
Nanotherapeutic Approaches to Treat COVID-19-Induced Pulmonary Fibrosis


Abstract
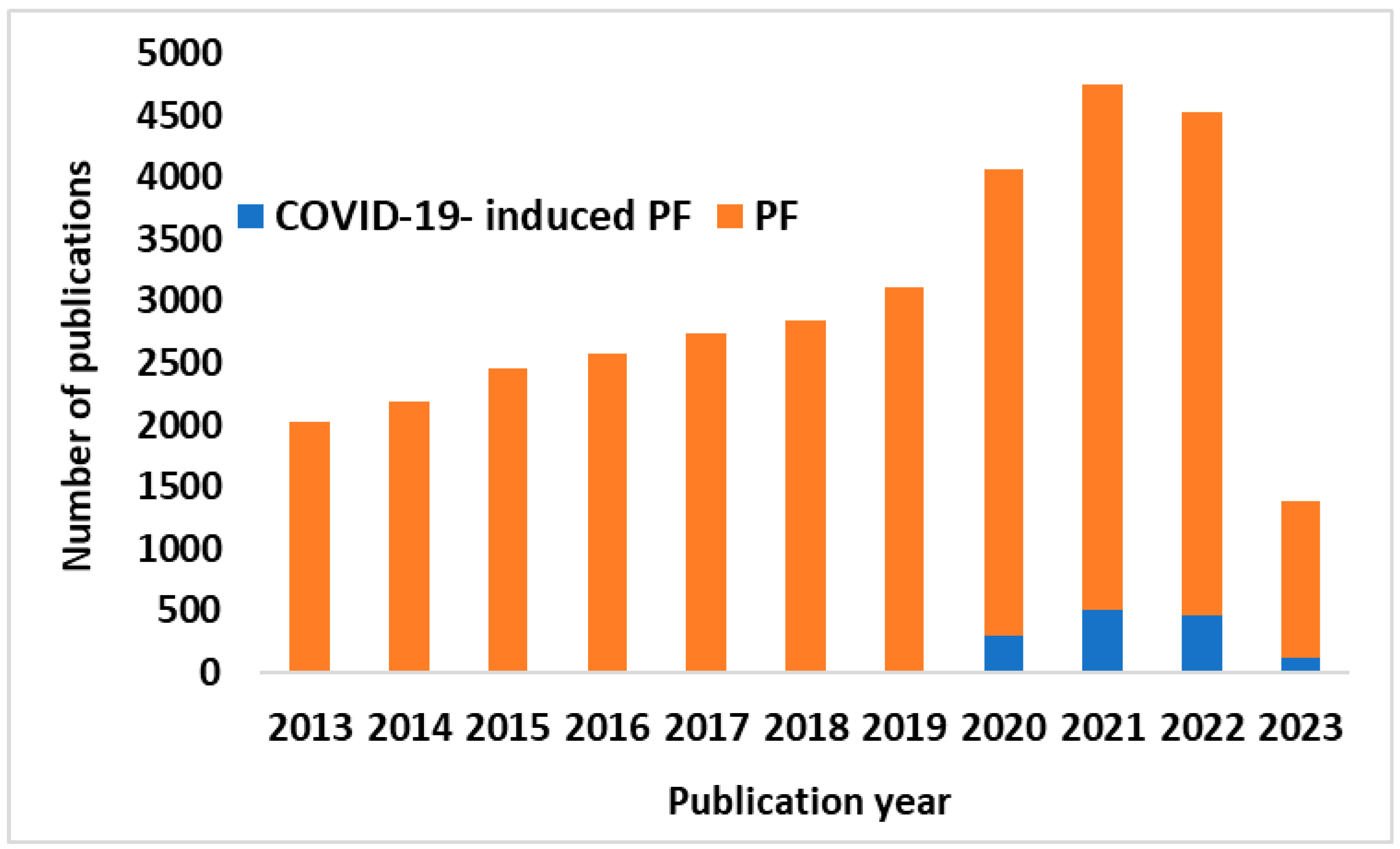
:1. Background
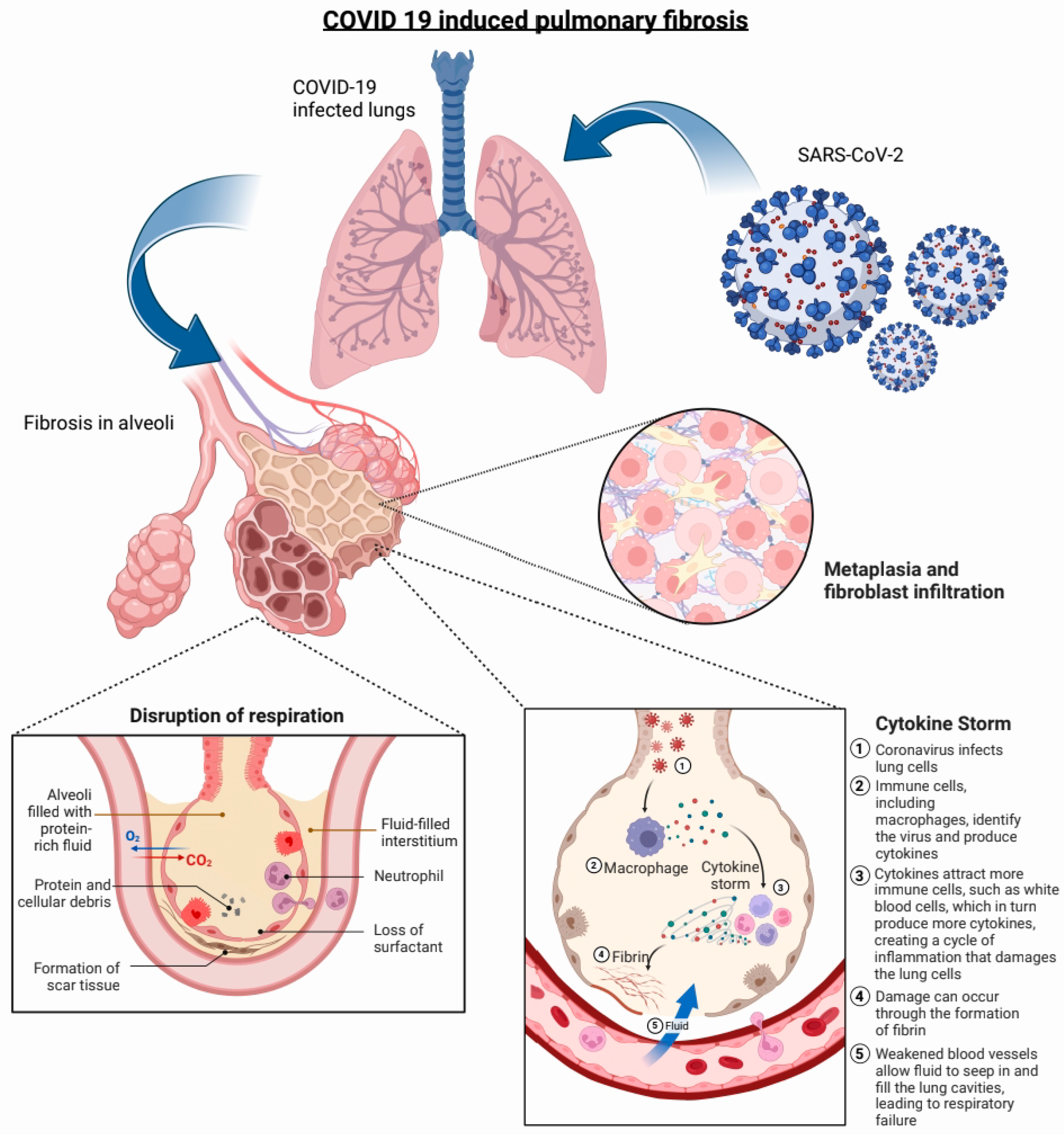
