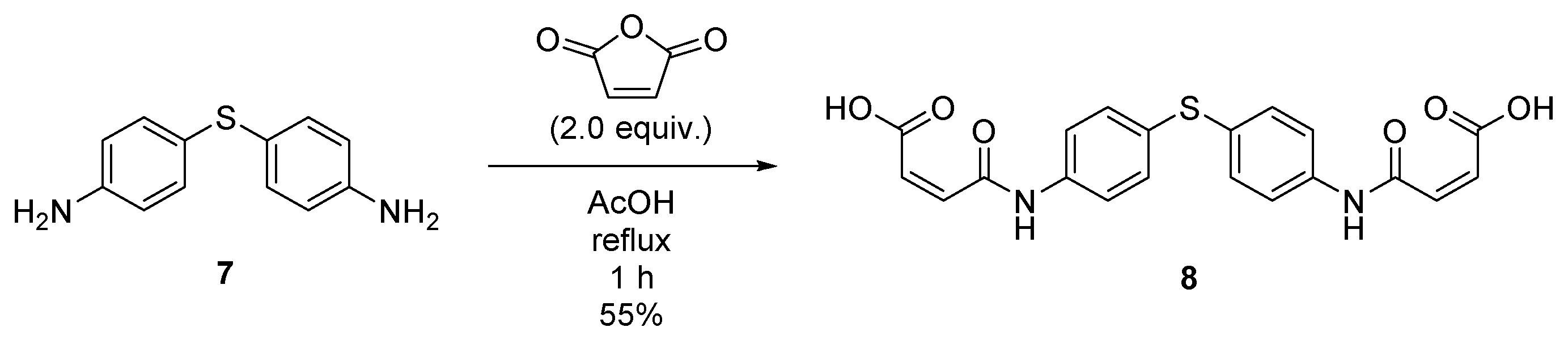
2.4. Synthesis of (2Z,2′Z)-4,4′-((thiobis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (8)
2.5. Synthesis of (2Z,2′Z)-4,4′-((sulfonylbis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (10)
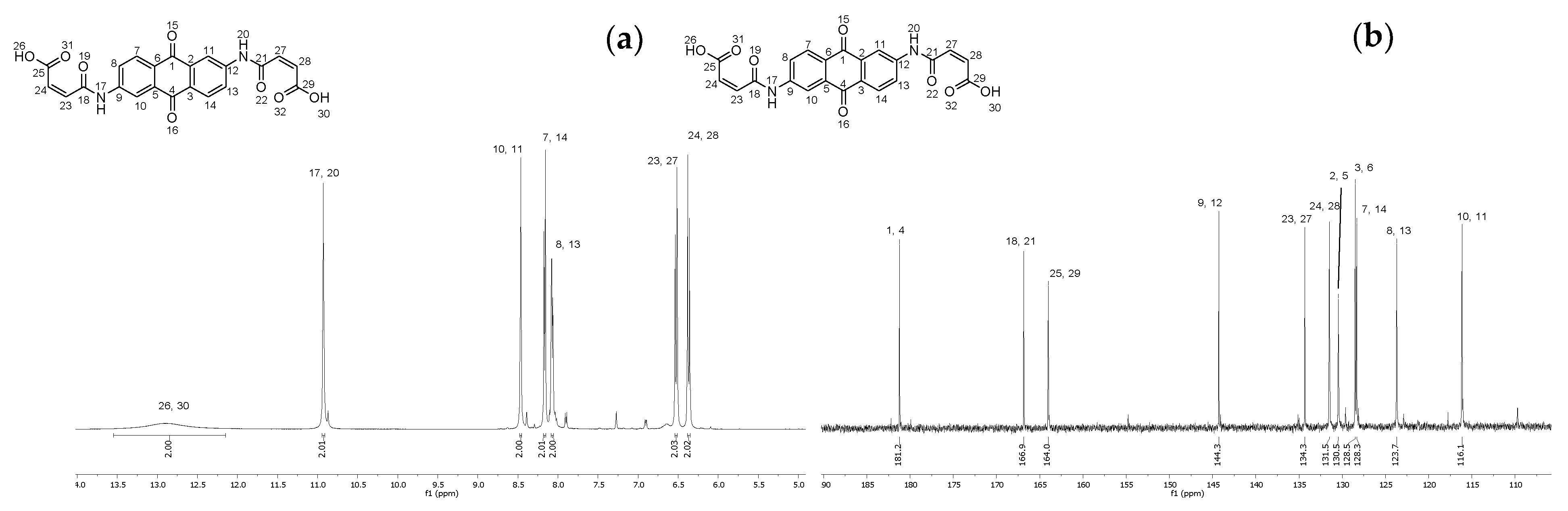
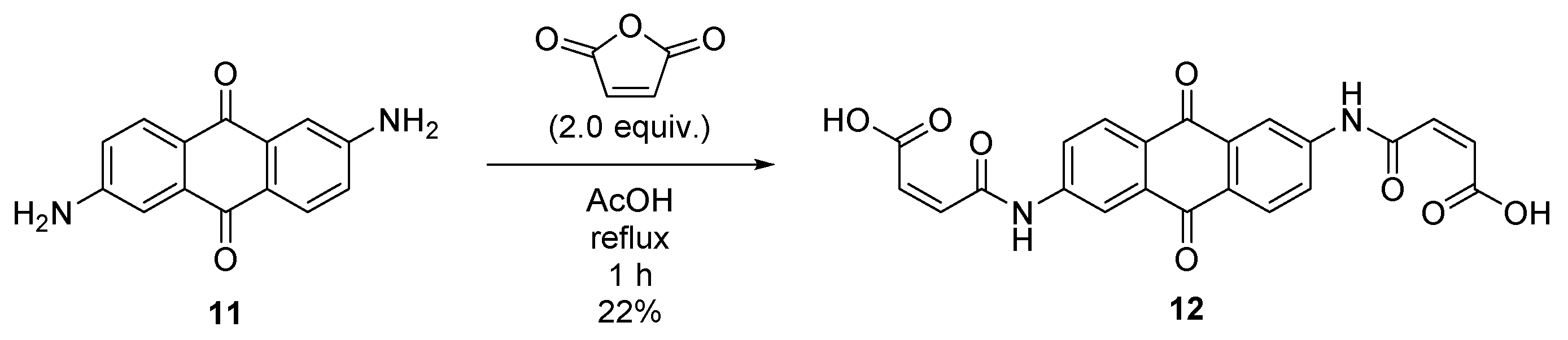
2.6. Synthesis of (2Z,2′Z)-4,4′-((9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl))bis(4-oxobut-2-enoic acid) (12)
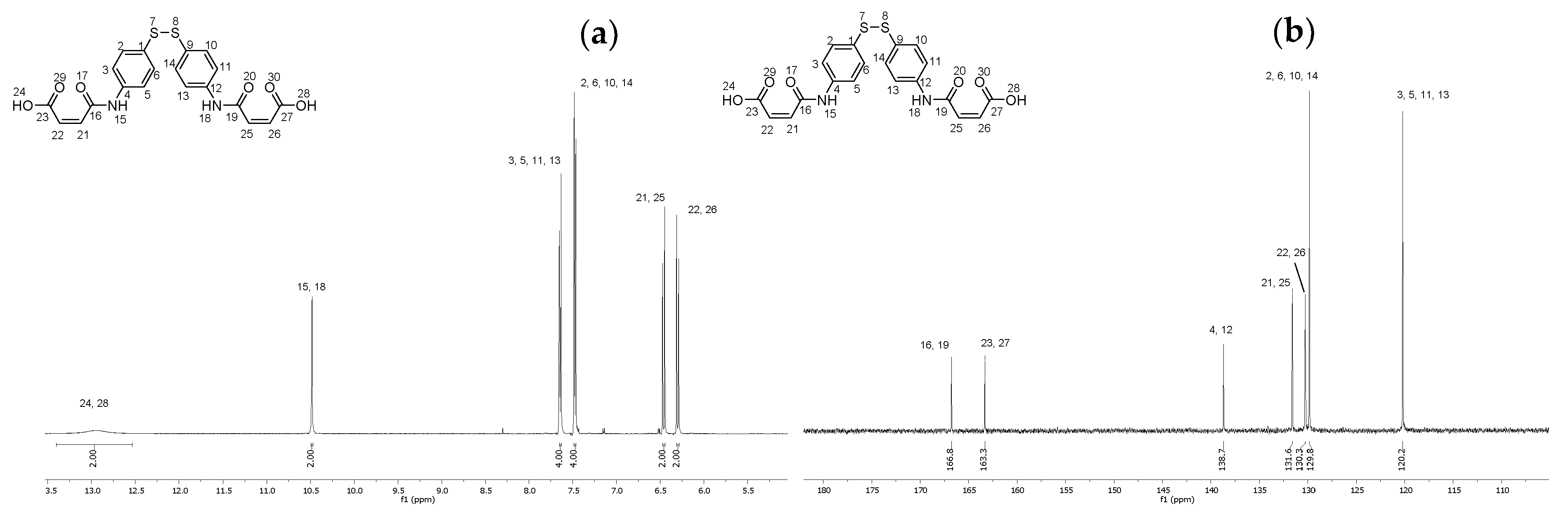
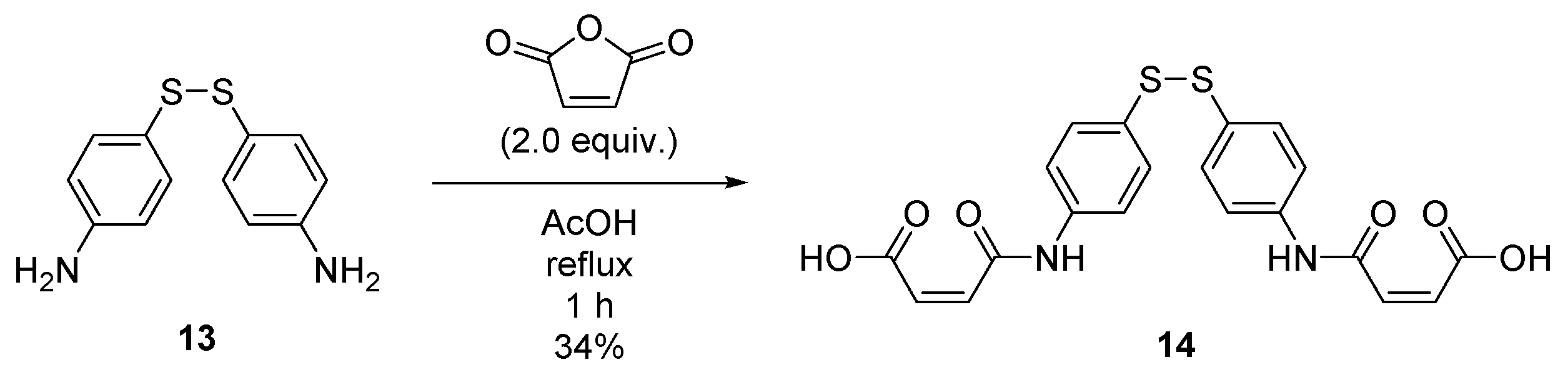
2.7. Synthesis of (2Z,2′Z)-4,4′-((disulfanediylbis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (14)
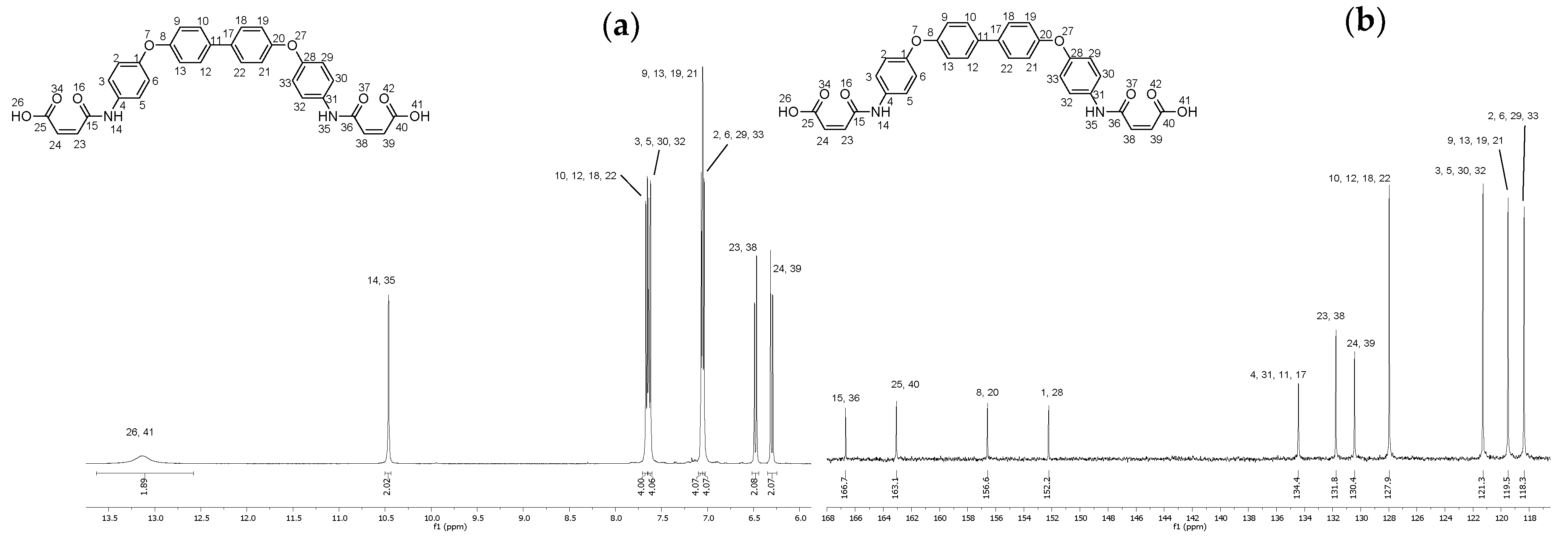
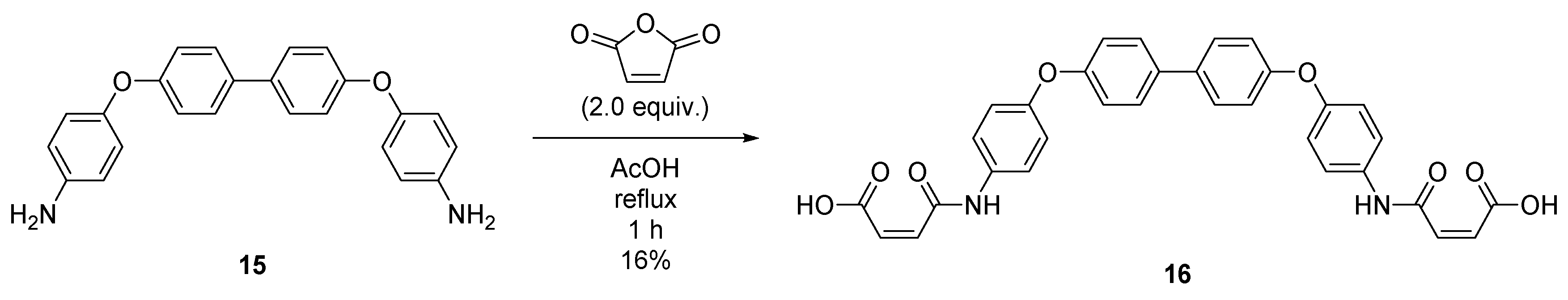
2.8. Synthesis of (2Z,2′Z)-4,4′-((([1,1′-biphenyl]-4,4′-diylbis(oxy))bis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (16)
3. Conclusions
4. Experimental Section
4.1. General Information, Instrumentation and Chemicals
4.2. Synthesis of (2E,2′E)-4,4′-(1,4-phenylenebis(azanediyl))bis(4-oxobut-2-enoic acid) (2)
4.3. Synthesis of (2Z,2′Z)-4,4′-((methylenebis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (4)
4.4. Synthesis of (2Z,2′Z)-4,4′-((oxybis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (6)
4.5. Synthesis of (2Z,2′Z)-4,4′-((thiobis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (8)
