
A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
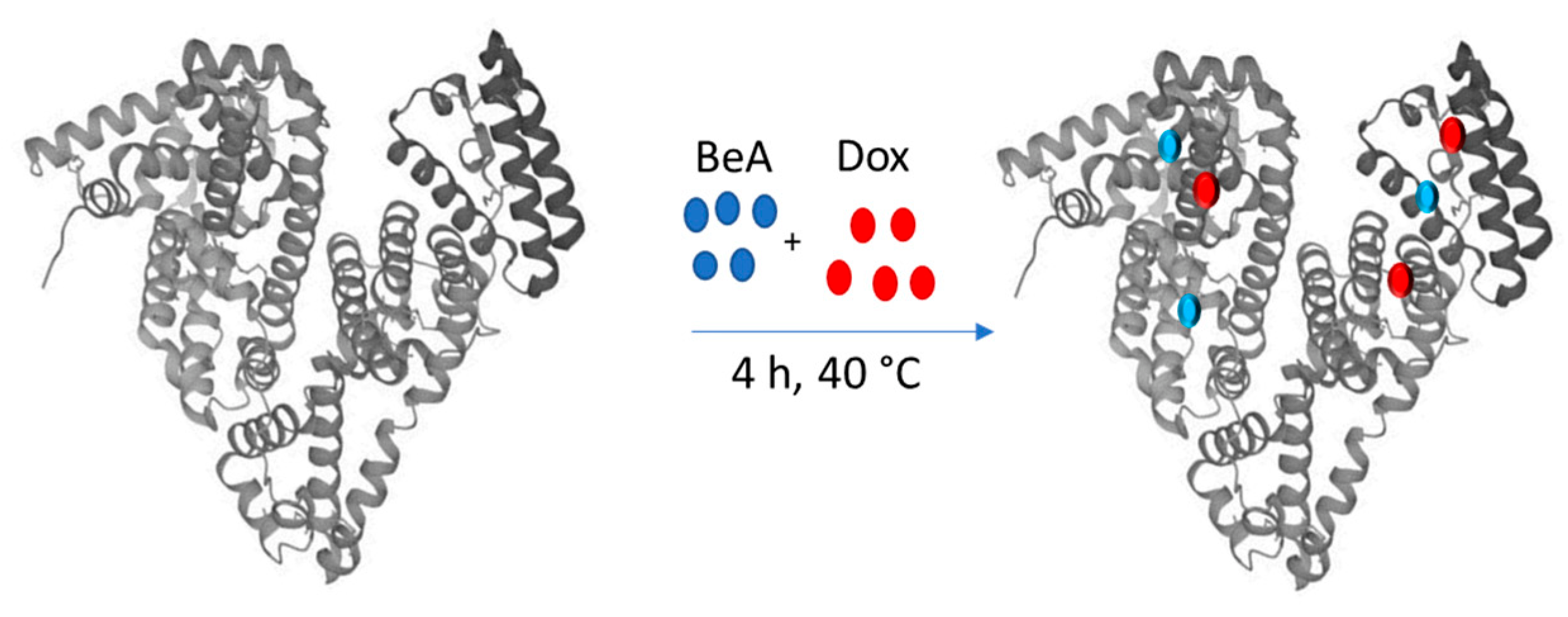
2.2. Preparation of the Nanosized DDS
2.3. Characterization of the DDS
2.3.1. Quantification of the DDS Components
2.3.2. Encapsulation Efficiency (EE) and NP Yield
2.3.3. Particle Size, Polydispersity, and Zeta Potential
2.3.4. In Vitro Drug Release Studies
2.4. Cell Culture Experiments
2.4.1. Cell Culture Conditions
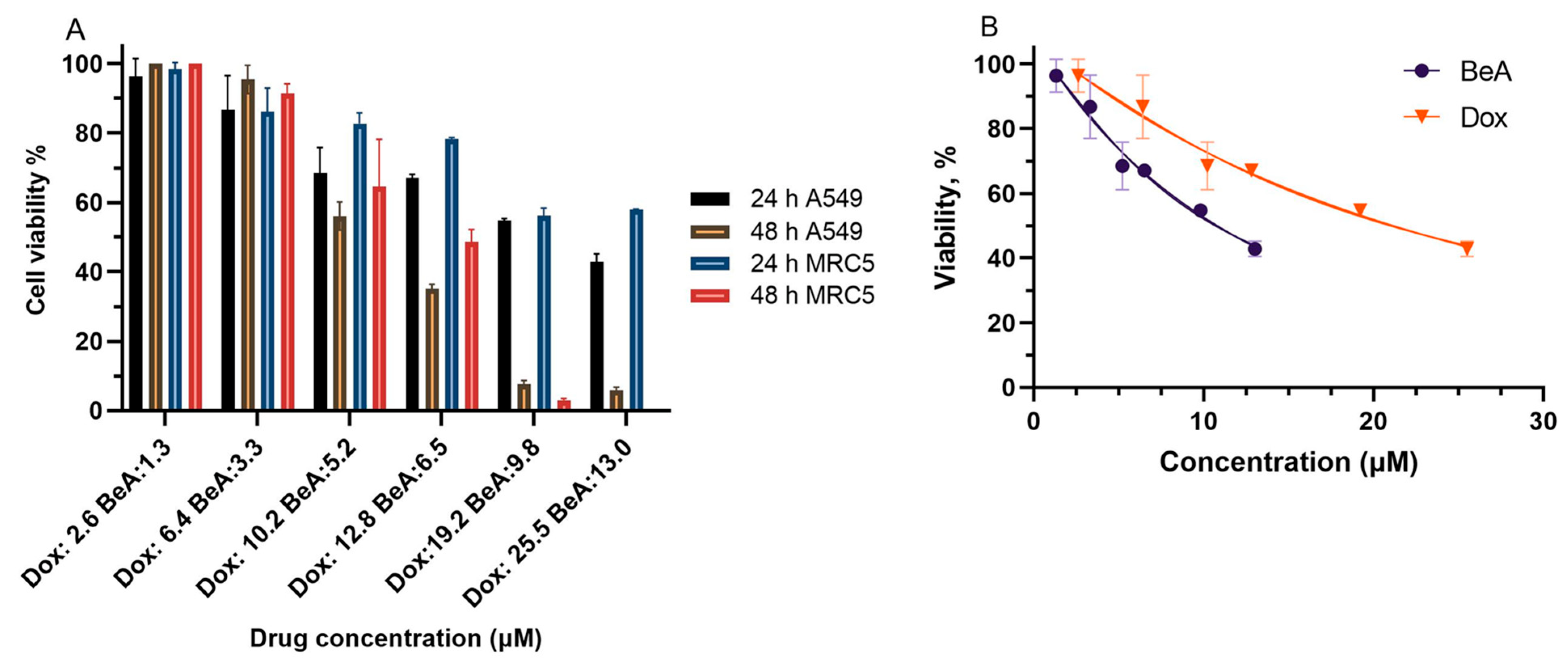
2.4.2. Cell Viability
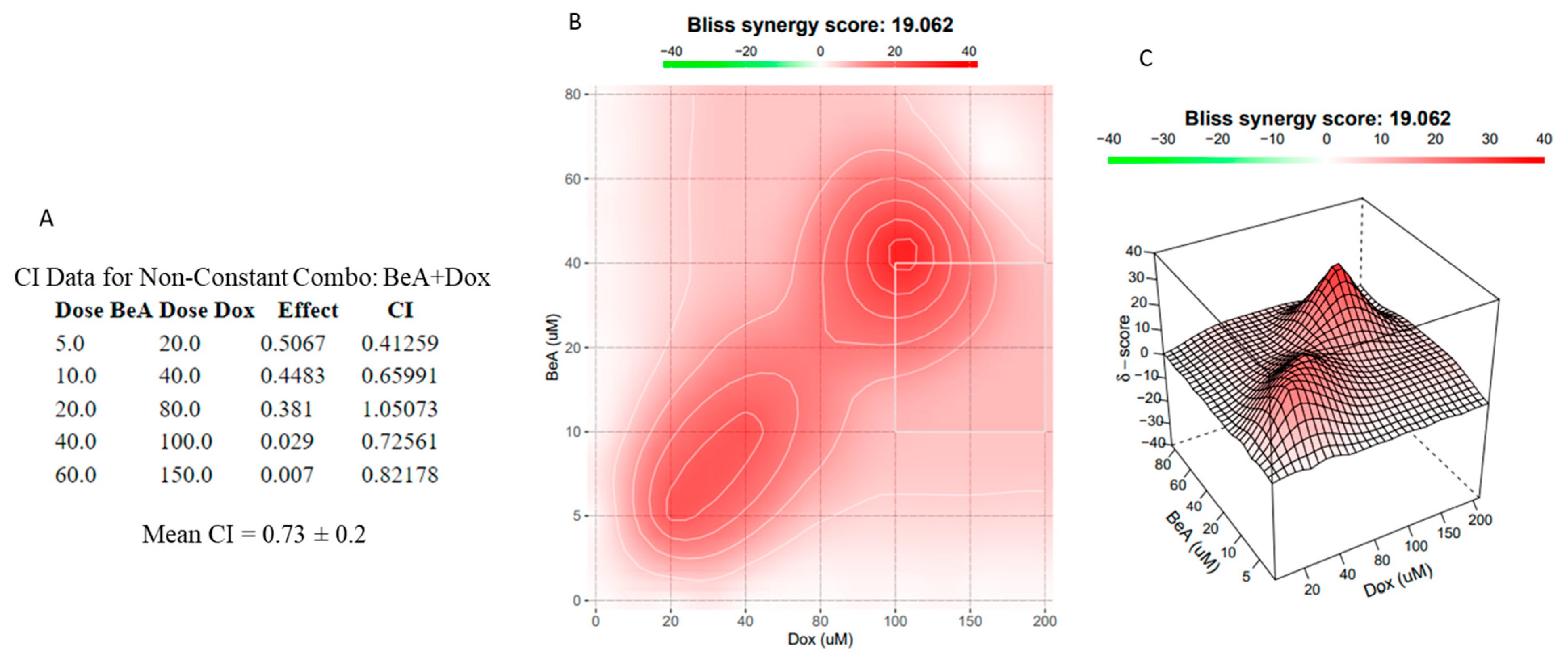
2.5. Synergism, Additive or Antagonistic Drug Interactions
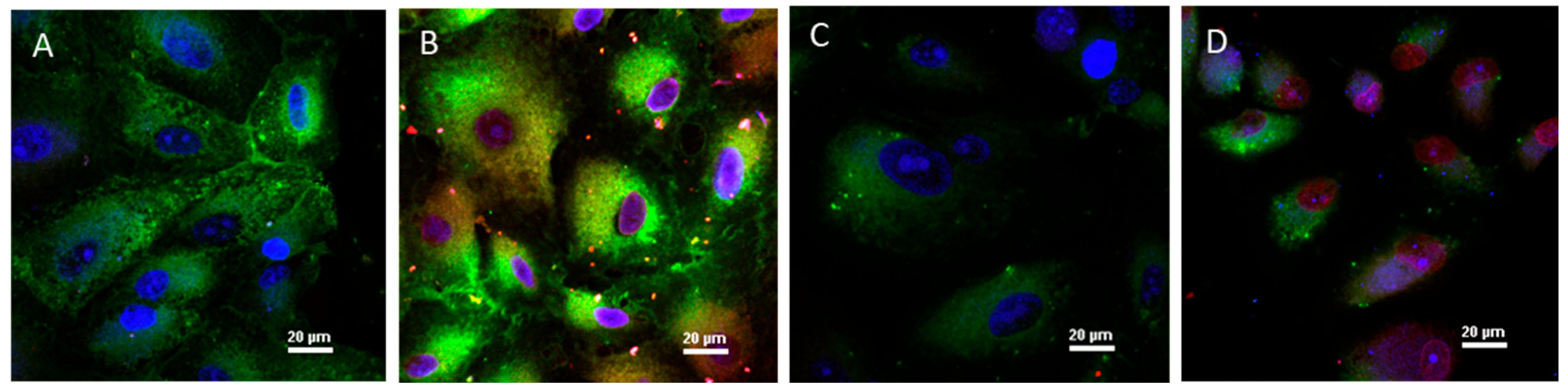
2.6. Cellular Internalization of DDS
2.6.1. DDS-FITC Labeling
2.6.2. DDS Uptake Visualization
2.7. Flow Cytometry Analysis
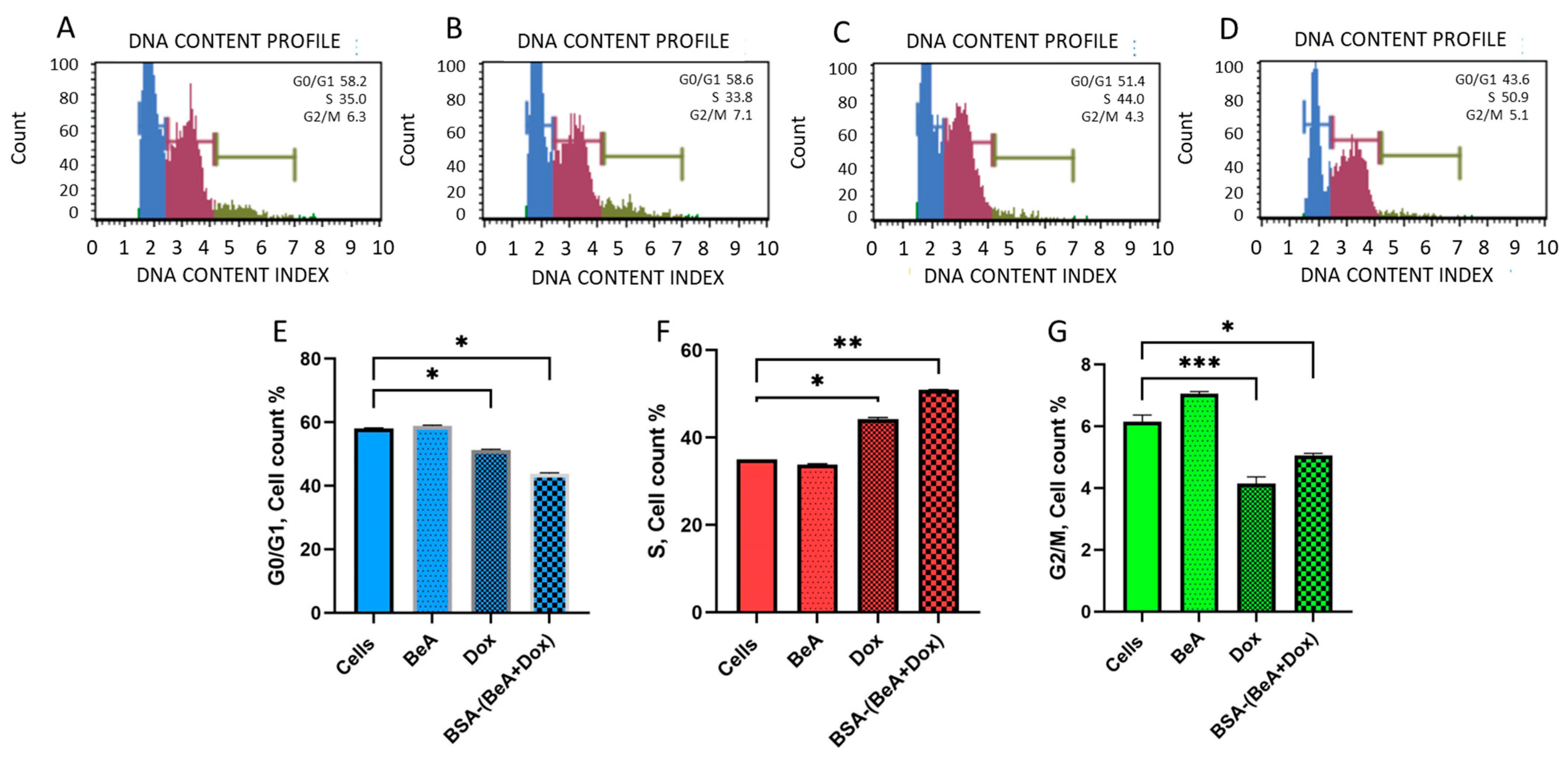
2.7.1. Cell Cycle Arrest
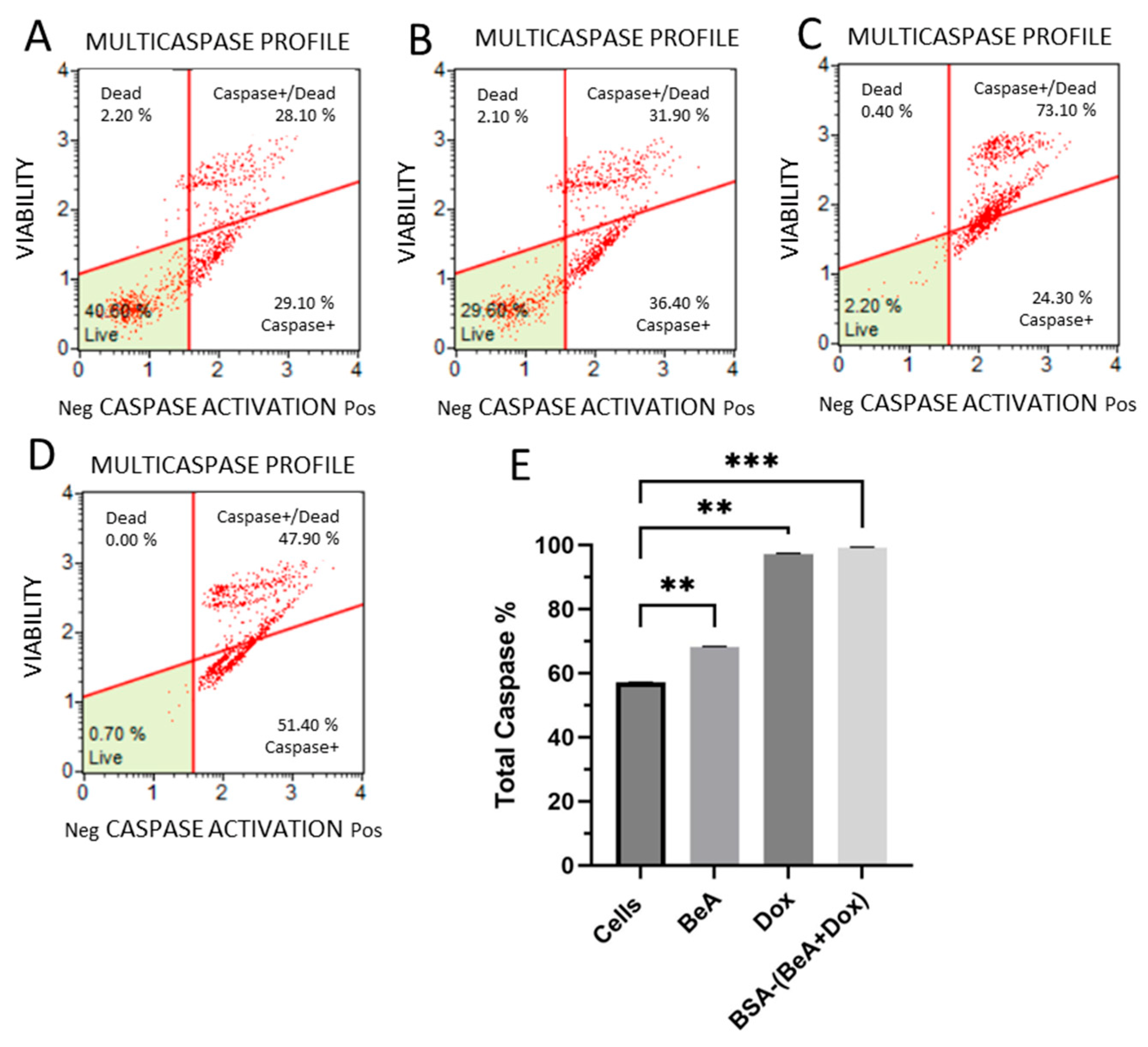
2.7.2. Multi-Caspase Activation
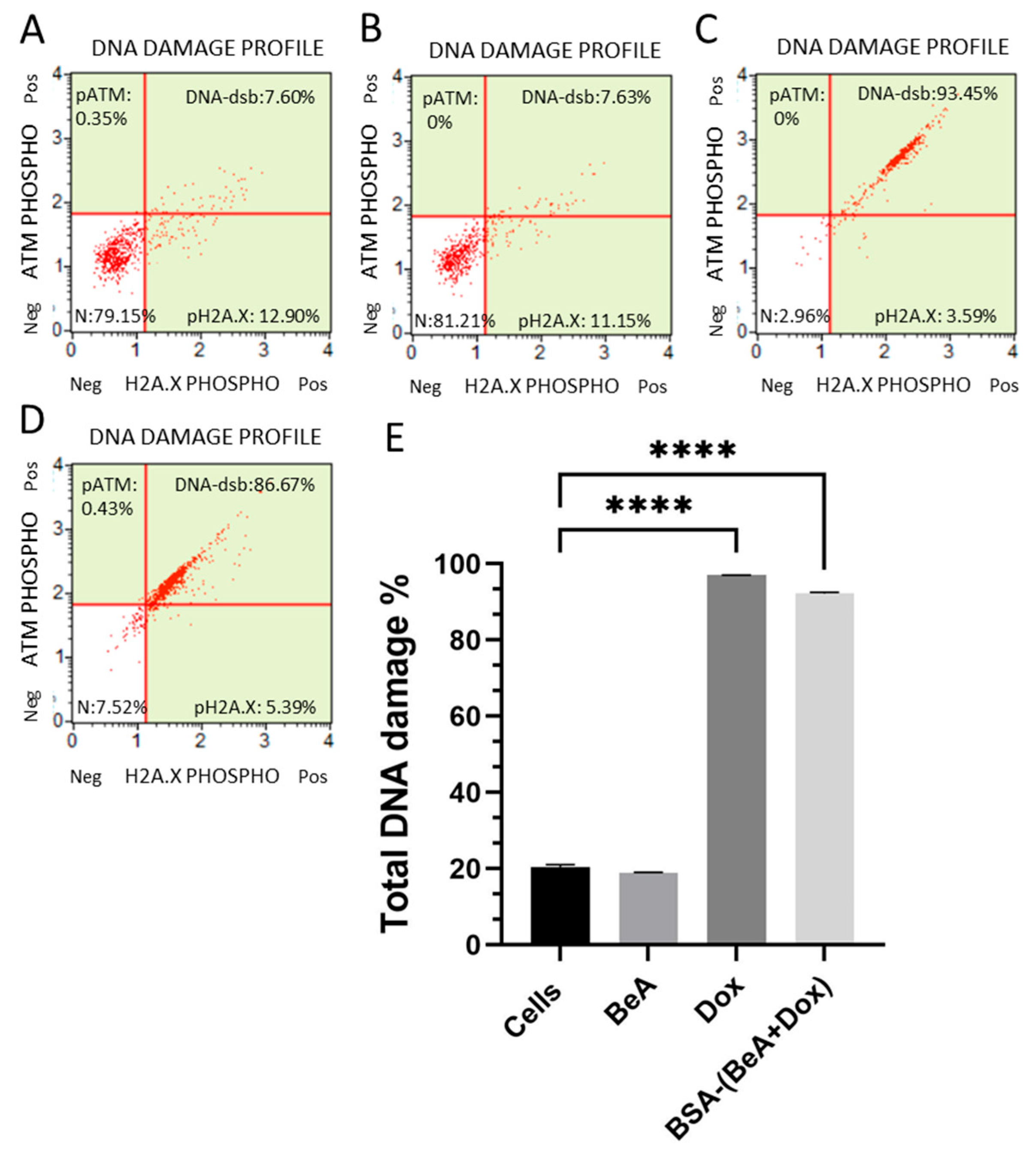
2.7.3. DNA Damage Induction
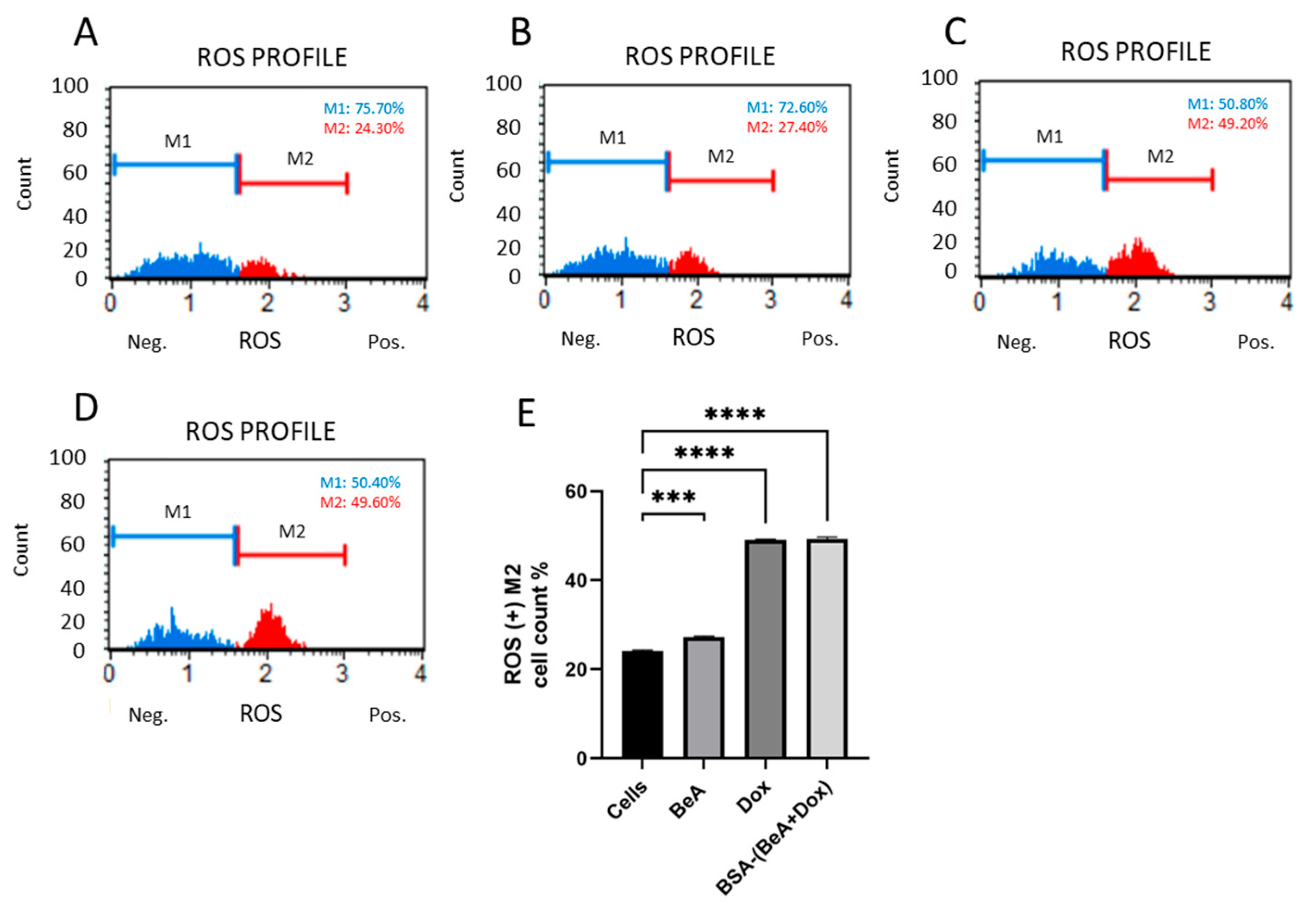
2.7.4. Oxidative Stress Production
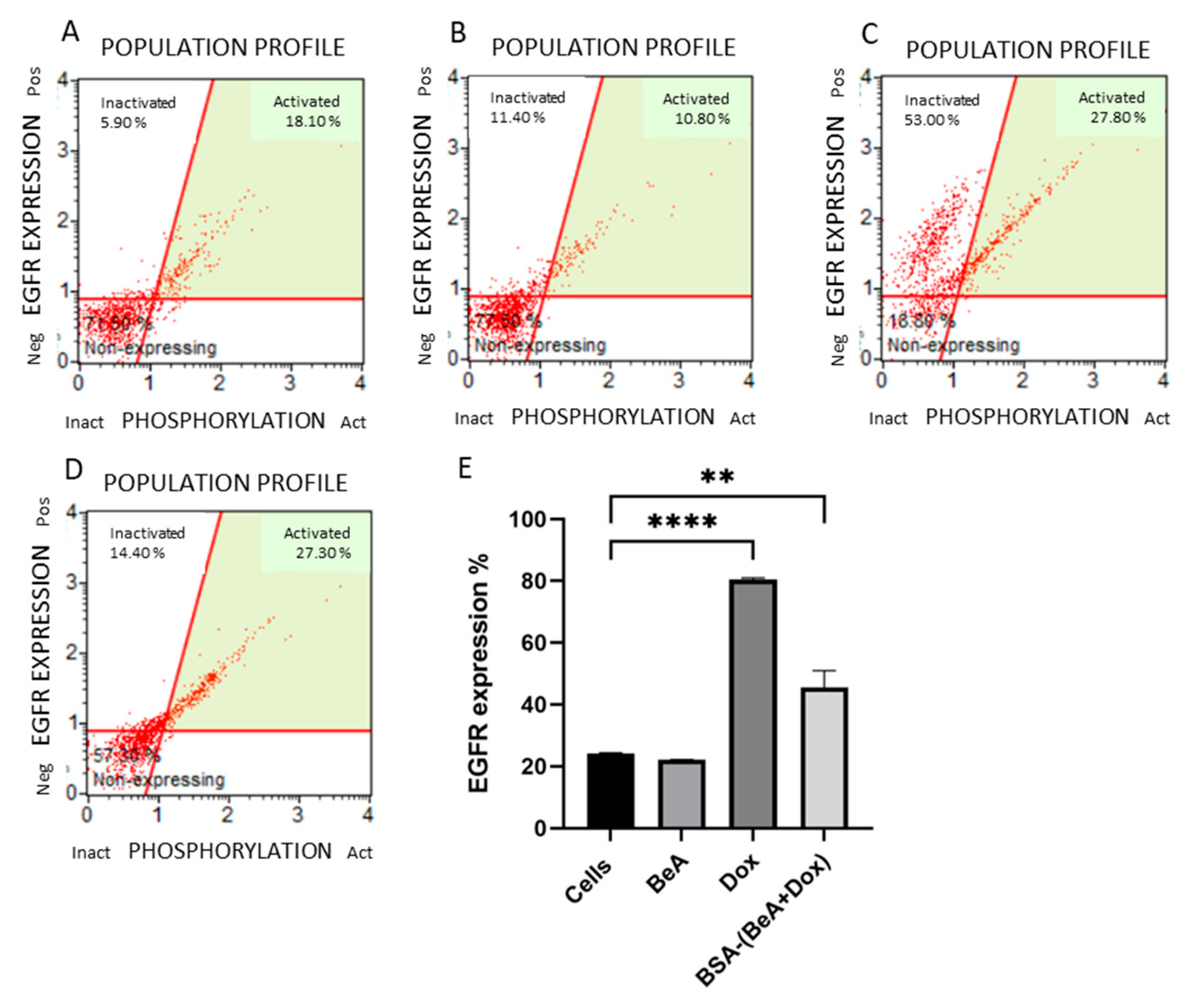
2.7.5. EGFR Expression
2.8. Statistical Analysis
3. Results
3.1. Preparation and Characterization of BSA-(BeA+Dox) DDS
3.1.1. Size and Charge of the DDS
3.1.2. Encapsulation Efficiency (EE) of BeA and Dox
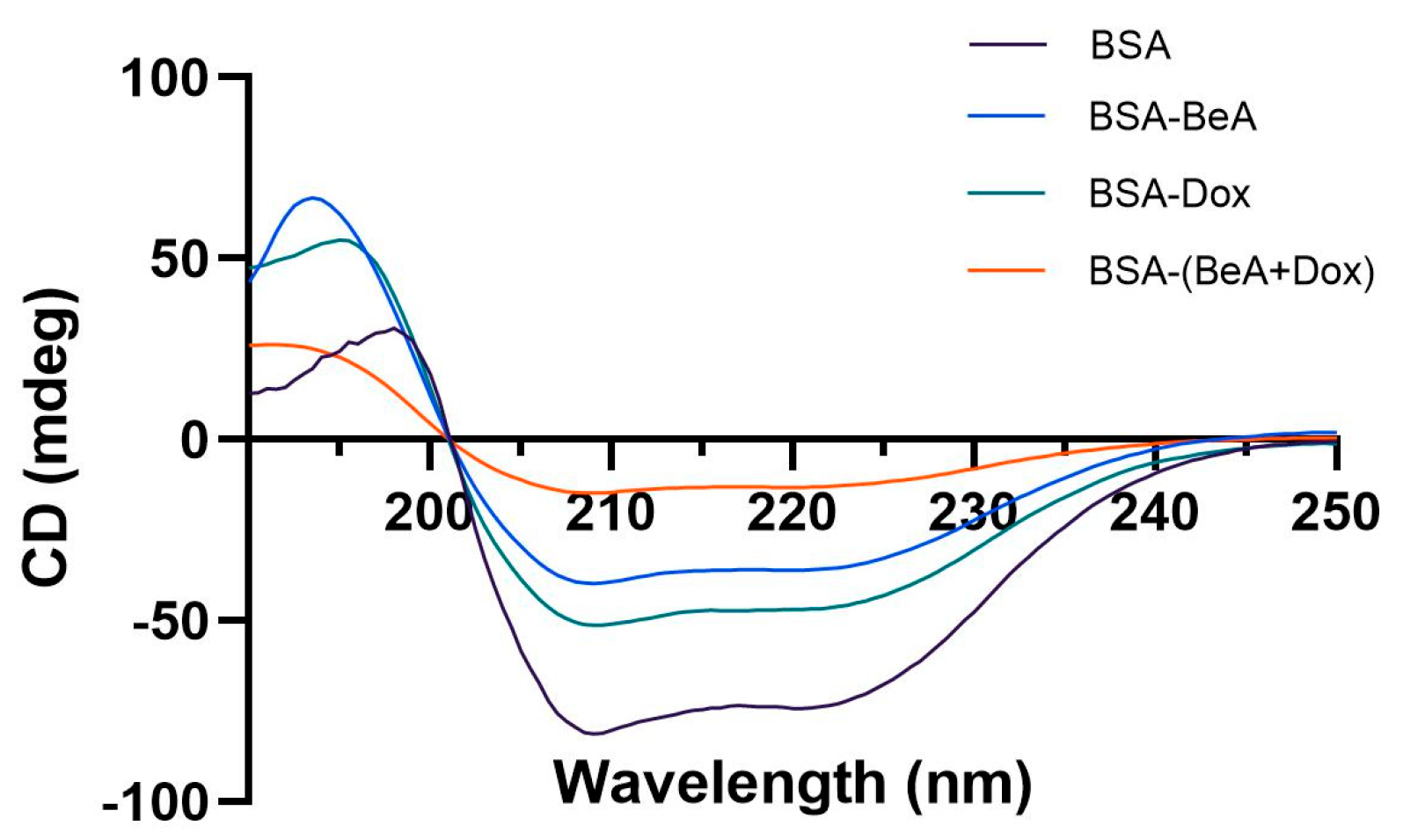
3.1.3. Circular Dichroism (CD) Analysis
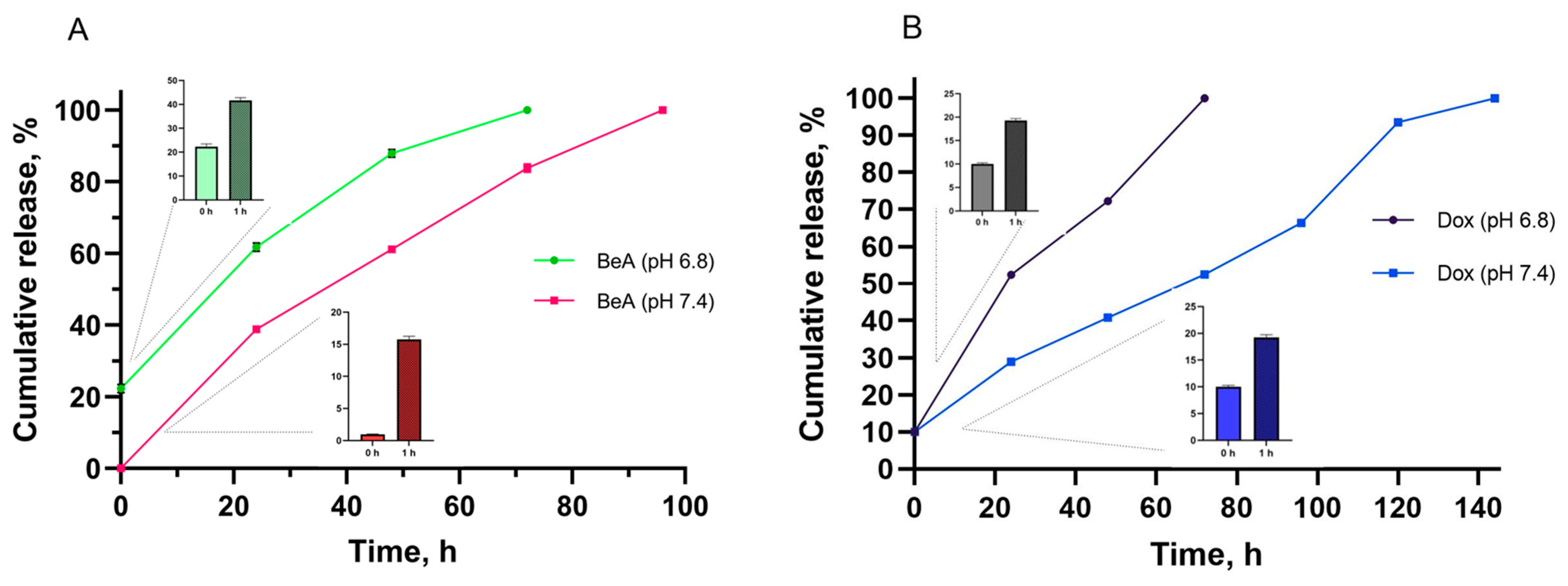
3.1.4. Cumulative Drug Release Profile
3.2. In Vitro Assays
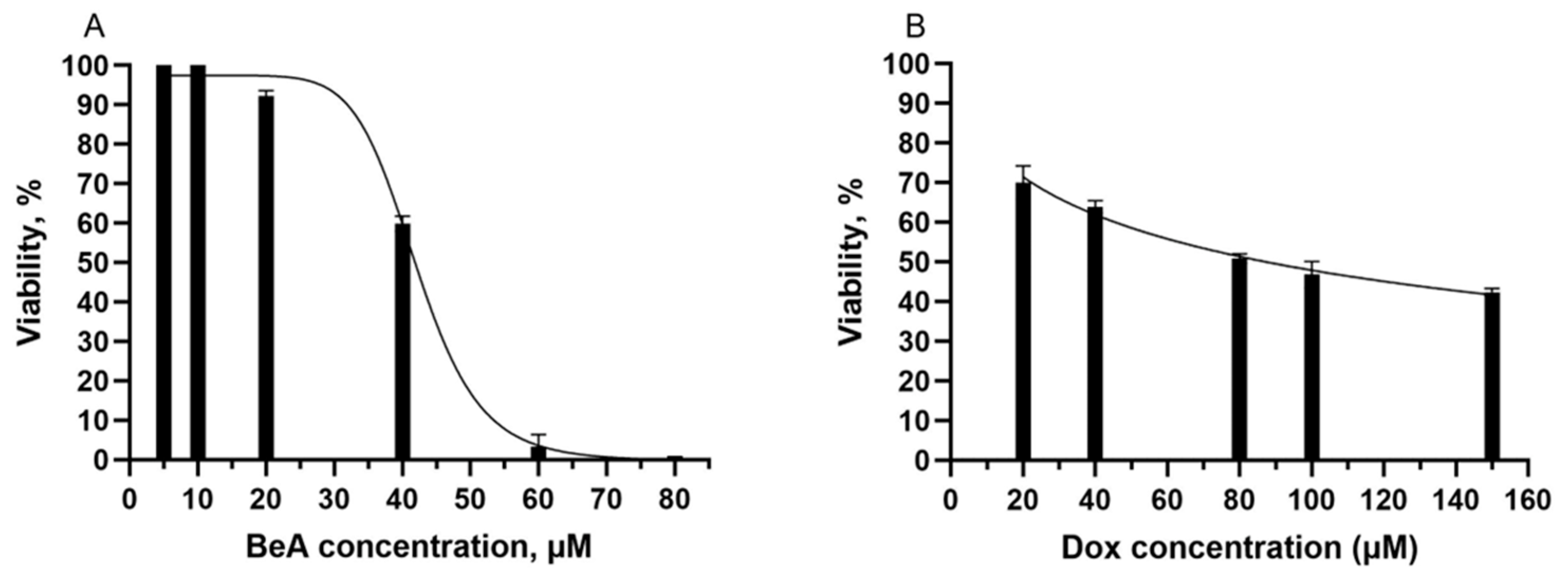
3.2.1. Cytotoxicity of BeA and Dox in A549 Cells
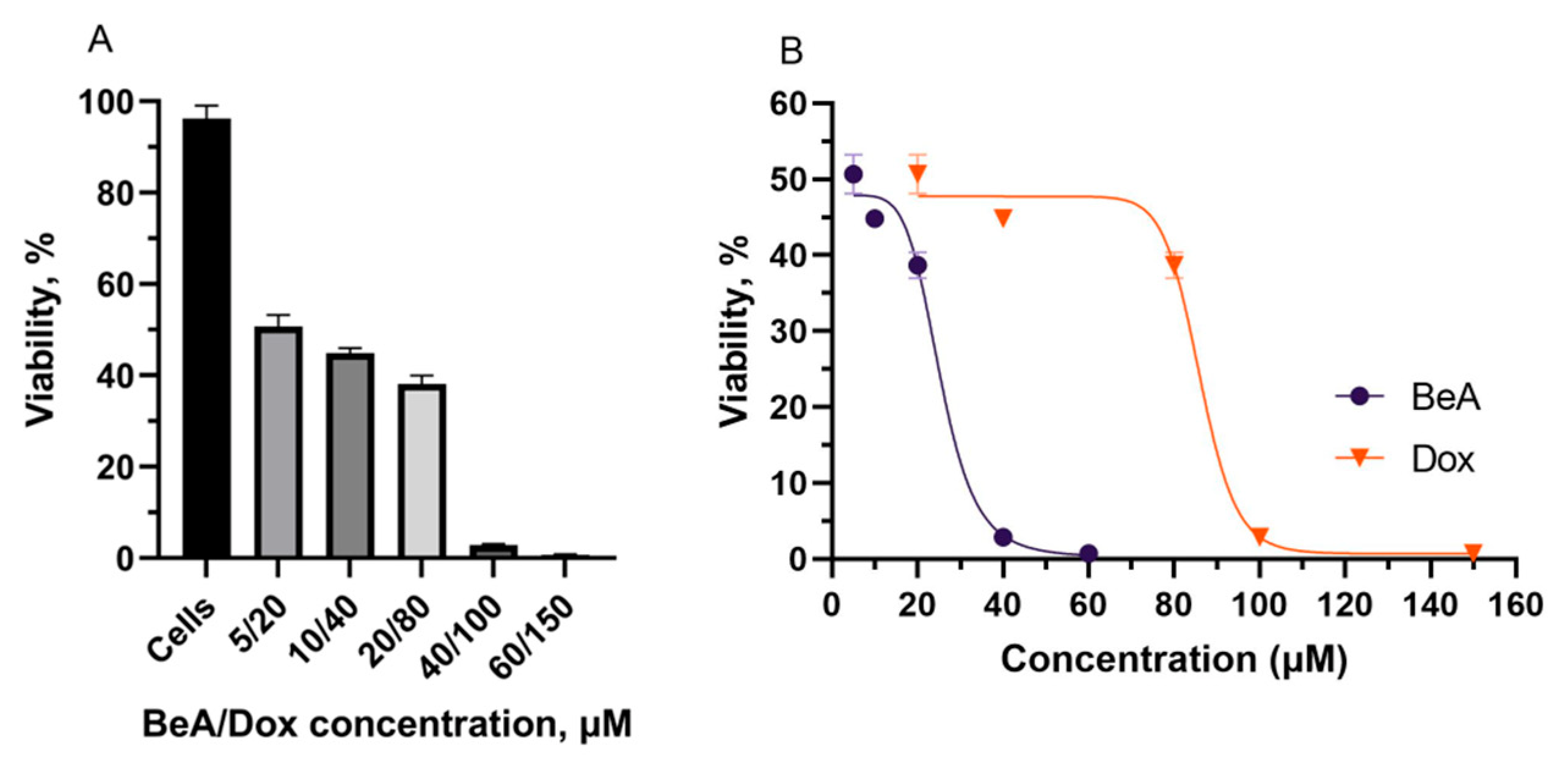
3.2.2. Molecular Interaction Effect of the BeA and Dox Combination
3.2.3. Cytotoxicity of the BSA-(BeA+Dox) DDS
3.2.4. Cellular Internalization of the BSA(BeA+Dox) DDS
3.3. Flow Cytometry Analysis
3.3.1. Effect of BSA-(BeA+Dox) DDS on the Cell Cycle
3.3.2. Effect of BSA-(BeA+Dox) on Caspase Activity
3.3.3. Effect of BSA-(BeA+Dox) on the DNA Processing Machinery
3.3.4. Effect of BSA-(BeA+Dox) on Oxidative Stress Production
