



Identification of Potential Antimalarial Drug Candidates Targeting Falcipain-2 Protein of Malaria Parasite—A Computational Strategy

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
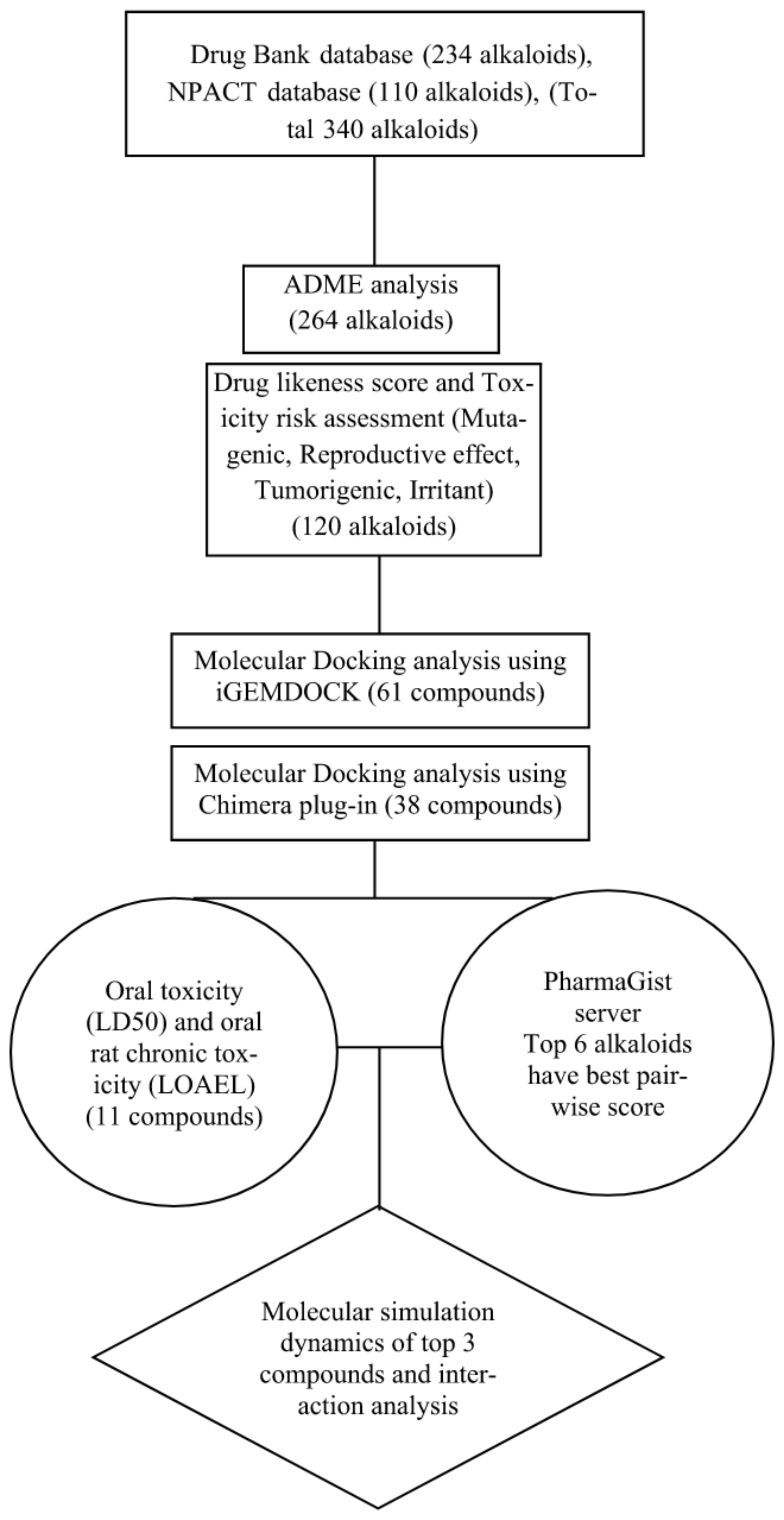