4.6. Synthesis of (2Z,2′Z)-4,4′-((sulfonylbis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (10)
4.7. Synthesis of (2Z,2′Z)-4,4′-((9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl))bis(4-oxobut-2-enoic acid) (12)
4.8. Synthesis of (2Z,2′Z)-4,4′-((disulfanediylbis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (14)
4.9. Synthesis of (2Z,2′Z)-4,4′-((([1,1′-biphenyl]-4,4′-diylbis(oxy))bis(4,1-phenylene))bis(azanediyl))bis(4-oxobut-2-enoic acid) (16)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tabăcăru, A.; Pettinari, C.; Galli, S. Coordination polymers and metal-organic frameworks built up with poly(tetrazolate) ligands. Coord. Chem. Rev. 2018, 372, 1–30. [Google Scholar] [CrossRef]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal-organic frameworks: Functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef] [PubMed]
- Lytvynenko, A.S.; Kolotilov, S.V. Electrochemically active coordination polymers: A review. Theor. Exp. Chem. 2016, 52, 197–211. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal–organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef]
- Giménez-Marquéz, M.; Hidalgo, T.; Serre, C.; Horcajada, P. Nanostructured metal-organic frameworks and their bio-related applications. Coord. Chem. Rev. 2016, 307, 342–360. [Google Scholar] [CrossRef]