3.3.5. Effect of BSA-(BeA+Dox) on Epidermal Growth Factor Receptor (EGFR) Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Figueroa, C.M.; Suarez, B.N.; Molina, A.M.; Fernandez, J.C.; Torres, Z.; Griebenow, K. Smart Release Nano-formulation of Cytochrome C and Hyaluronic Acid Induces Apoptosis in Cancer Cells. J. Nanomed. Nanotechnol. 2017, 8, 427. [Google Scholar]
- López-González, A.; Diz, P.; Gutierrez, L.; Almagro, E.; Palomo, A.G.; Provencio, M. The role of anthracyclines in small cell lung cancer. Ann. Transl. Med. 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Rathos, M.J.; Khanwalkar, H.; Joshi, K.; Manohar, S.M.; Joshi, K.S. Potentiation of in vitro and in vivoantitumor efficacy of doxorubicin by cyclin-dependent kinase inhibitor P276-00 in human non-small cell lung cancer cells. BMC Cancer 2013, 13, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christowitz, C.; Davis, T.; Isaacs, A.; Van Niekerk, G.; Hattingh, S.; Engelbrecht, A.-M. Mechanisms of doxorubicin-induced drug resistance and drug resistant tumour growth in a murine breast tumour model. BMC Cancer 2019, 19, 757. [Google Scholar] [CrossRef] [Green Version]
- Ijäs, H.; Shen, B.; Heuer-Jungemann, A.; Keller, A.; Kostiainen, M.A.; Liedl, T.; Ihalainen, J.A.; Linko, V. Unraveling the interaction between doxorubicin and DNA origami nanostructures for customizable chemotherapeutic drug release. Nucleic Acids Res. 2021, 49, 3048–3062. [Google Scholar] [CrossRef] [PubMed]
- Kaowinn, S.; Jun, S.W.; Kim, C.S.; Shin, D.-M.; Hwang, Y.-H.; Kim, K.; Shin, B.; Kaewpiboon, C.; Jeong, H.H.; Koh, S.S. Increased EGFR expression induced by a novel oncogene, CUG2, confers resistance to doxorubicin through Stat1-HDAC4 signaling. Cell Oncol. 2017, 40, 549–561. [Google Scholar] [CrossRef]
- Yu, X.; Xieripu, A.; Xu, Q.; Zulipikaer, A.; Song, Y.; Cai, L.; Chen, J. GSH-responsive curcumin/doxorubicin encapsulated Bactrian camel serum albumin nanocomposites with synergistic effect against lung cancer cells. J. Biomed. Res. 2020, 34, 54. [Google Scholar] [CrossRef]