2.1. Dataset
2.2. Absorption, Distribution, Metabolism, and Excretion (ADME) Analysis
2.3. Toxicity Risk Analysis and Drug-Likeness Prediction
2.4. Protein-Ligand Docking Study
2.5. Oral Toxicity Assessment
2.6. 3D Pharmacophore Analysis
2.7. Molecular Dynamics (MD) Simulation
2.8. Binding Free Energy Calculations Using MM-PBSA Approach
3. Results
3.1. Pharmacokinetics Analysis
3.2. Toxicity Risk and Drug-Likeness
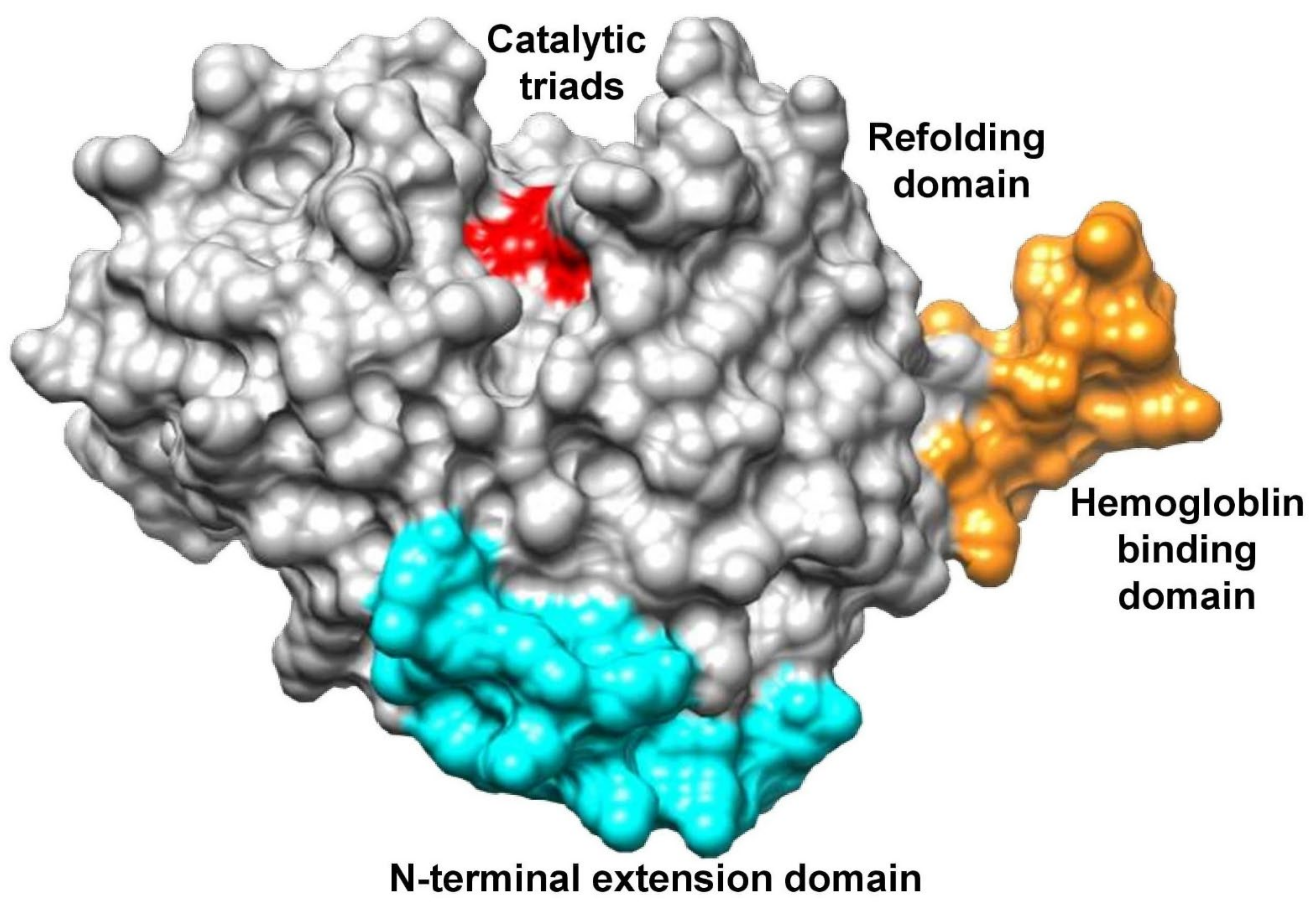