- Robin, A.Y.; Fromm, K.M. Coordination polymer networks with O- and N-donors: What they are, why and how they are made. Coord. Chem. Rev. 2006, 250, 2127–2157. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design Analysis and Application, 6th ed.; Royal Society of Chemistry: Cambridge, UK, 2009; pp. 172–173. [Google Scholar]
- Huang, Z.; Song, H.-B.; Du, M.; Chen, S.-T.; Bu, X.-H. Coordination Polymers Assembled from Angular Dipyridyl Ligands and CuII, CdII, CoII Salts: Crystal Structures and Properties. Inorg. Chem. 2004, 43, 931–944. [Google Scholar] [CrossRef]
- Niu, L.-E.; Yang, Y.; Qi, X.-H.; Liang, X. Synthesis and spectroscopic studies of Lanthanide(III) complexes with (Z)-4-oxo-4-(phenylamino) but-2-enoic acid. Synth. React. Inorg. Met. Org. Chem. 2009, 39, 1–5. [Google Scholar] [CrossRef]
- Li, M.; Liang, Y.-D.; Wu, Y.-X.; Li, K.-S. Synergistic effect of complexes of ethylenediamine double maleamic acid radical and lanthanum (III) with pentaerythritol on the thermal stability of poly(vinyl chloride). Polym. Degrad. Stabil. 2017, 140, 176–193. [Google Scholar] [CrossRef]