- Suresh, C.; Zhao, H.; Gumbs, A.; Chetty, C.S.; Bose, H.S. New ionic derivatives of betulinic acid as highly potent anti-cancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Costa, J.F.; Meira, C.S.; Neves, M.V.G.d.; Dos Reis, B.P.Z.C.; Soares, M.B.P. Anti-Inflammatory Activities of Betulinic Acid: A Review. Front. Pharm. 2022, 13, 1866. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, X.; Dong, S.; Zhou, W. Betulinic acid in the treatment of tumour diseases: Application and research progress. Biomed. Pharm. 2021, 142, 111990. [Google Scholar] [CrossRef]
- Bravo-Alfaro, D.A.; Ochoa-Rodríguez, L.R.; Villaseñor-Ortega, F.; Luna-Barcenas, G.; García, H.S. Self-nanoemulsifying drug delivery system (SNEDDS) improves the oral bioavailability of betulinic acid. J. Mol. Liq. 2022, 364, 119946. [Google Scholar] [CrossRef]
- Saneja, A.; Arora, D.; Kumar, R.; Dubey, R.D.; Panda, A.K.; Gupta, P.N. Therapeutic applications of betulinic acid nanoformulations. Ann. N. Y. Acad. Sci. 2018, 1421, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short Iii, G.F.; Staunton, J.E.; Jin, X. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Ma, W.; Pei, J.; Ouyang, Q.; Tang, C.; Lai, L. Synergistic and antagonistic drug combinations depend on network topology. PLoS ONE 2014, 9, e93960. [Google Scholar] [CrossRef]
- Karami, K.; Jamshidian, N.; Hajiaghasi, A.; Amirghofran, Z. BSA nanoparticles as controlled release carriers for isophethalaldoxime palladacycle complex; synthesis, characterization, in vitro evaluation, cytotoxicity and release kinetics analysis. New J. Chem. 2020, 44, 4394–4405. [Google Scholar] [CrossRef]
- Topală, T.; Bodoki, A.; Oprean, L.; Oprean, R. Bovine serum albumin interactions with metal complexes. Clujul Med. 2014, 87, 215. [Google Scholar] [CrossRef] [Green Version]
- Motevalli, S.M.; Eltahan, A.S.; Liu, L.; Magrini, A.; Rosato, N.; Guo, W.; Bottini, M.; Liang, X.-J. Co-encapsulation of curcumin and doxorubicin in albumin nanoparticles blocks the adaptive treatment tolerance of cancer cells. Biophys. Rep. 2019, 5, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, R.; Garcia-Garrido, E.; Somoza, Á. Albumin-based nanoparticles for the delivery of doxorubicin in breast cancer. Cancers 2021, 13, 3011. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Guo, H.; Luan, X.; He, M.; Li, F.; Burnett, J.; Truchan, N.; Sun, D. Albumin nanoparticle of paclitaxel (Abraxane) decreases while taxol increases breast cancer stem cells in treatment of triple negative breast cancer. Mol. Pharm. 