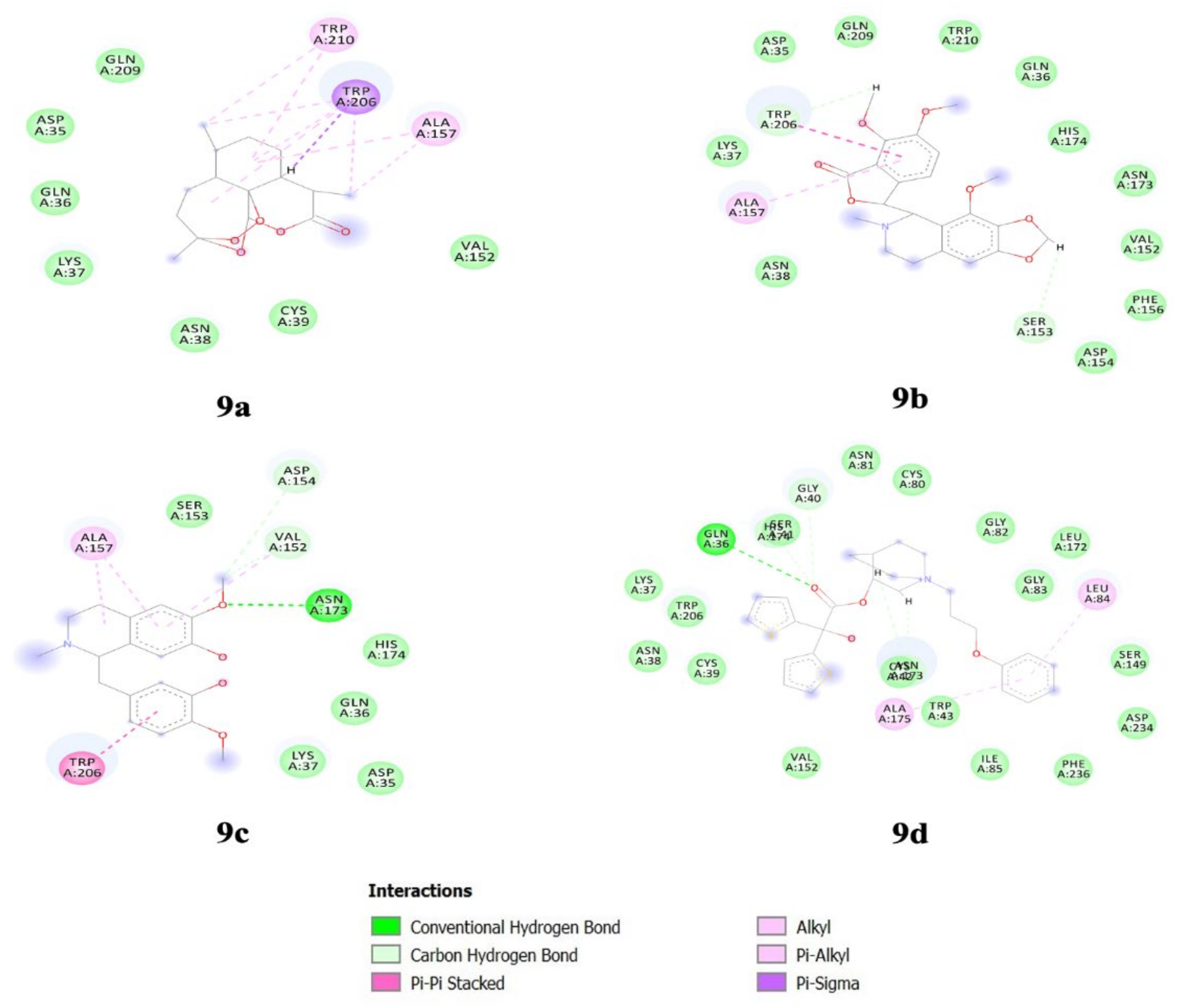
3.3. Biomolecules Docking Analysis
3.4. Oral Toxicity Assessment
3.5. 3D Pharmacophore Study
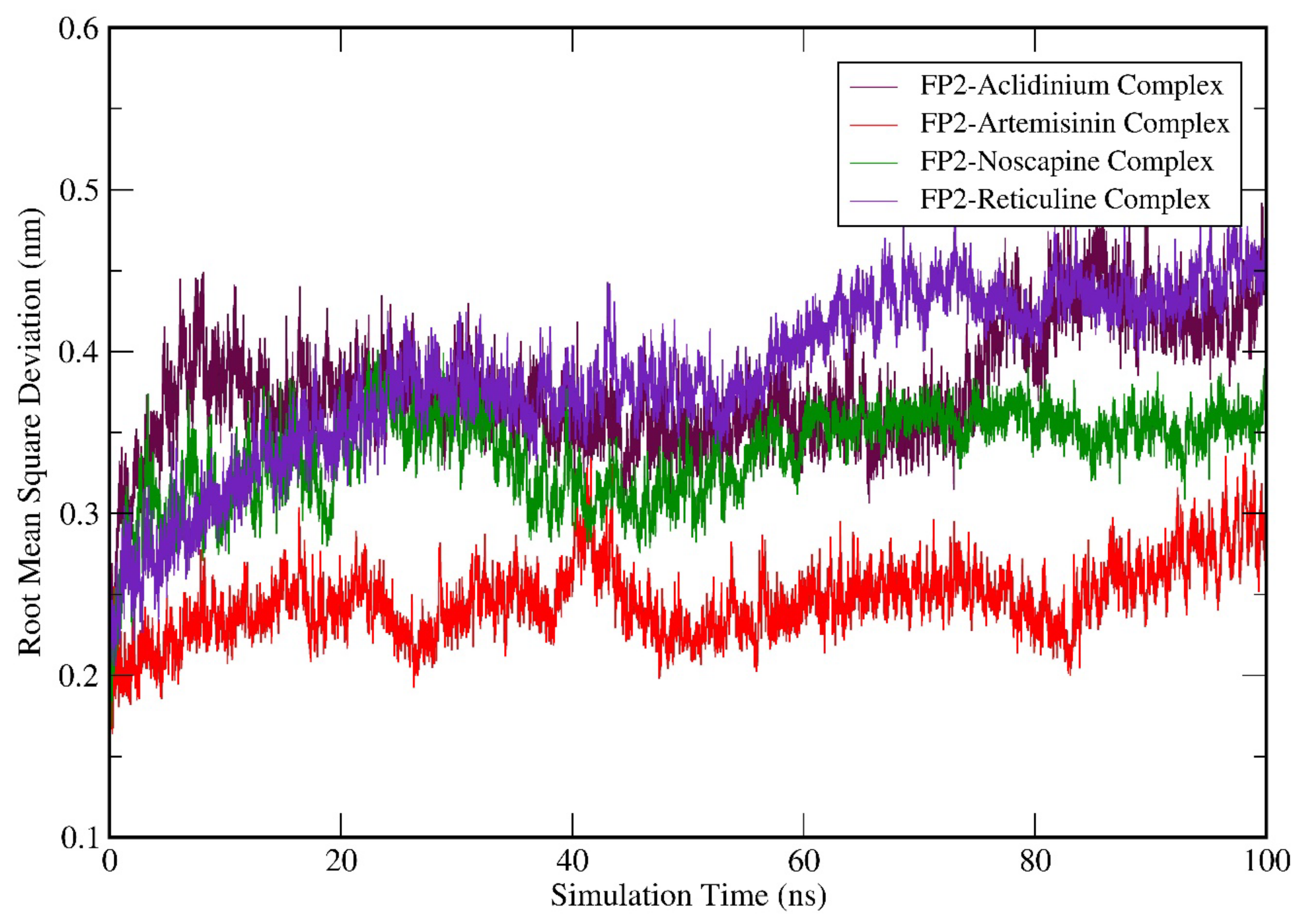
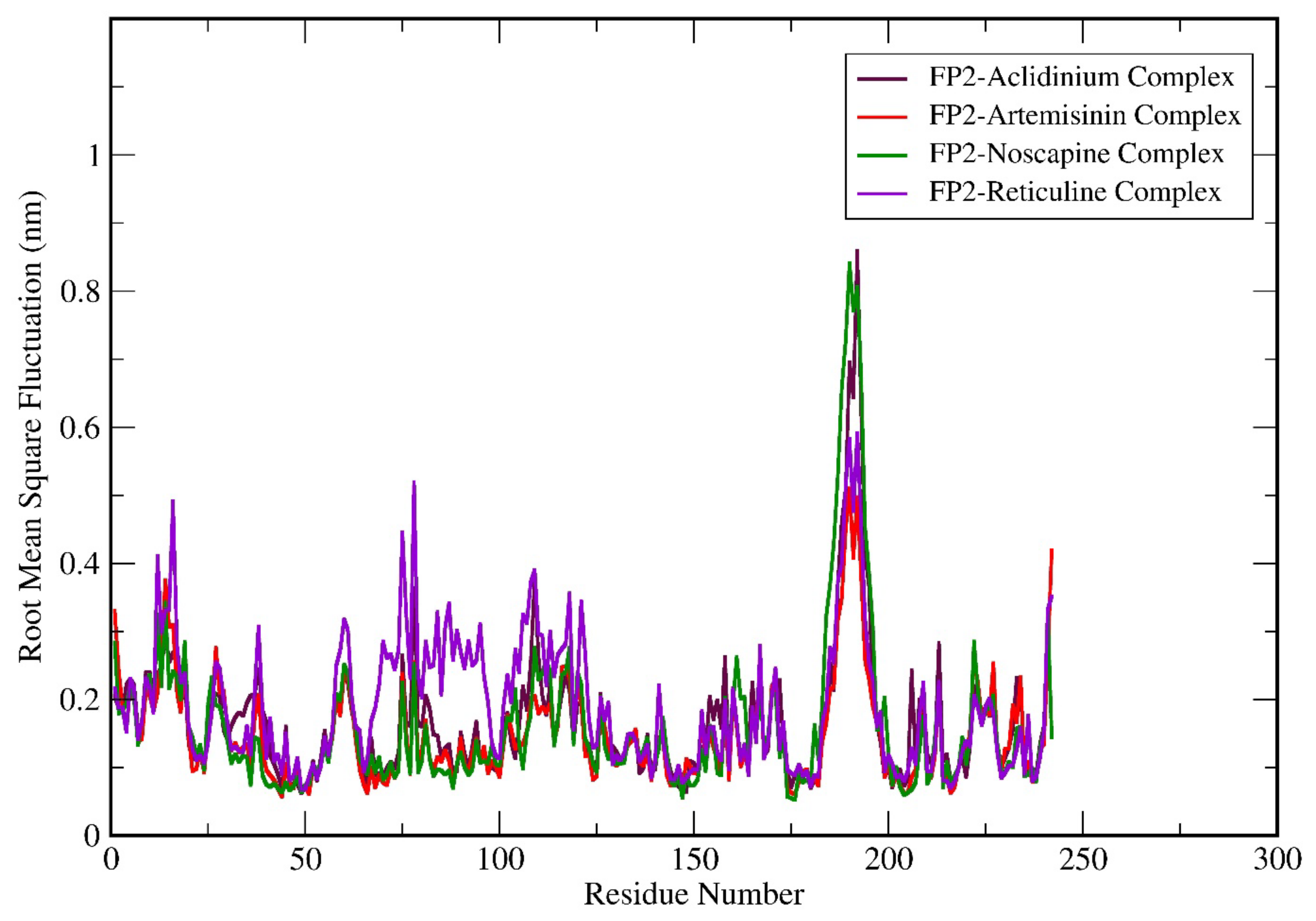
