
VirusLab: A Tool for Customized SARS-CoV-2 Data Analysis



Abstract
:1. Introduction
2. Related Work
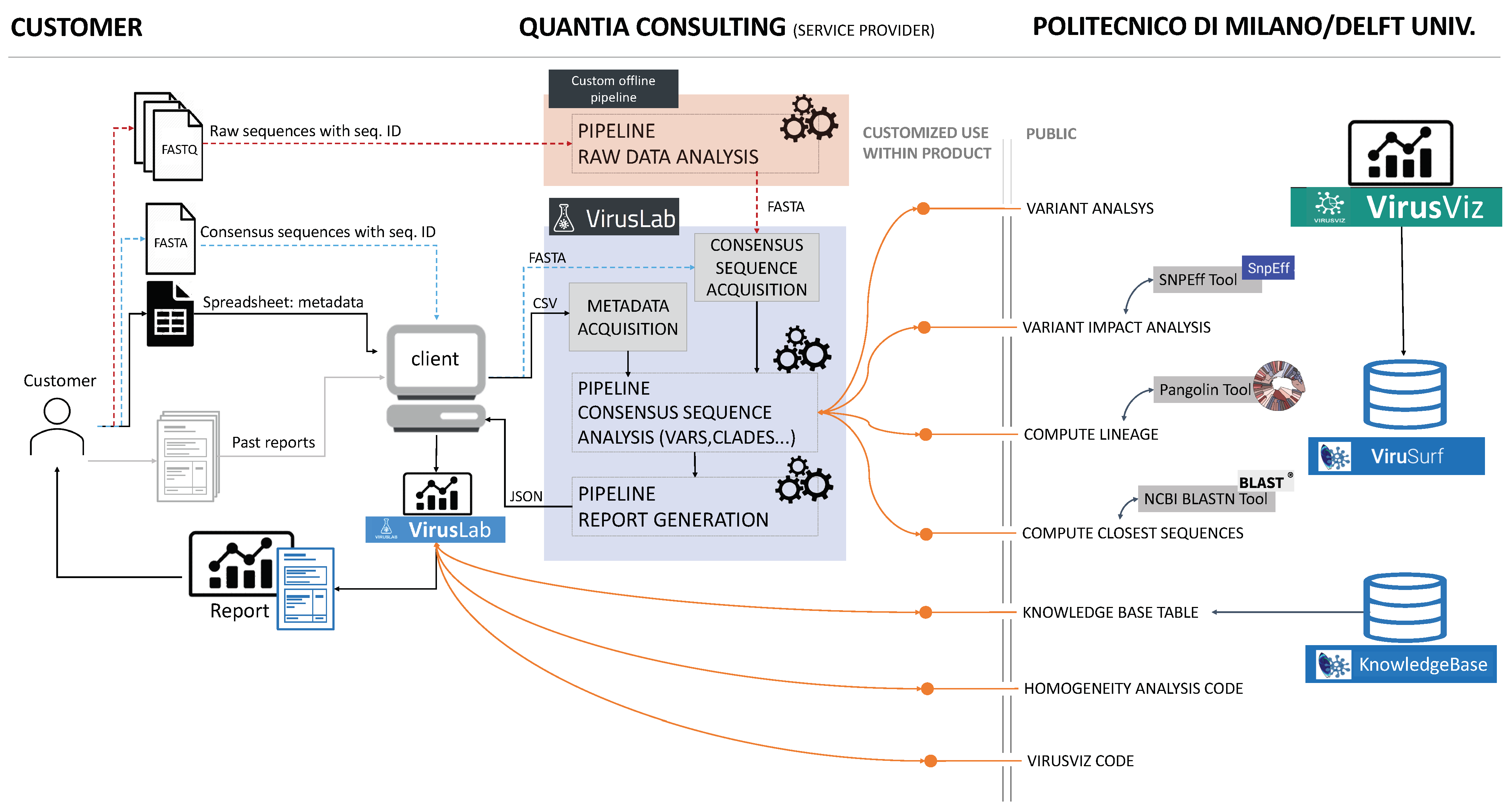
3. System Overview
3.1. Open-Source Component
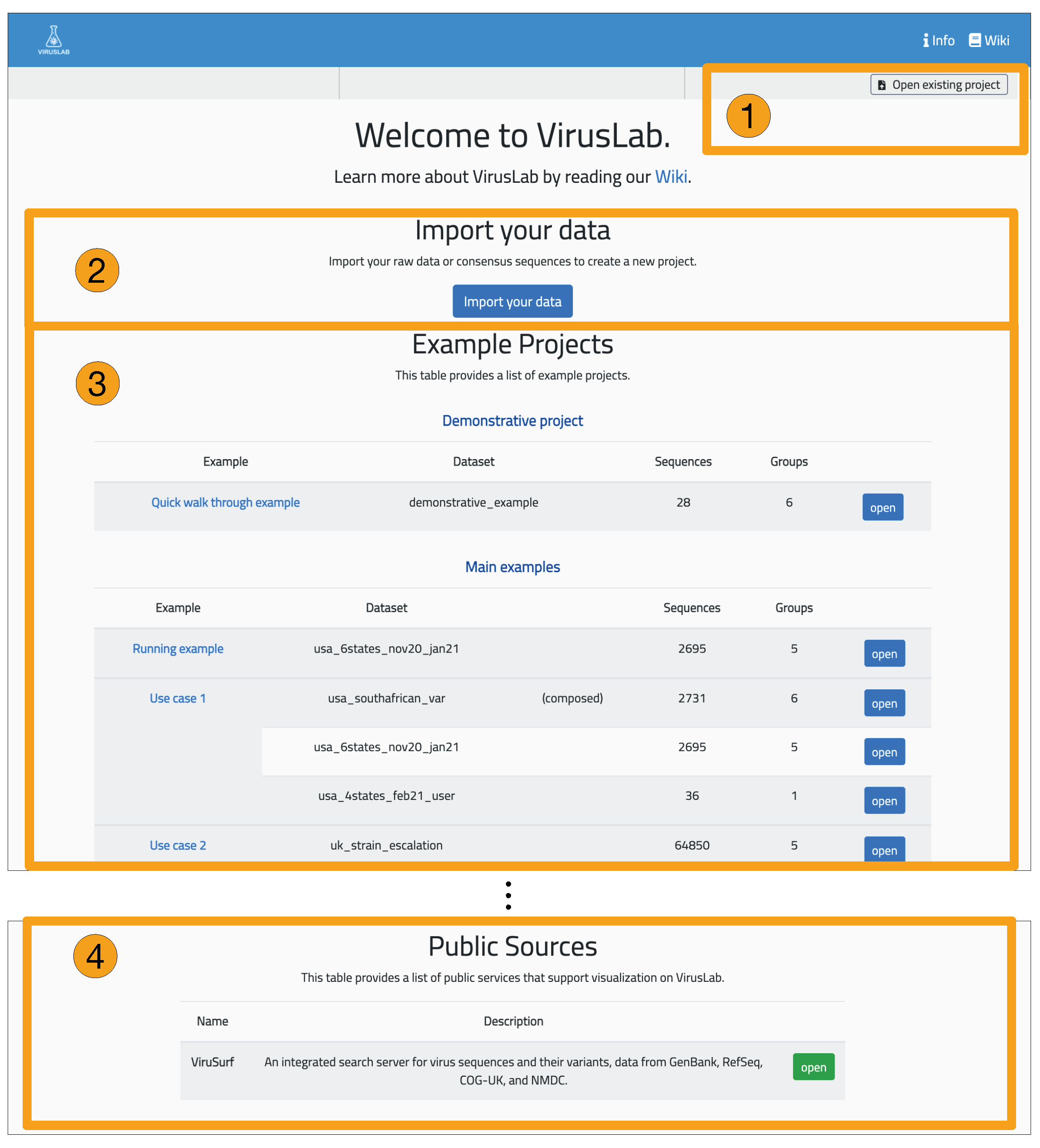
3.2. User Interaction
3.3. VirusLab Pipelines
4. Methods
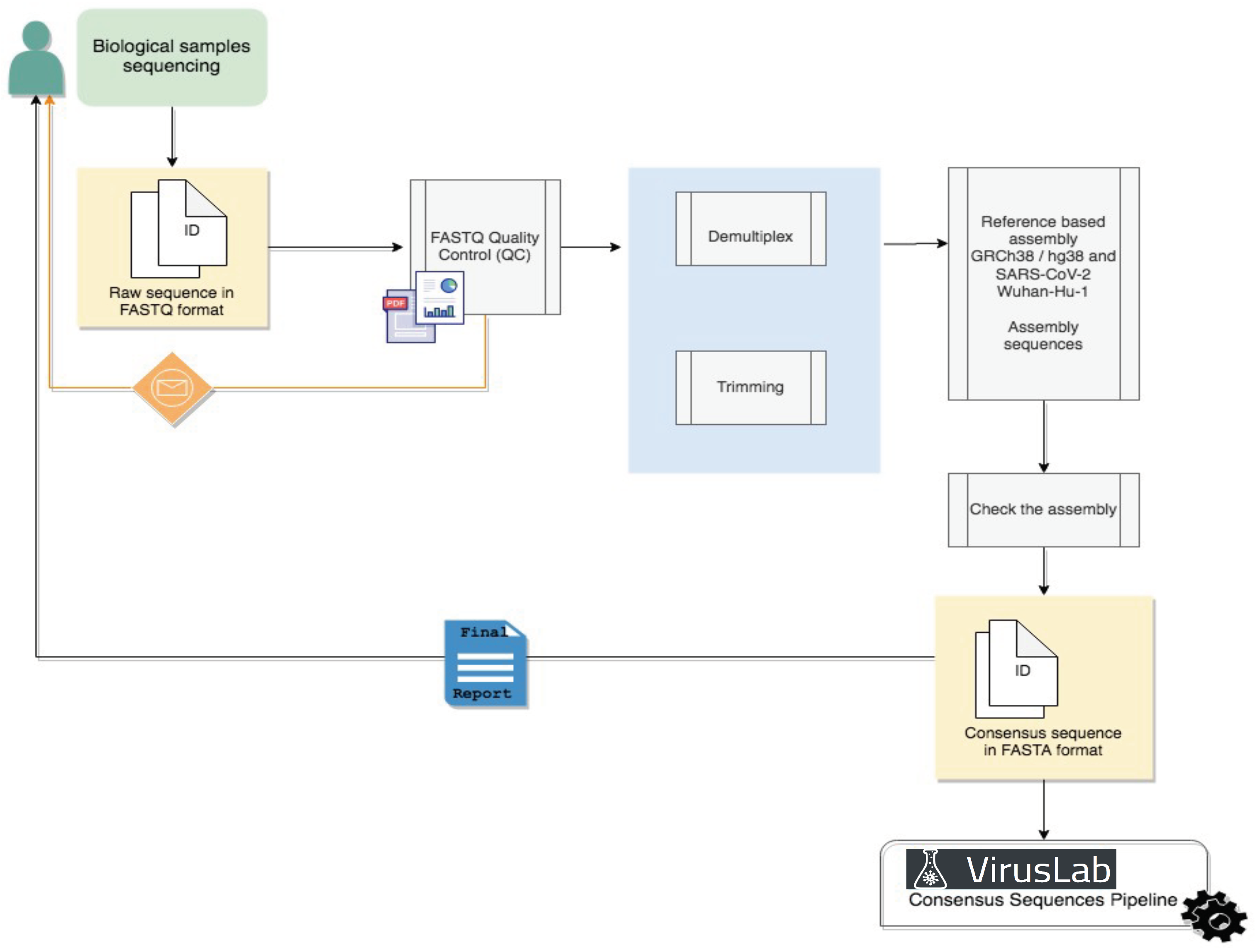
4.1. Raw Data Analysis
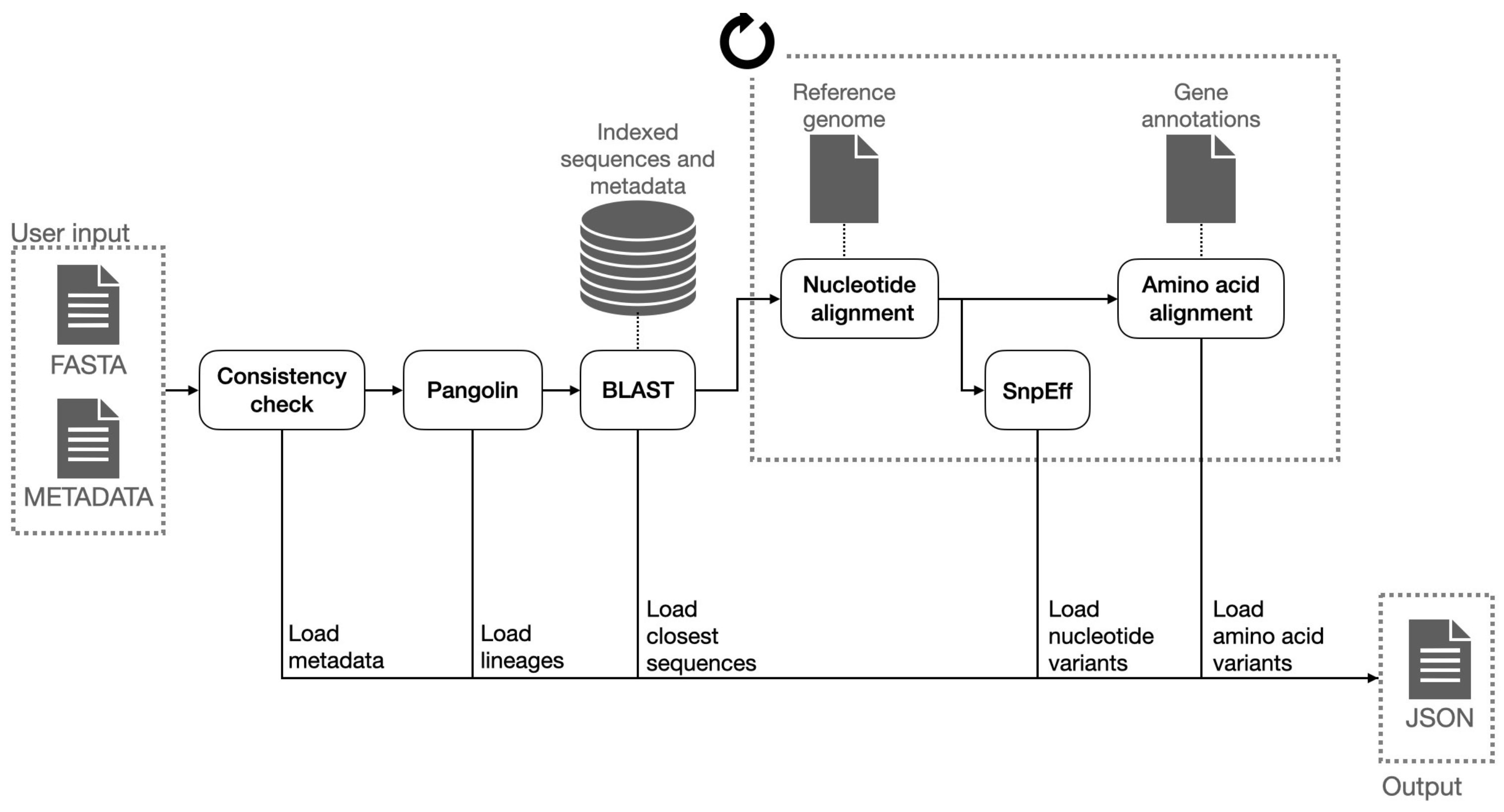
4.2. Consensus Sequence Pipeline
- Lineage assignment for each sequence (for Sars-CoV-2 sequences). This operation is performed using Pangolin, a third-party open source software, which is considered the de facto standard tool for lineage assignment of Sars-CoV-2 sequences;