2020, 17, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Elbatanony, R.S.; Goyal, M.; Chavan, T.; Vega, N.; Kolluru, S.; Muth, A.; Gupta, V.; Kunda, N.K. Repurposing bedaquiline for effective non-small cell lung Cancer (NSCLC) therapy as inhalable cyclodextrin-based molecular inclusion complexes. Int. J. Mol. Sci. 2021, 22, 4783. [Google Scholar] [CrossRef] [PubMed]
- Skupin-Mrugalska, P.; Minko, T. Development of liposomal vesicles for osimertinib delivery to egfr mutation—Positive lung cancer cells. Pharmaceutics 2020, 12, 939. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, L.; Li, Y.; Song, S.; Qu, J.; He, R.; Ren, S.; Gong, P. An activatable, carrier-free, triple-combination nanomedicine for ALK/EGFR-mutant non-small cell lung cancer highly permeable targeted chemotherapy. New J. Chem. 2022, 46, 17673–17677. [Google Scholar] [CrossRef]
- Huang, X.; Chen, Q.; Li, X.; Lin, C.; Wang, K.; Luo, C.; Le, W.; Pi, X.; Liu, Z.; Chen, B. CKAP4 Antibody-Conjugated Si Quantum Dot Micelles for Targeted Imaging of Lung Cancer. Nanoscale Res. Lett. 2021, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.X. Development of high-drug-loading nanoparticles. Chem. Plus Chem. 2020, 85, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Z. Natural products, alone or in combination with FDA-approved drugs, to treat COVID-19 and lung cancer. Biomedicines 2021, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.; Lightner, C.; Elbai, S.; Brady, L.; Nicholson, S.; Ryan, R.; O’Sullivan, K.E.; O’Byrne, K.J.; Blanco-Aparicio, C.; Cuffe, S. Co-Targeting PIM Kinase and PI3K/mTOR in NSCLC. Cancers 2021, 13, 2139. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.M.; Morales-Cruz, M.; Benitez, M.; Berrios, K.; Figueroa, C.M.; Griebenow, K. Redox-sensitive cross-linking enhances albumin nanoparticle function as delivery system for photodynamic cancer therapy. J. Nanomed. Nanotechnol. 