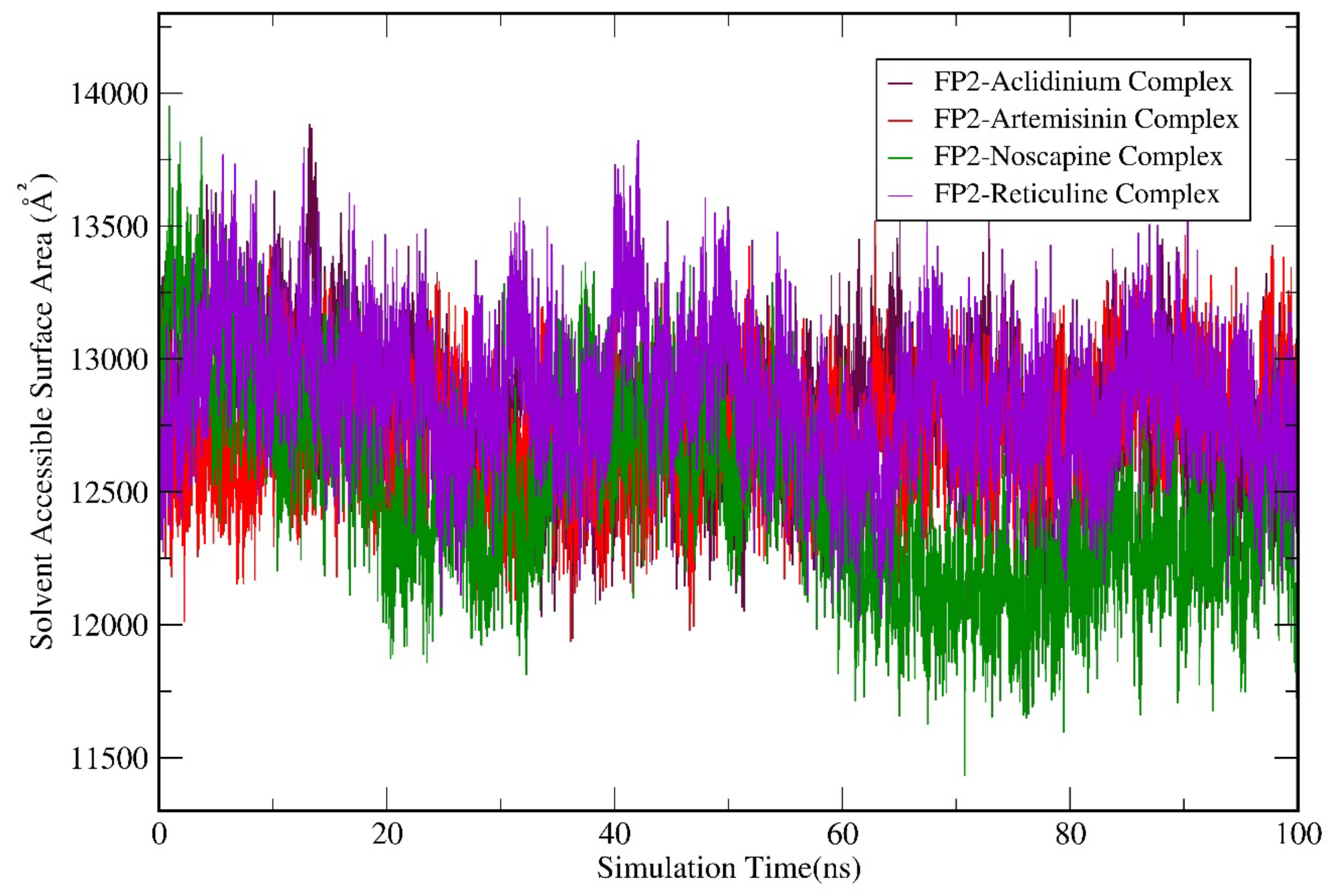
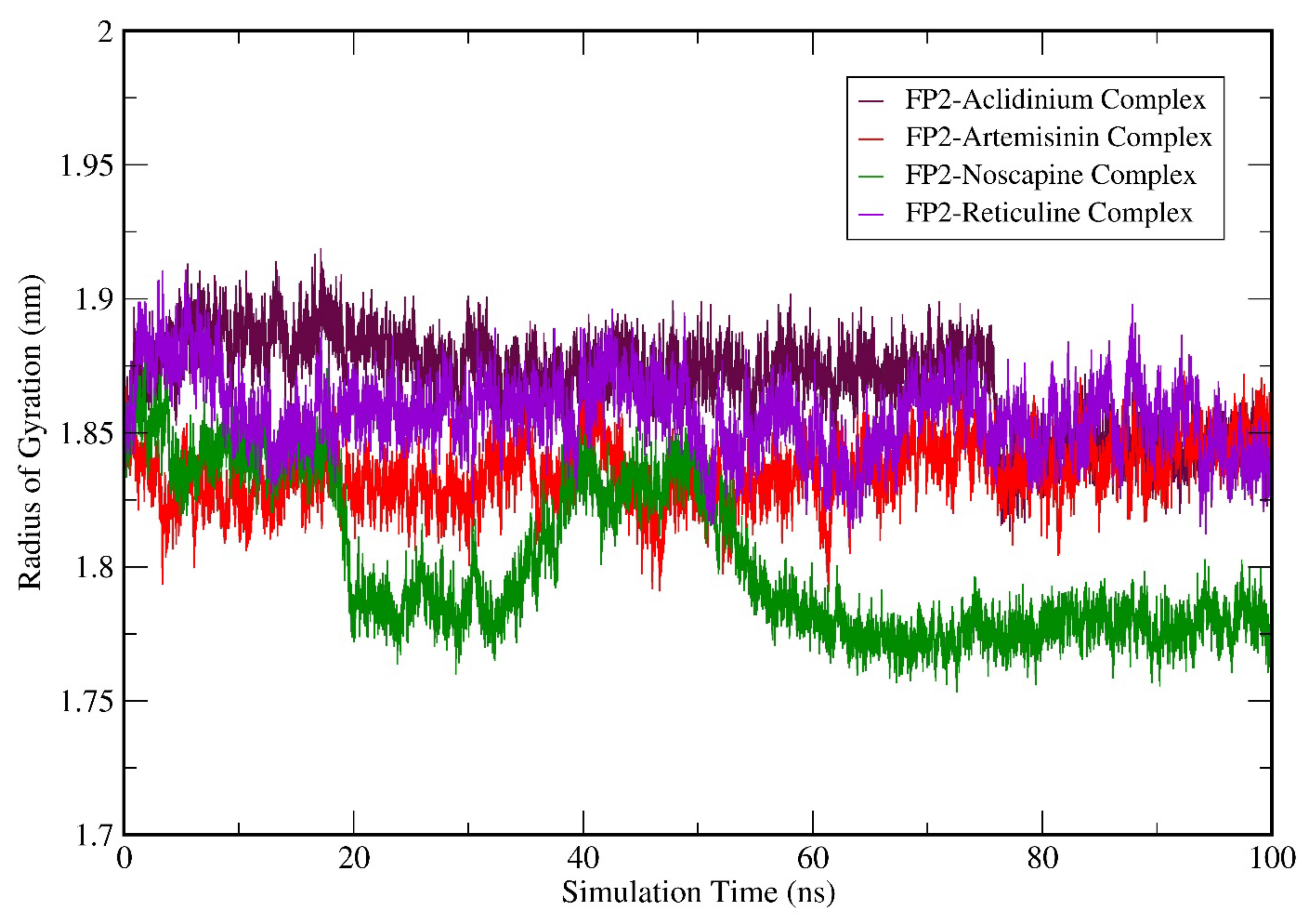
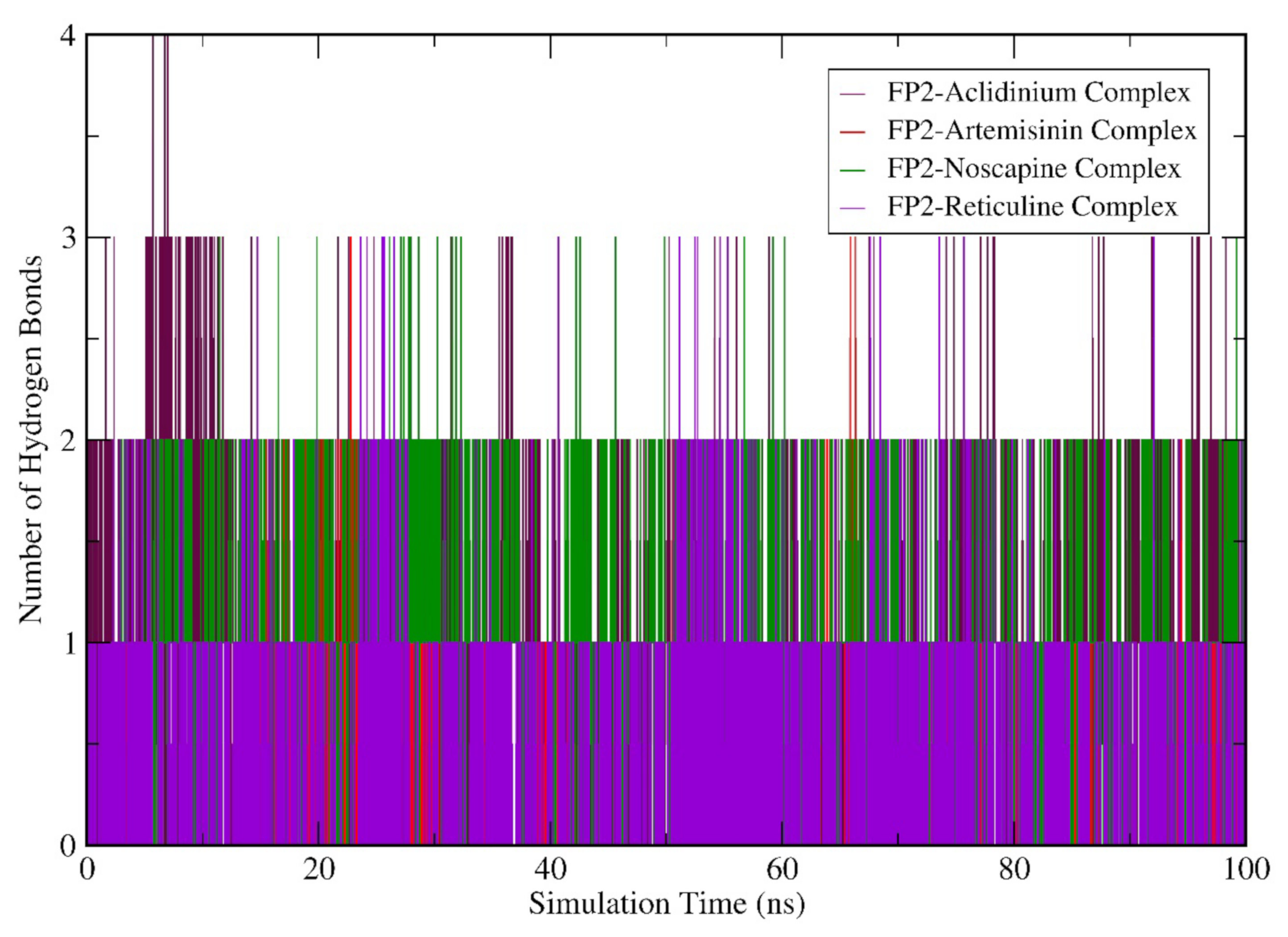
3.6. Molecular Dynamics (MD) Simulation Analysis
3.7. Binding Free Energy Calculations of FP-2 Complex Proteins
3.8. Alkaloids Interaction Study with FP-2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Malaria Report 2021. Available online: https://www.who.int/publications-detail-redirect/9789240040496 (accessed on 30 December 2021).