- Nucleotide variants with respect reference sequence. In order to do so, input sequences are aligned to the reference genome of the related species; the alignment is performed with a variant of the Needleman-Wunsh algorithm [30] with affine gap penality, designed to favor gaps at ends of the sequences, as this regions are usually less conserved and more subject to sequencing errors. The aligned sequence is finally scanned and nucleotide substitutions, insertions and deletions are identified. Deletions at the very end of the input sequences are removed from the output, as those are related with incomplete sequencing rather than with actual genetic differences;
- Putative impact of each nucleotide variant. Nucleotide variants are analyzed with SnpEff and annotated with their putative effect. This tool associates to each variant the indication of its position with respect to coding and structural regions of the genome and the impact on the translated protein (e.g., synonymous mutation, frameshift mutation, etc.);
- Amino acid variants with respect to the reference sequence. The list of coding sequences is retrieved from a collection of structural annotations. Each user input nucleotide sequence is converted into the corresponding amino acid sequences (one for each coding region) and each of them is aligned with the corresponding amino acid reference sequences, using a global alignment algorithm (Needleman-Wunsch); the amino acid variants are inferred from the pairwise alignments;
- List of the N (e.g., 20) most similar nucleotide sequences found in ViruSurf database and associated metadata. those sequences are retrieved using BLASTN and sorted on the identity percentage and the e-value (i.e., the number of expected hits of similar quality that could be found just by chance).
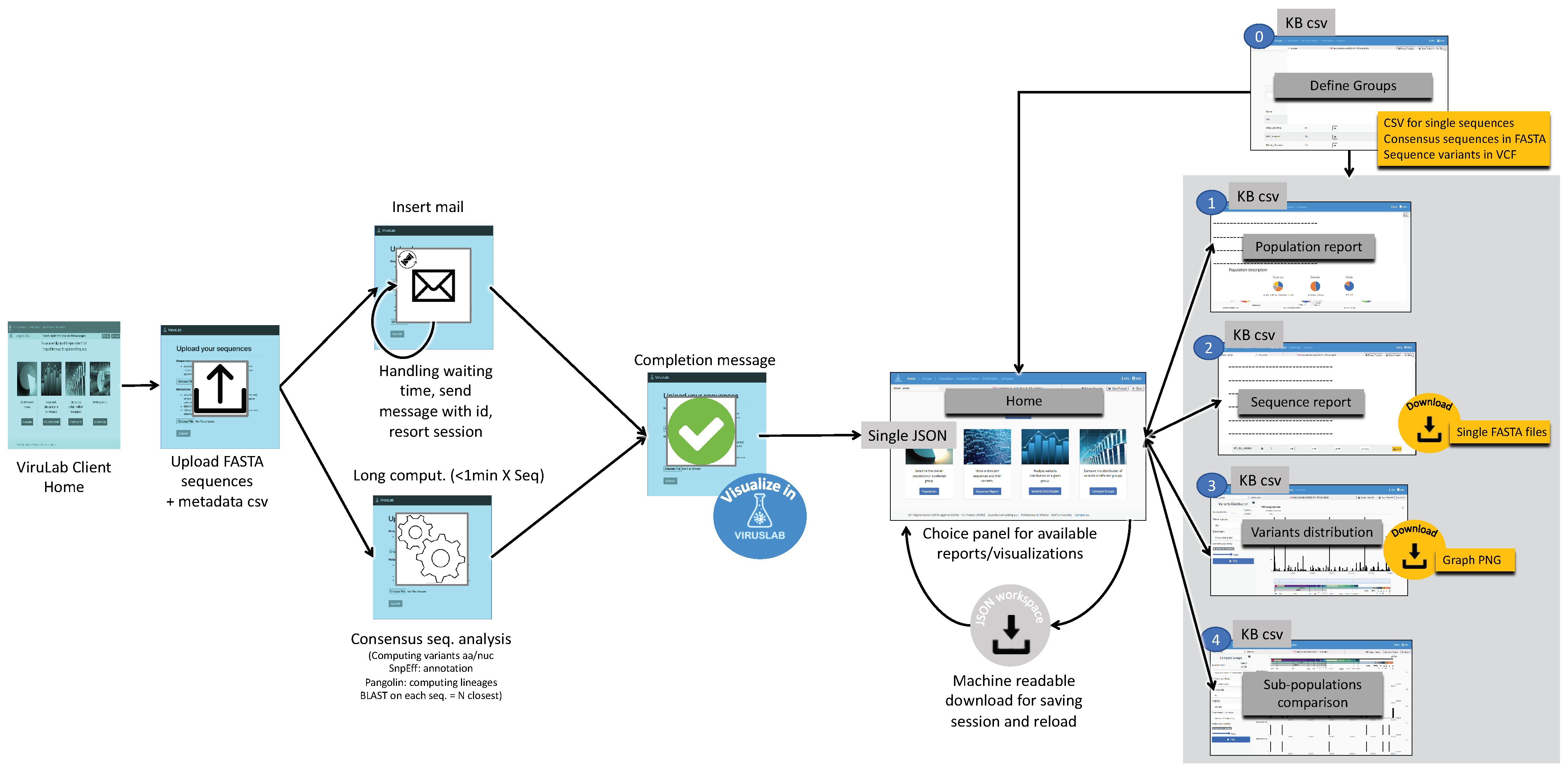
4.3. Report Generation Pipeline
5. Results
- Manage groups: define sub-populations starting from the full set of uploaded sequences.