2016, 6, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, Y.; Morales-Cruz, M.; Hernández-Román, J.; Hernández, G.; Griebenow, K. Development of HAMLET-like cytochrome c-oleic acid nanoparticles for cancer therapy. J. Nanomed. Nanotechnol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Bradford assay for determining protein concentration. Cold Spring Harb. Protoc. 2020, 2020, pdb-prot102269. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Chandra, A.; Kaur, A.; Sabnis, N.; Lacko, A.; Gryczynski, Z.; Fudala, R.; Gryczynski, I. Fluorescence properties of doxorubicin in PBS buffer and PVA films. J. Photochem. Photobiol. B 2017, 170, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiai, S.; Oura, H.; Nakajima, T. Color reaction of some sapogenins and saponins with vanillin and sulfuric acid. Planta. Med. 1976, 29, 116–122. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Flores-Fernandez, G.M.; Morales-Cruz, M.; Orellano, E.A.; Rodriguez-Martinez, J.A.; Ruiz, M.; Griebenow, K. Two-step nanoprecipitation for the production of protein-loaded PLGA nanospheres. Results Pharm. Sci. 2012, 2, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Decuzzi, P.; Godin, B.; Tanaka, T.; Lee, S.-Y.; Chiappini, C.; Liu, X.; Ferrari, M. Size and shape effects in the biodistribution of intravascularly injected particles. J. Control. Release 2010, 141, 320–327. [Google Scholar] [CrossRef]
- Augustine, R.; Hasan, A.; Primavera, R.; Wilson, R.J.; Thakor, A.S.; Kevadiya, B.D. Cellular uptake and retention of nanoparticles: Insights on particle properties and interaction with cellular components. Mater. Today Commun. 2020, 25, 101692. [Google Scholar] [CrossRef]
- Larsson, M.; Hill, A.; Duffy, J. Suspension stability; why particle size, zeta potential and rheology are important. Annu. Trans. Nord. Rheol. Soc. 2012, 20, 209–214. [Google Scholar]
- Clogston, J.D.; Patri, A.K. Zeta potential measurement. In Characterization of Nanoparticles Intended for Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2011; pp. 63–70. [Google Scholar]
- Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191. [Google Scholar] [CrossRef] [Green Version]
- Harush-Frenkel, O.; Rozentur, E.; Benita, S.; Altschuler, Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules 2008, 9, 435–443. [Google Scholar] [CrossRef]