2. Pathophysiology of COVID-19-Induced PF
3. Current Status of COVID-19-Induced PF Therapy
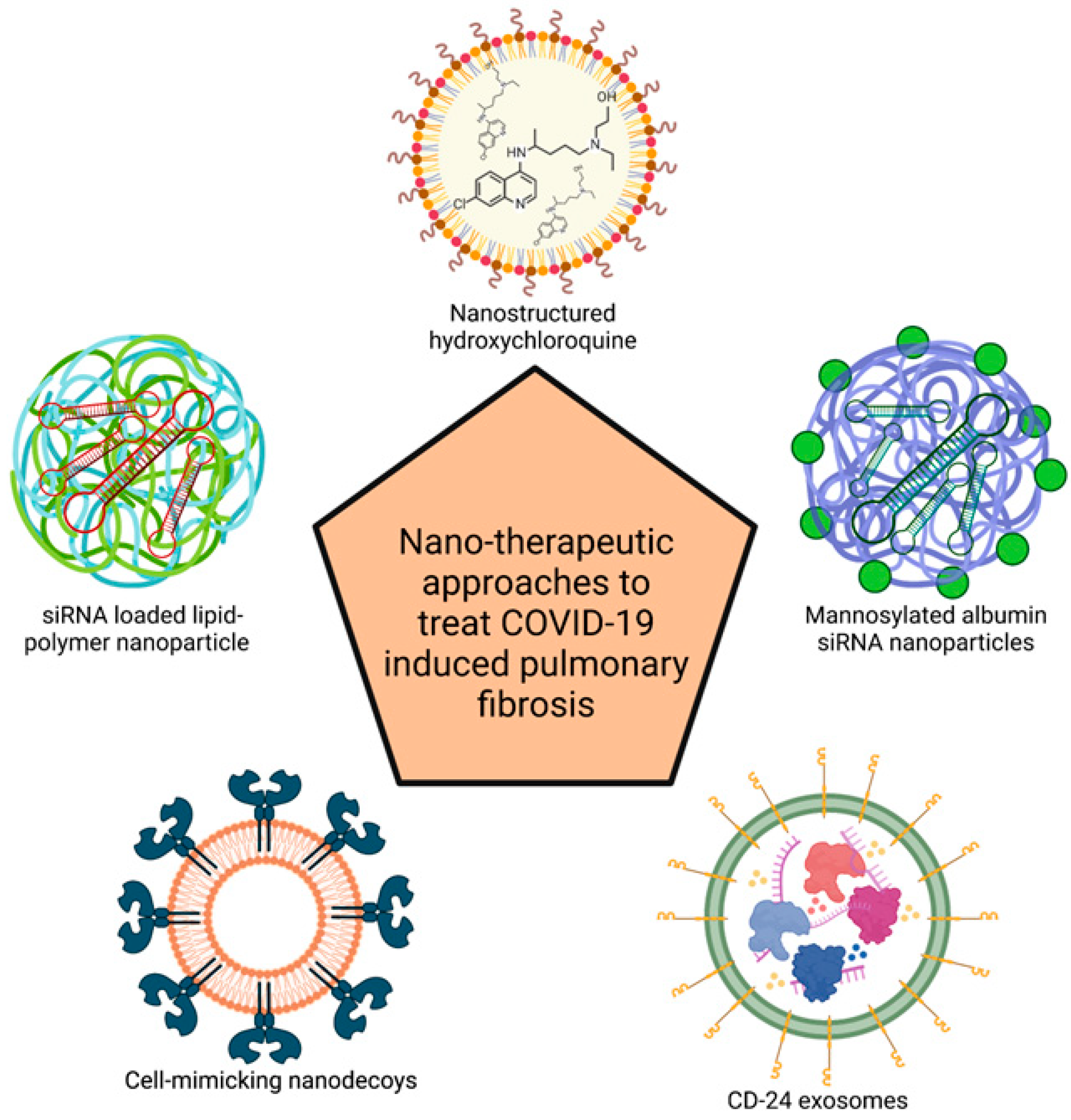
4. Nanotherapeutic Approaches to Treat COVID-19-Induced PF