- Inspect the description of a population or of its groups.
- Visualize and export a sequence report, providing details on single sequences.
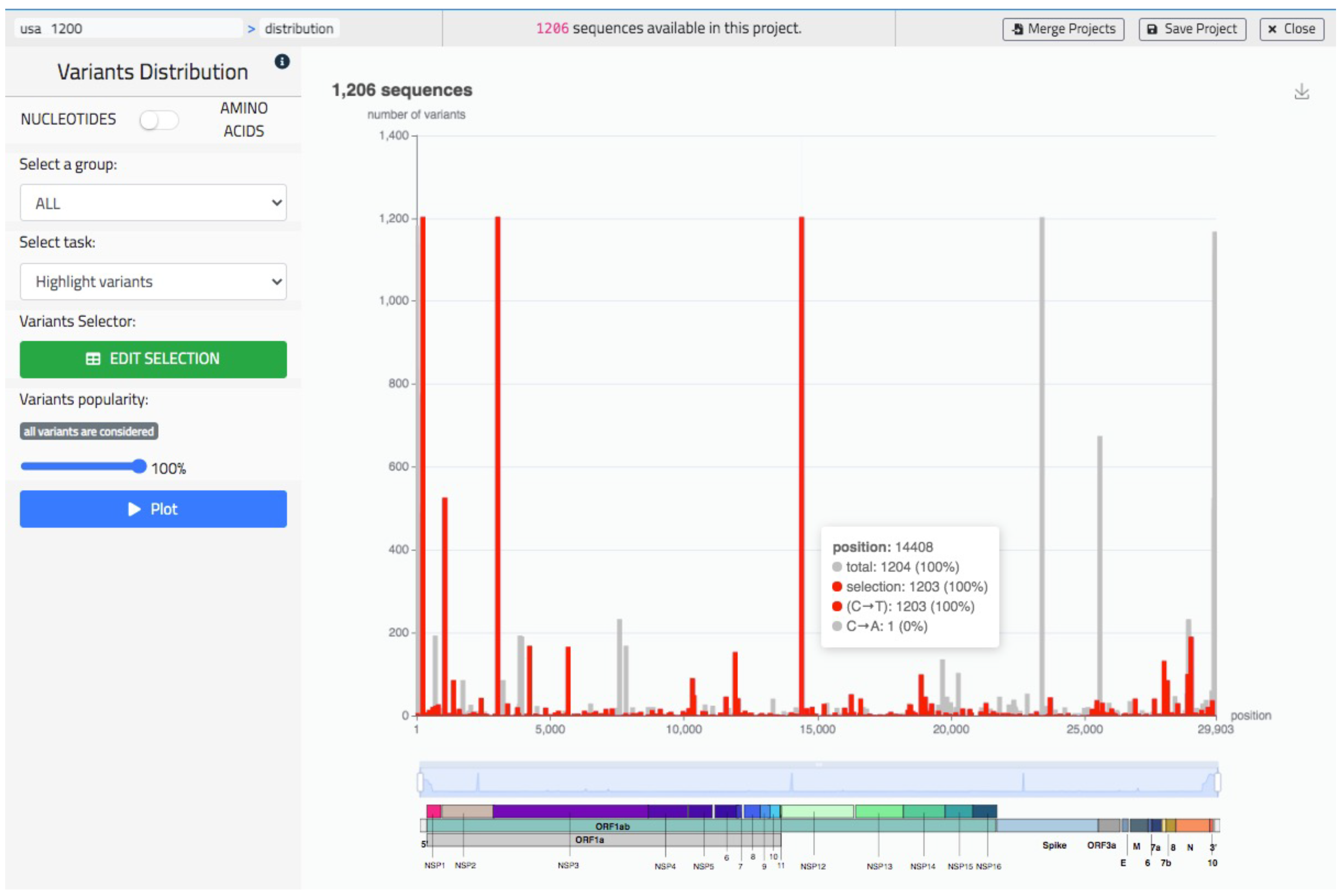
- Visualize the distribution of variants in the full population or its groups.