- Lazarou, K.N.; Psycharis, V.; Perlepes, S.P.; Raptopoulou, C.P. Complexes derived from the copper(II) perchlorate/maleamic acid/2,20-bipyridine and copper(II) perchlorate/maleic acid/2,20-bipyridine reaction systems: Synthetic, reactivity, structural and spectroscopic studies. Polyhedron 2009, 28, 1085–1096. [Google Scholar] [CrossRef]
- Lazarou, K.N.; Boudalis, A.K.; Perlepes, S.P.; Terzis, A.; Raptopoulou, C.P. Maleamate(–1) and maleate(–2) copper(II)–2,2′-bipyridine complexes: Synthesis, reactivity and structural and physical studies Eur. J. Inorg. Chem. 2009, 4554–4563. [Google Scholar] [CrossRef]
- Li, B.; Shen, Y.; Wu, H.; Wu, X.; Yuan, L.; Ji, Q. Design, synthesis and biological evaluation of novel 3,4-dihydro- 2(1H)-quinolinone derivatives as potential chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 2020, 195, 112278. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, G. Photoinduced decarboxylative amino-fluoroalkylation of maleic anhydride. Chem. Eur. J. 2020, 26, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Badru, R.; Singh, B. Triethylamine-Catalyzed Synthesis of Oxazepine from Maleamic Acids. J. Heterocycl. Chem. 2015, 52, 635–640. [Google Scholar] [CrossRef]
- Guevara, J.A.; Trujillo, J.G.; Quintana, D.; Jiménez, H.A.; Arellano, M.G.; Bahena, J.R.; Tamay, F.; Ciprés, F.J. Acetylcholinesterase inhibition by products generated in situ from the transformation of N-arylisomaleimides. Med. Chem. Res. 2018, 27, 989–1003. [Google Scholar] [CrossRef]