- Nema, S.; Ghanghoria, P.; Bharti, P.K. Malaria Elimination in India: Bridging the Gap Between Control and Elimination. Indian Pediatr. 2020, 57, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, A.; Zammarchi, L. Clinical Aspects of Uncomplicated and Severe Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.E.; Sullivan, D.J.; Goldberg, D.E. Hemoglobin Metabolism in the Malaria Parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997, 51, 97–123. [Google Scholar] [CrossRef] [PubMed]
- Chugh, M.; Sundararaman, V.; Kumar, S.; Reddy, V.S.; Siddiqui, W.A.; Stuart, K.D.; Malhotra, P. Protein Complex Directs Hemoglobin-to-Hemozoin Formation in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2013, 110, 5392–5397. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Hardt, M.; Choe, Y.; Niles, R.K.; Johansen, E.B.; Legac, J.; Gut, J.; Kerr, I.D.; Craik, C.S.; Rosenthal, P.J. Hemoglobin Cleavage Site-Specificity of the Plasmodium falciparum Cysteine Proteases Falcipain-2 and Falcipain-3. PLoS ONE 2009, 4, e5156. [Google Scholar] [CrossRef] [Green Version]
- Hanspal, M.; Dua, M.; Takakuwa, Y.; Chishti, A.H.; Mizuno, A. Plasmodium falciparum Cysteine Protease Falcipain-2 Cleaves Erythrocyte Membrane Skeletal Proteins at Late Stages of Parasite Development: Presented in Part in Abstract Form at the 43rd Annual Meeting of the American Society of Hematology, Orlando, FL, 2001. Blood 2002, 100, 1048–1054. [Google Scholar] [CrossRef]
- Pandey, K.C.; Dixit, R. Structure-Function of Falcipains: Malarial Cysteine Proteases. J. Trop. Med. 2012, 2012, 5195. [Google Scholar] [CrossRef] [Green Version]
- Hogg, T.; Nagarajan, K.; Herzberg, S.; Chen, L.; Shen, X.; Jiang, H.; Wecke, M.; Blohmke, C.; Hilgenfeld, R.; Schmidt, C.L. Structural and Functional Characterization of Falcipain-2, a Hemoglobinase from the Malarial Parasite Plasmodium falciparum. J. Biol. Chem. 2006, 281, 25425–25437. [Google Scholar] [CrossRef] [Green Version]
- Sijwali, P.S.; Rosenthal, P.J. Gene Disruption Confirms a Critical Role for the Cysteine Protease Falcipain-2 in Hemoglobin Hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2004, 101, 4384–4389. [Google Scholar] [CrossRef] [Green Version]
- Nema, S.; Verma, A.K.; Bharti, P.K. Strengthening Diagnosis Is Key to Eliminating Malaria in India. Lancet Infect. Dis. 2019, 19, 1277–1278. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, W.-Q.; Cui, K.-Q.; Luo, W.; Wang, J.; Guo, C. Synthesis and Biological Activities of Novel Artemisinin Derivatives as Cysteine Protease Falcipain-2 Inhibitors. Arch. Pharm. Res. 2012, 35, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthivarakul, S.; Wong, M.H.L.; Hemingway, J.; Biagini, G.A.; O’Neill, P.M.; Ward, S.A. Artemisinin Activity-Based Probes Identify Multiple Molecular Targets within the Asexual Stage of the Malaria Parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. USA 2016, 113, 2080–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilley, L.; Straimer, J.; Gnädig, N.F.; Ralph, S.A.; Fidock, D.A. Artemisinin Action and Resistance in Plasmodium falciparum. Trends Parasitol. 2016, 32, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, K.; Lahariya, A.K.; Verma, G.; Kumari, M.; Gupta, D.; Maurya, N.; Verma, A.K.; Mani, A.; Schneider, K.A.; Bharti, P.K. Screening of Potential Antiplasmodial Agents Targeting Cysteine Protease-Falcipain 2: A Computational Pipeline. J. Biomol. Struct. Dyn. 2022. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Pousset, J.-L.; Martin, M.-T.; Jossang, A.; Bodo, B. Isocryptolepine from Cryptolepis sanguinolenta. Phytochemistry 1995, 39, 735–736. [Google Scholar] [CrossRef]
- Kubo, M.; Yatsuzuka, W.; Matsushima, S.; Harada, K.; Inoue, Y.; Miyamoto, H.; Matsumoto, M.; Fukuyama, Y. Antimalarial Phenanthroindolizine Alkaloids from Ficus septica. Chem. Pharm. Bull. 2016, 64, 957–960. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Sarmah, S.; Lyndem, S.; Roy, A.S. An Investigation into the Identification of Potential Inhibitors of SARS-CoV-2 Main Protease Using Molecular Docking Study. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Verma, K.; Kannan, K.; Shanti, V.; Sethumadhavan, R.; Karthick, V.; Ramanathan, K. Exploring β-Tubulin Inhibitors from Plant Origin Using Computational Approach. Phytochem. Anal. 2017, 28, 230–241. [Google Scholar] [CrossRef]
- Fadaeinasab, M.; Taha, H.; Fauzi, P.N.M.; Ali, H.M.; Widyawaruyanti, A. Anti-Malarial Activity of Isoquinoline Alkaloids from the Stem Bark of Actinodaphne macrophylla. Nat. Prod. Commun. 2015, 10, 1934578X1501000913. [Google Scholar] [CrossRef]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.S.; Agarwal, S.M. NPACT: Naturally Occurring Plant-Based Anti-Cancer Compound-Activity-Target Database. Nucleic Acids Res. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. Selection Criteria for Drug-like Compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef] [PubMed]
- Von Korff, M.; Sander, T. Toxicity-Indicating Structural Patterns. J. Chem. Inf. Model. 2006, 46, 536–544. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Walum, E. Acute Oral Toxicity. Environ. Health Perspect. 1998, 106, 497–503. [Google Scholar] [CrossRef]
- Didziapetris, R.; Reynolds, D.P.; Japertas, P.; Zmuidinavicius, D.; Petrauskas, A. In Silico Technology for Identification of Potentially Toxic Compounds in Drug Discovery. Available online: https://www.eurekaselect.com/56095/article (accessed on 5 November 2020).