- Sun, C.; Yang, J.; Wu, X.; Huang, X.; Wang, F.; Liu, S. Unfolding and refolding of bovine serum albumin induced by cetylpyridinium bromide. Biophys J. 2005, 88, 3518–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, S.H.; Choi, Y.; Choi, J. Stimuli-responsive nanomaterials for application in antitumor therapy and drug delivery. Pharmaceutics 2020, 12, 630. [Google Scholar] [CrossRef]
- Lee, C.; Kim, Y.-J.; Kim, K.S.; Lee, J.Y.; Kim, D.-N. Modulating the chemo-mechanical response of structured DNA assemblies through binding molecules. Nucleic Acids Res. 2021, 49, 12591–12599. [Google Scholar] [CrossRef]
- Nasmyth, K. Putting the cell cycle in order. Science 1996, 274, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Gao, L.; Tan, Y.; Cai, J.; Ye, Z.; Chen, A.T.; Xu, Y.; Zhao, L.; Tong, S. Betulinic acid self-assembled nanoparticles for effective treatment of glioblastoma. J. Nanobiotechnol. 2022, 20, 39. [Google Scholar] [CrossRef]
- Park, E.-J.; Kwon, H.-K.; Choi, Y.-M.; Shin, H.-J.; Choi, S. Doxorubicin induces cytotoxicity through upregulation of perk–dependent ATF3. PLoS ONE 2012, 7, e44990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-S.; Lee, Y.-S.; Kim, D.-K. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef]
- Gamen, S.; Anel, A.; Pérez-Galán, P.; Lasierra, P.; Johnson, D.; Piñeiro, A.; Naval, J. Doxorubicin treatment activates a Z-VAD-sensitive caspase, which causes Δψm loss, caspase-9 activity, and apoptosis in Jurkat cells. Exp. Cell Res. Suppl. 2000, 258, 223–235. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Chen, Y.-M. Influence of chemotherapy on EGFR mutation status. Transl. Lung Cancer Res. 2013, 2, 442. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Protein-based nanocarriers as promising drug and gene delivery systems. J. Control. Release 2012, 161, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.M.K.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the interaction between FcRn and albumin: Opportunities for design of albumin-based therapeutics. Front. Immunol. 2015, 5, 682. [Google Scholar] [CrossRef] [Green Version]