- Long, Y.L.; Jiang, W.; Wu, X.; Wang, Q. Grafted copolymerization of N-phenylmaleimide and styrene in porous polyvinyl chloride particles suspended in aqueous solution. Des. Monomers Polym. 2019, 22, 66–78. [Google Scholar] [CrossRef]
- Kumar, P.P.; Devi, B.R.; Dubey, P.K.; Mohiuddin, S.M.G. PEG-600 mediated simple, efficient and eco-friendly synthesis of N-substituted imides and chemoselective C=C reduction. Green Chem. Lett. Rev. 2011, 4, 341–348. [Google Scholar] [CrossRef]
- Chen, Y.; Tssao, K.; De Francesco, E.; Keillor, J.W. Ring substituent effects on the thiol addition and hydrolysis reactions of N-arylmaleimides. J. Org. Chem. 2015, 80, 12182–12192. [Google Scholar] [CrossRef]
- Kiselev, V.D.; Kashaeva, H.A.; Potapova, L.N.; Kornilov, D.A.; Latypova, L.I.; Konovalov, A.I. Hydrophobic acceleration in the Diels—Alder reaction of 9-hydroxymethylanthracene with N-phenylmaleimide. Russ. Chem. Bull. Int. Ed. 2016, 65, 2202–2205. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Reyes, J.C.; Sosa-Juárez, J.L.; Sánchez-Serratos, M.; Islas-Jácome, P.; Gutiérrez-Carrillo, A.; Méndez, F.; Suárez-Moreno, G.V.; Islas-Jácome, A.; González-Zamora, E. Green Synthesis of Symmetric Dimaleamic Acids from Dianilines and Maleic Anhydride: Behind New Bidentate Ligands for MOFs. Chem. Proc. 2021, 3, 92. https://doi.org/10.3390/ecsoc-24-08379