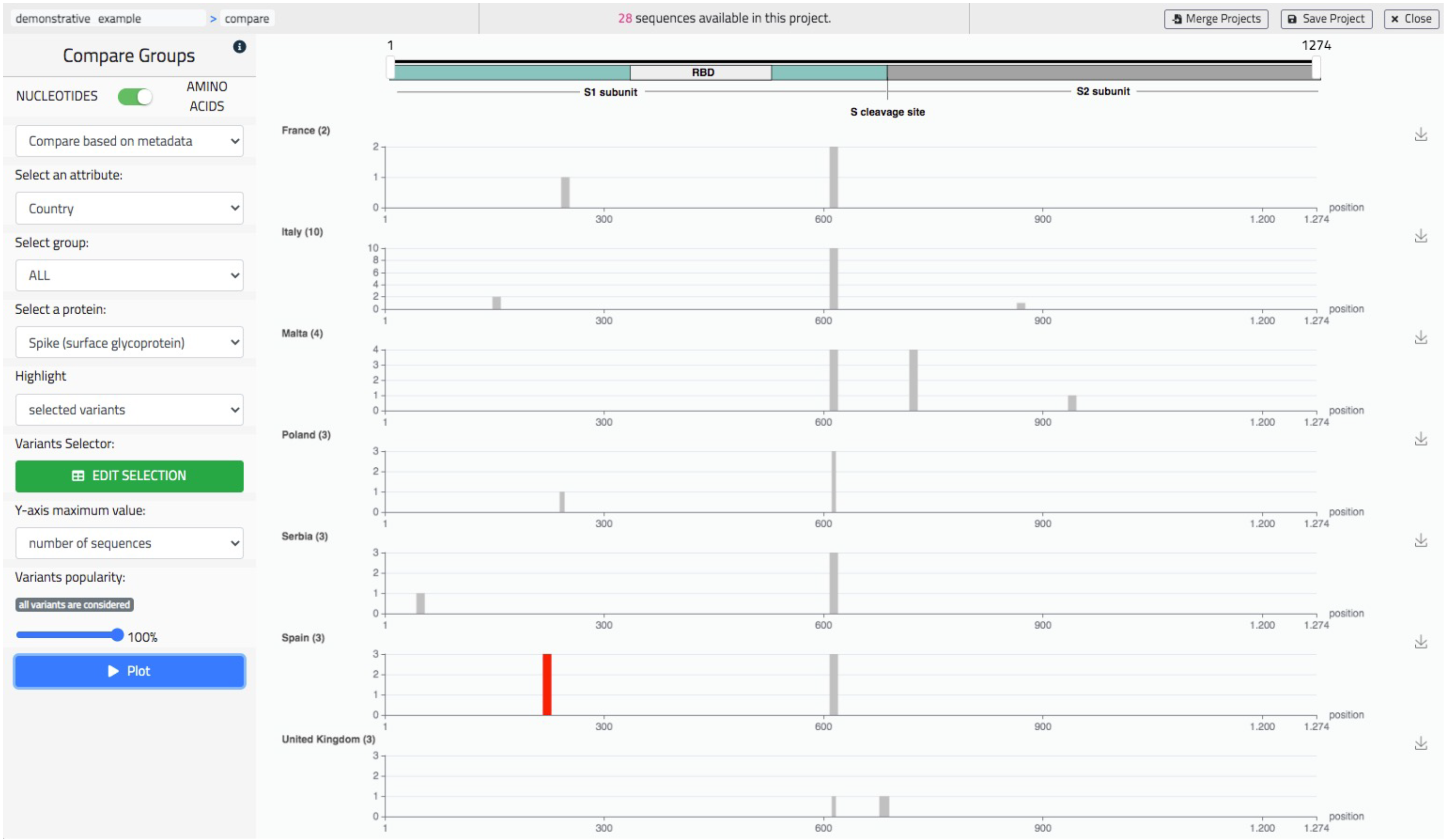
- Comparatively visualize the variants distributions of different groups, either user-defined or defined using sequences metadata.