- Malleda, C.; Ahalawat, N.; Gokara, M.; Subramanyam, R. Molecular dynamics simulation studies of betulinic acid with human serum albumin. J. Mol. Model 2012, 18, 2589–2597. [Google Scholar] [CrossRef]
- Agudelo, D.; Bourassa, P.; Bruneau, J.; Berube, G.; Asselin, E.; Tajmir-Riahi, H.-A. Probing the binding sites of antibiotic drugs doxorubicin and N-(trifluoroacetyl) doxorubicin with human and bovine serum albumins. PLoS ONE 2012, 7, e43814. [Google Scholar] [CrossRef] [Green Version]
- Kamran, S.; Sinniah, A.; Chik, Z.; Alshawsh, M.A. Diosmetin Exerts Synergistic Effects in Combination with 5-Fluorouracil in Colorectal Cancer Cells. Biomedicines 2022, 10, 531. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, N.; Huang, Y.; Dou, X.; Peng, X.; Wang, W.; Zhang, Z.; Wang, R.; Qiu, Y.; Jin, M. Lansoprazole Alone or in Combination With Gefitinib Shows Antitumor Activity Against Non-small Cell Lung Cancer A549 Cells in vitro and in vivo. Front. Cell Dev. Biol. 2021, 9, 655559. [Google Scholar] [CrossRef]
- Park, J.; Baek, S.H. Combination therapy with cinnamaldehyde and hyperthermia induces apoptosis of A549 non-small cell lung carcinoma cells via regulation of reactive oxygen species and mitogen-activated protein kinase family. Int. J. Mol. Sci. 2020, 21, 6229. [Google Scholar] [CrossRef]
- Han, Y.; Ma, R.; Cao, G.; Liu, H.; He, L.; Tang, L.; Li, H.; Luo, Q. Combined treatment of cinobufotalin and gefitinib exhibits potent efficacy against lung cancer. Evid. Based Complement. Altern. Med. 2021, 2021, 6612365. [Google Scholar] [CrossRef]
- Kang, S.; Kim, J.H.; Hong, J.; Moon, J.H.; Kwon, Y.Y.; Ko, S.-G. SH005S7 Overcomes Primary and Acquired Resistance of Non-Small Cell Lung Cancer by Combined MET/EGFR/HER3 Inhibition. BioMed Res. Int. 2022, 2022, 1840541. [Google Scholar] [CrossRef]
- Lee, S.-H.; Griffiths, J.R. How and why are cancers acidic? Carbonic anhydrase IX and the homeostatic control of tumour extracellular pH. Cancers 2020, 12, 1616. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.-M.; Lu, J.-F.; Edoo, M.I.A.; Zhou, L.; Xie, H.-Y.; Zheng, S.-S.; Li, Q.