Flores-Reyes JC, Sosa-Juárez JL, Sánchez-Serratos M, Islas-Jácome P, Gutiérrez-Carrillo A, Méndez F, Suárez-Moreno GV, Islas-Jácome A, González-Zamora E. Green Synthesis of Symmetric Dimaleamic Acids from Dianilines and Maleic Anhydride: Behind New Bidentate Ligands for MOFs. Chemistry Proceedings. 2021; 3(1):92. https://doi.org/10.3390/ecsoc-24-08379
Chicago/Turabian StyleFlores-Reyes, Julio C., José L. Sosa-Juárez, Mayra Sánchez-Serratos, Perla Islas-Jácome, Atilano Gutiérrez-Carrillo, Francisco Méndez, Galdina V. Suárez-Moreno, Alejandro Islas-Jácome, and Eduardo González-Zamora. 2021. "Green Synthesis of Symmetric Dimaleamic Acids from Dianilines and Maleic Anhydride: Behind New Bidentate Ligands for MOFs" Chemistry Proceedings 3, no. 1: 92. https://doi.org/10.3390/ecsoc-24-08379
APA StyleFlores-Reyes, J. C., Sosa-Juárez, J. L., Sánchez-Serratos, M., Islas-Jácome, P., Gutiérrez-Carrillo, A., Méndez, F., Suárez-Moreno, G. V., Islas-Jácome, A., & González-Zamora, E. (2021). Green Synthesis of Symmetric Dimaleamic Acids from Dianilines and Maleic Anhydride: Behind New Bidentate Ligands for MOFs. Chemistry Proceedings, 3(1), 92. https://doi.org/10.3390/ecsoc-24-08379