- Olaniyan, J.M.; Muhammad, H.L.; Makun, H.A.; Busari, M.B.; Abdullah, A.S. Acute and Sub-Acute Toxicity Studies of Aqueous and Methanol Extracts of Nelsonia campestris in Rats. J. Acute Dis. 2016, 5, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Mazzatorta, P.; Estevez, M.D.; Coulet, M.; Schilter, B. Modeling Oral Rat Chronic Toxicity. J. Chem. Inf. Model. 2008, 48, 1949–1954. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A Webserver for Ligand-Based Pharmacophore Detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [Green Version]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.C. Molecular Dynamics Simulations at Constant Pressure and/or Temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Soman, S.S.; Sivakumar, K.C.; Sreekumar, E. Molecular Dynamics Simulation Studies and In Vitro Site Directed Mutagenesis of Avian Beta-Defensin Apl_AvBD2. BMC Bioinform. 2010, 11, S7. [Google Scholar] [CrossRef] [Green Version]
- Gogoi, B.; Chowdhury, P.; Goswami, N.; Gogoi, N.; Naiya, T.; Chetia, P.; Mahanta, S.; Chetia, D.; Tanti, B.; Borah, P.; et al. Identification of Potential Plant-Based Inhibitor against Viral Proteases of SARS-CoV-2 through Molecular Docking, MM-PBSA Binding Energy Calculations and Molecular Dynamics Simulation. Mol. Divers. 2021, 25, 1963–1977. [Google Scholar] [CrossRef]
- Choudhury, M.; Dhanabalan, A.K.; Goswami, N. Understanding the Binding Mechanism for Potential Inhibition of SARS-CoV-2 Mpro and Exploring the Modes of ACE2 Inhibition by Hydroxychloroquine. J. Cell. Biochem. 2022, 123, 347–358. [Google Scholar] [CrossRef]
- Salawu, E.O. In Silico Study Reveals How E64 Approaches, Binds to, and Inhibits Falcipain-2 of Plasmodium falciparum That Causes Malaria in Humans. Sci. Rep. 2018, 8, 16380. [Google Scholar] [CrossRef] [Green Version]
- Rosmalena; Prasasty, V.D.; Hanafi, M. Novel Cinchona Alkaloid Derivatives as Potential Antimalarial Agents through Receptor–Inhibitor Interaction Fingerprint and Biosynthesis Design. Orient. J. Chem. 2018, 34, 2643–2650. [Google Scholar] [CrossRef] [Green Version]
- Uddin, A.; Gupta, S.; Mohammad, T.; Shahi, D.; Hussain, A.; Alajmi, M.F.; El-Seedi, H.R.; Hassan, I.; Singh, S.; Abid, M. Target-Based Virtual Screening of Natural Compounds Identifies a Potent Antimalarial With Selective Falcipain-2 Inhibitory Activity. Front. Pharmacol. 2022, 13, 850176. [Google Scholar]
- Fogel, D.B. Factors Associated with Clinical Trials That Fail and Opportunities for Improving the Likelihood of Success: A Review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, R.Y.; Faratisha, I.F.; Mardhiyyah, K.; Ariel, D.G.; Putri, F.F.; Nafisatuzzamrudah; Winarsih, S.; Sardjono, T.W.; Fitri, L.E. Antimalarial Properties of Isoquinoline Derivative from Streptomyces hygroscopicus Subsp. Hygroscopicus: An In Silico Approach. BioMed Res. Int. 2020, 2020, e6135696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, R.U.; Roy, N. Recent Trends in Drug-Likeness Prediction: A Comprehensive Review of In Silico Methods. Indian J. Pharm. Sci. 2007, 69, 609. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, S. Toxicological Screening. J. Pharmacol. Pharmacother. 2011, 2, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Zavodszky, M.I.; Kuhn, L.A. Side-Chain Flexibility in Protein–Ligand Binding: The Minimal Rotation Hypothesis. Protein Sci. Publ. Protein Soc. 2005, 14, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Nagasundaram, N.; George Priya, D.C.; Chakraborty, C.; Karthick, V.; Thirumal, K.D.; Balaji, V.; Siva, R.; Aiping, L.; Zhang, G.; Hailong, H. Mechanism of Artemisinin Resistance for Malaria PfATP6 L263 Mutations and Discovering Potential Antimalarials: An Integrated Computational Approach. Sci. Rep. 2016, 6, 30106. [Google Scholar] [CrossRef] [Green Version]
- Kerr, I.D.; Lee, J.H.; Pandey, K.C.; Harrison, A.; Sajid, M.; Rosenthal, P.J.; Brinen, L.S. Structures of Falcipain-2 and Falcipain-3 Bound to Small Molecule Inhibitors: Implications for Substrate Specificity. J. Med. Chem. 2009, 52, 852–857. [Google Scholar] [CrossRef]
- Rida, P.C.G.; LiVecche, D.; Ogden, A.; Zhou, J.; Aneja, R. The Noscapine Chronicle: A Pharmaco-Historic Biography of the Opiate Alkaloid Family and Its Clinical Applications. Med. Res. Rev. 2015, 35, 1072–1096. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nema, S.; Verma, K.; Mani, A.; Maurya, N.S.; Tiwari, A.; Bharti, P.K. Identification of Potential Antimalarial Drug Candidates Targeting Falcipain-2 Protein of Malaria Parasite—A Computational Strategy. BioTech 2022, 11, 54. https://doi.org/10.3390/biotech11040054
Nema S, Verma K, Mani A, Maurya NS, Tiwari A, Bharti PK. Identification of Potential Antimalarial Drug Candidates Targeting Falcipain-2 Protein of Malaria Parasite—A Computational Strategy. BioTech. 2022; 11(4):54. https://doi.org/10.3390/biotech11040054
Chicago/Turabian StyleNema, Shrikant, Kanika Verma, Ashutosh Mani, Neha Shree Maurya, Archana Tiwari, and Praveen Kumar Bharti. 2022. "Identification of Potential Antimalarial Drug Candidates Targeting Falcipain-2 Protein of Malaria Parasite—A Computational Strategy" BioTech 11, no. 4: 54. https://doi.org/10.3390/biotech11040054
APA StyleNema, S., Verma, K., Mani, A., Maurya, N. S., Tiwari, A., & Bharti, P. K. (2022). Identification of Potential Antimalarial Drug Candidates Targeting Falcipain-2 Protein of Malaria Parasite—A Computational Strategy. BioTech, 11(4), 54. https://doi.org/10.3390/biotech11040054