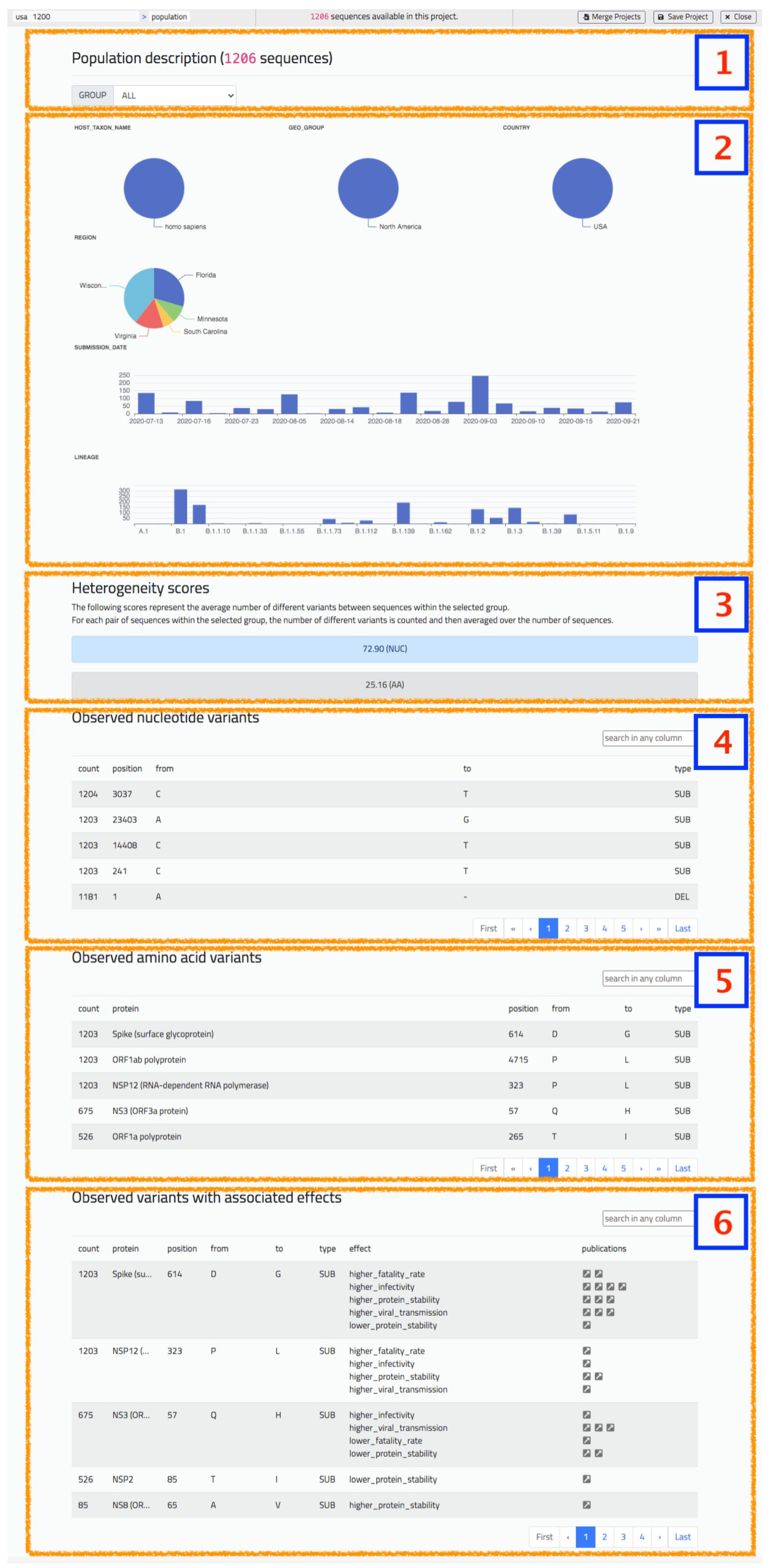
5.1. Population Report
- Piecharts for categorical metadata, with value and corresponding percentage on the selected group.
- Barplots for numerical or date metadata.
- Heterogeneity score indication, considering both nucleotides and amino acids.
- Table with observed nucleotide variants in the group, ordered by descending count.
- Table with observed amino acid variants in the group, ordered by descending count.
- Table with observed interesting variants in the group, present in the CoV2K knowledge base [29] with effects documented in literature.
5.2. Sequence Data Analysis
5.3. Comparative Data Analysis
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, A.; Canakoglu, A.; Masseroli, M.; Pinoli, P.; Ceri, S. A review on viral data sources and search systems for perspective mitigation of COVID-19. Briefings Bioinform. 2021, 22, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Holding, A.N.; Giorgi, F.M. Web tools to fight pandemics: The COVID-19 experience. Briefings Bioinform. 2021, 22, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Briefings Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints 2020. [Google Scholar] [CrossRef]
- Gong, Z.; Zhu, J.W.; Li, C.P.; Jiang, S.; Ma, L.N.; Tang, B.X.; Zou, D.; Chen, M.L.; Sun, Y.B.; Song, S.H.; et al. An online coronavirus analysis platform from the National Genomics Data Center. Zool Res. 2020, 41, 705–708. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Alam, I.; Radovanovic, A.; Incitti, R.; Kamau, A.A.; Alarawi, M.; Azhar, E.I.; Gojobori, T. CovMT: An interactive SARS-CoV-2 mutation tracker, with a focus on critical variants. Lancet Infect. Dis. 2021, 21, 602. [Google Scholar] [CrossRef]
- Fang, S.; Li, K.; Shen, J.; Liu, S.; Liu, J.; Yang, L.; Hu, C.D.; Wan, J. GESS: A database of global evaluation of SARS-CoV-2/hCoV-19 sequences. Nucleic Acids Res. 2021, 49, D706–D714. [Google Scholar] [CrossRef]
- Mullen, J.L.; Tsueng, G.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Matteson, N.; Andersen, K.G.; et al. Outbreak.info. Available online: https://outbreak.info/ (accessed on 30 September 2021).