-Y. MRC-5 Cancer-associated Fibroblasts Influence Production of Cancer Stem Cell Markers and Inflammation-associated Cell Surface Molecules, in Liver Cancer Cell Lines. Int. J. Med. Sci. 2019, 16, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukourakis, M.I.; Kalamida, D.; Mitrakas, A.G.; Liousia, M.; Pouliliou, S.; Sivridis, E.; Giatromanolaki, A. Metabolic cooperation between co-cultured lung cancer cells and lung fibroblasts. Lab. Investig. 2017, 97, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDS | Z Potential (mV) | %PDI | Size (Hydrodynamic Radius, nm) | DLS Graph |
|---|---|---|---|---|
| BSA | −16 ± 5 | 57 ± 0 | 71 ± 14 |  |
| BSA-BeA | −4.6 ± 0.4 | 27 ± 2 | 97 ± 1 |  |
| BSA-Dox | −3 ± 1 | 51 ± 14 | 138 ± 13 |  |
| BSA-(BeA+Dox) | −2.1 ± 0.7 | 23.2 ± 0.4 | 181 ± 2 |  |
| DDS | BSA (µM) | Dox (µM) | BeA (µM) |
|---|---|---|---|
| BSA-BeA | 77 ± 11 | - | 18 ± 6 |
| BSA-Dox | 131 ± 24 | 43 ± 6 | - |
| BSA-(BeA+Dox) | 110 ± 3 | 61 ± 6 | 28 ± 12 |
| DDS | Drug EE (%) | Carrier Yield (%) | |
|---|---|---|---|
| BeA | Dox | BSA | |
| BSA-BeA | 10 ± 2 | - | 58 ± 2 |
| BSA-Dox | - | 53 ± 7 | 37 ± 5 |
| BSA-(BeA+Dox) | 18 ± 4 | 77 ± 15 | 80 ± 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Martinez, Z.; Pérez, D.; Torres, G.; Estrada, S.; Correa, C.; Mederos, N.; Velazquez, K.; Castillo, B.; Griebenow, K.; Delgado, Y. A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC. BioTech 2023, 12, 13. https://doi.org/10.3390/biotech12010013
Torres-Martinez Z, Pérez D, Torres G, Estrada S, Correa C, Mederos N, Velazquez K, Castillo B, Griebenow K, Delgado Y. A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC. BioTech. 2023; 12(1):13. https://doi.org/10.3390/biotech12010013
Chicago/Turabian StyleTorres-Martinez, Zally, Daraishka Pérez, Grace Torres, Sthephanie Estrada, Clarissa Correa, Natasha Mederos, Kimberly Velazquez, Betzaida Castillo, Kai Griebenow, and Yamixa Delgado. 2023. "A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC" BioTech 12, no. 1: 13. https://doi.org/10.3390/biotech12010013
APA StyleTorres-Martinez, Z., Pérez, D., Torres, G., Estrada, S., Correa, C., Mederos, N., Velazquez, K., Castillo, B., Griebenow, K., & Delgado, Y. (2023). A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC. BioTech, 12(1), 13. https://doi.org/10.3390/biotech12010013