- Chen, A.T.; Altschuler, K.; Zhan, S.H.; Chan, Y.A.; Deverman, B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. Elife 2021, 10, e63409. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, X.; Gao, X.; Dong, Q. MicroGMT: A Mutation Tracker for SARS-CoV-2 and Other Microbial Genome Sequences. Front. Microbiol. 2020, 11, 1502. [Google Scholar] [CrossRef]
- Chiara, M.; Zambelli, F.; Tangaro, M.A.; Mandreoli, P.; Horner, D.S.; Pesole, G. CorGAT: A tool for the functional annotation of SARS-CoV-2 genomes. Bioinformatics 2020, 36, 5522–5523. [Google Scholar] [CrossRef]
- Cleemput, S.; Dumon, W.; Fonseca, V.; Abdool Karim, W.; Giovanetti, M.; Alcantara, L.C.; Deforche, K.; de Oliveira, T. Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics 2020, 36, 3552–3555. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Bray, S.; van den Beek, M.; Bouvier, D.; Coraor, N.; Miladi, M.; Singh, B.; De Argila, J.R.; Baker, D.; Roach, N.; et al. Freely accessible ready to use global infrastructure for SARS-CoV-2 monitoring. bioRxiv 2021, 1–35. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.; Rowe, W.; Rambaut, A. nCoV-2019 Novel Coronavirus Bioinformatics Protocol; Artic Network: Wasilla, AK, USA, 2020. [Google Scholar]
- Kumar, A.; Bangash, A.H.; Gruening, B. Community Research Amid COVID-19 Pandemic: Genomics Analysis of SARS-CoV-2 over Public GALAXY server. Preprints 2020, 2020050343. [Google Scholar] [CrossRef]
- Canakoglu, A.; Pinoli, P.; Bernasconi, A.; Alfonsi, T.; Melidis, D.P.; Ceri, S. ViruSurf: An integrated database to investigate viral sequences. Nucleic Acids Res. 2021, 49, D817–D824. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The COVID-19 Genomics UK (COG-UK) Consortium. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 2020, 1, e99. [Google Scholar] [CrossRef]
- Bernasconi, A.; Canakoglu, A.; Pinoli, P.; Ceri, S. Empowering Virus Sequence Research Through Conceptual Modeling. In Conceptual Modeling; Dobbie, G., Frank, U., Kappel, G., Liddle, S.W., Mayr, H.C., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 388–402. [Google Scholar]
- Cingolani, P.; Platts, A.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.; Lu, X.; Ruden, D. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 30, veab064. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Al Khalaf, R.; Alfonsi, T.; Ceri, S.; Bernasconi, A. CoV2K: A Knowledge Base of SARS-CoV-2 Variant Impacts. In Research Challenges in Information Science; Cherfi, S., Perini, A., Nurcan, S., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 274–282. [Google Scholar]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Bernasconi, A.; Gulino, A.; Alfonsi, T.; Canakoglu, A.; Pinoli, P.; Sandionigi, A.; Ceri, S. VirusViz: Comparative analysis and effective visualization of viral nucleotide and amino acid variants. Nucleic Acids Res. 2021, 49, e90. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | CoV-GLUE | 2019nCoVR | CoVMT | GESS | Outbreak | CoV Genetics | VirusLab |
|---|---|---|---|---|---|---|---|
| User-input FASTA/metadata support | ✓ | ✓ | - | - | - | - | ✓ |
| Non-SARS-CoV-2 viruses support | - | - | - | - | - | - | ✓ |
| Nucleotide analysis | ✓ | ✓ | ✓ | ✓ | - | - | ✓ |
| Amino acid analysis | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Mutation co-occurrence analysis | - | - | - | ✓ | ✓ | ✓ | ✓ |
| External knowledge integration | - | - | - | - | ✓ | - | ✓ |
| Population definition by metadata/mut/var | - | ✓ | - | - | ✓ | ✓ | ✓ |
| Mutations visualization on genome | - | ✓ | ✓ | ✓ | - | ✓ | ✓ |
| Mutation visualization through time | - | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| Comparison of distributions | - | - | - | - | - | - | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinoli, P.; Bernasconi, A.; Sandionigi, A.; Ceri, S. VirusLab: A Tool for Customized SARS-CoV-2 Data Analysis. BioTech 2021, 10, 27. https://doi.org/10.3390/biotech10040027
Pinoli P, Bernasconi A, Sandionigi A, Ceri S. VirusLab: A Tool for Customized SARS-CoV-2 Data Analysis. BioTech. 2021; 10(4):27. https://doi.org/10.3390/biotech10040027
Chicago/Turabian StylePinoli, Pietro, Anna Bernasconi, Anna Sandionigi, and Stefano Ceri. 2021. "VirusLab: A Tool for Customized SARS-CoV-2 Data Analysis" BioTech 10, no. 4: 27. https://doi.org/10.3390/biotech10040027
APA StylePinoli, P., Bernasconi, A., Sandionigi, A., & Ceri, S. (2021). VirusLab: A Tool for Customized SARS-CoV-2 Data Analysis. BioTech, 10(4), 27. https://doi.org/10.3390/biotech10040027