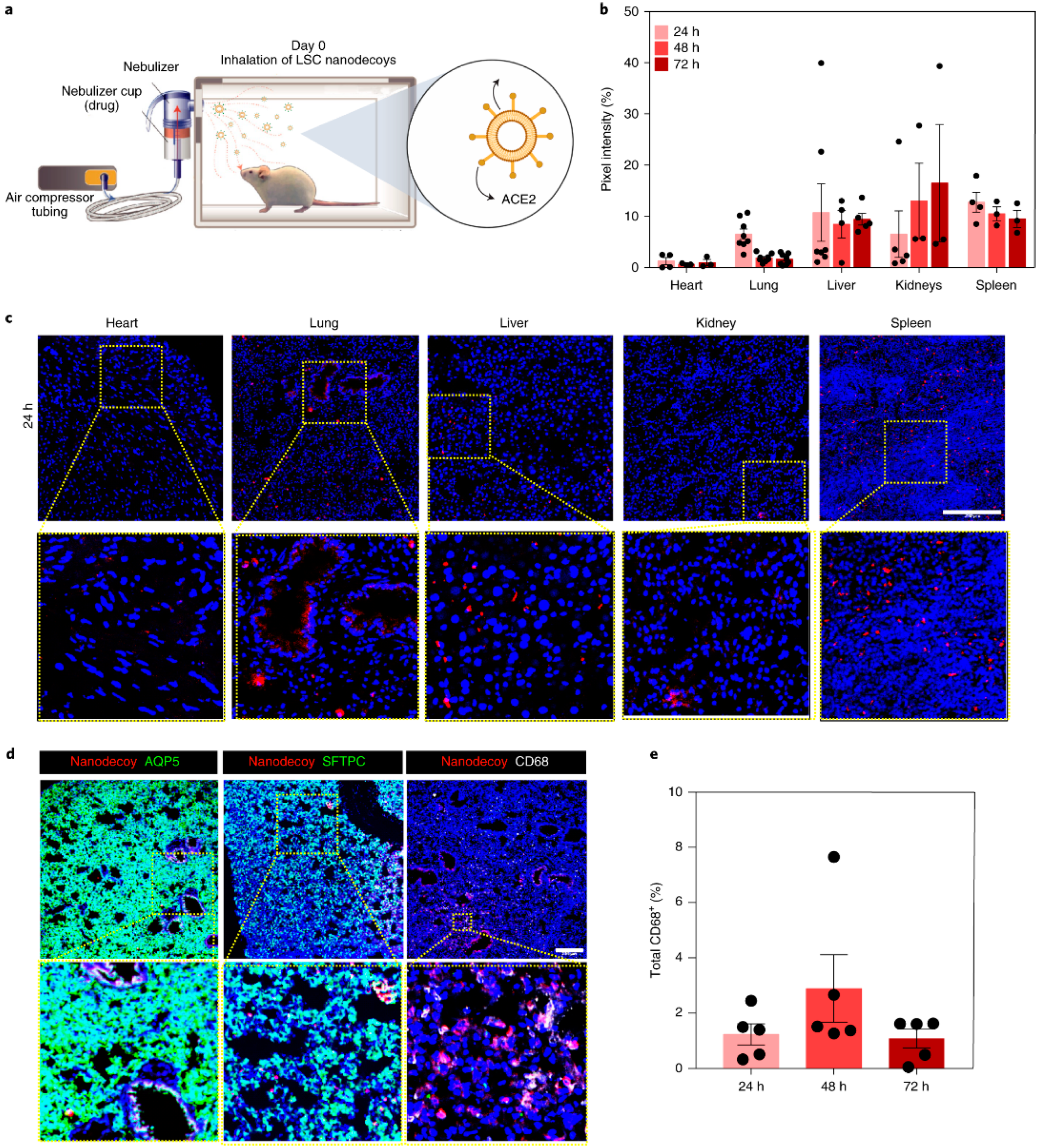
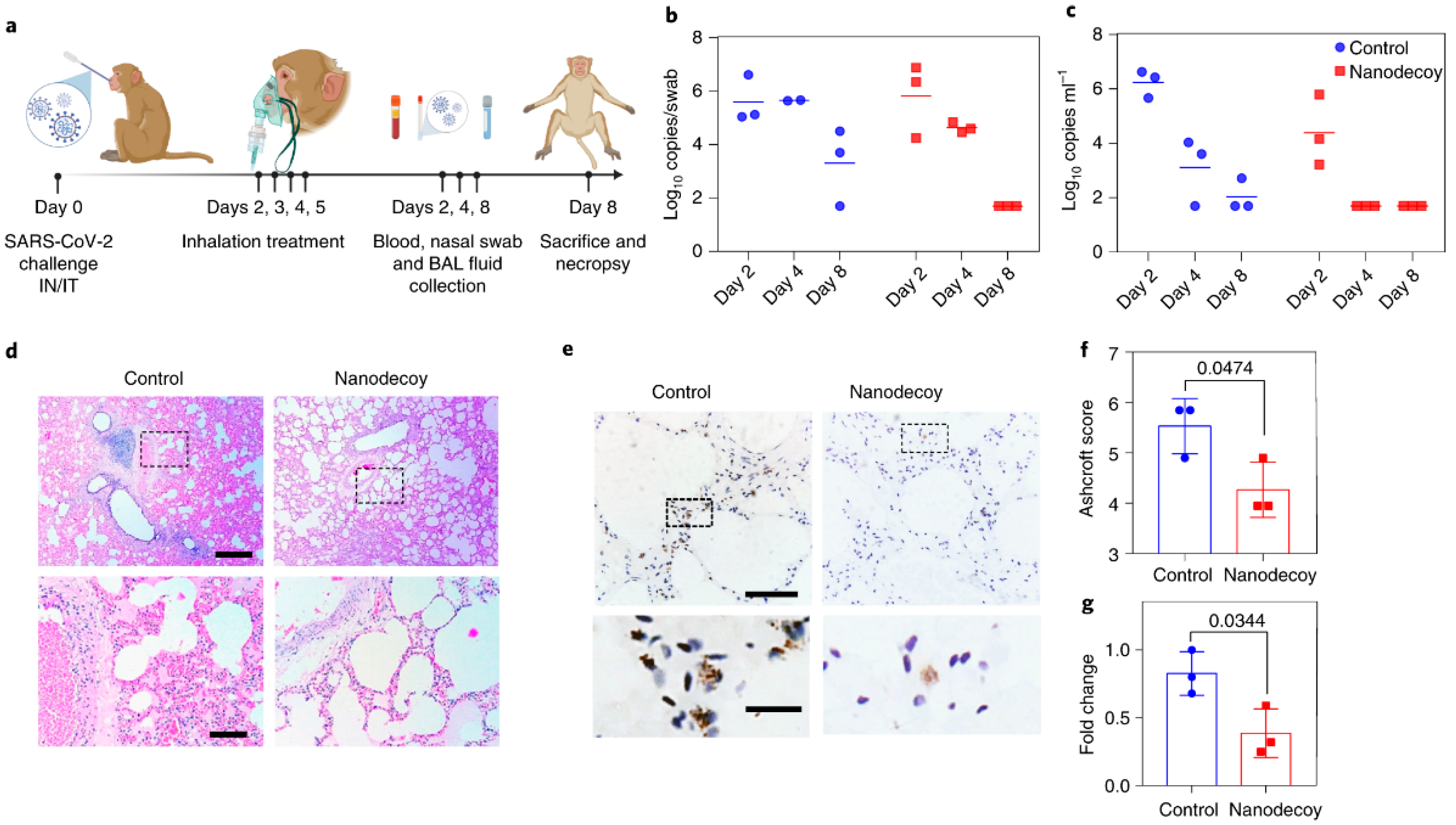
4.1. Cell-Mimicking Nanodecoys
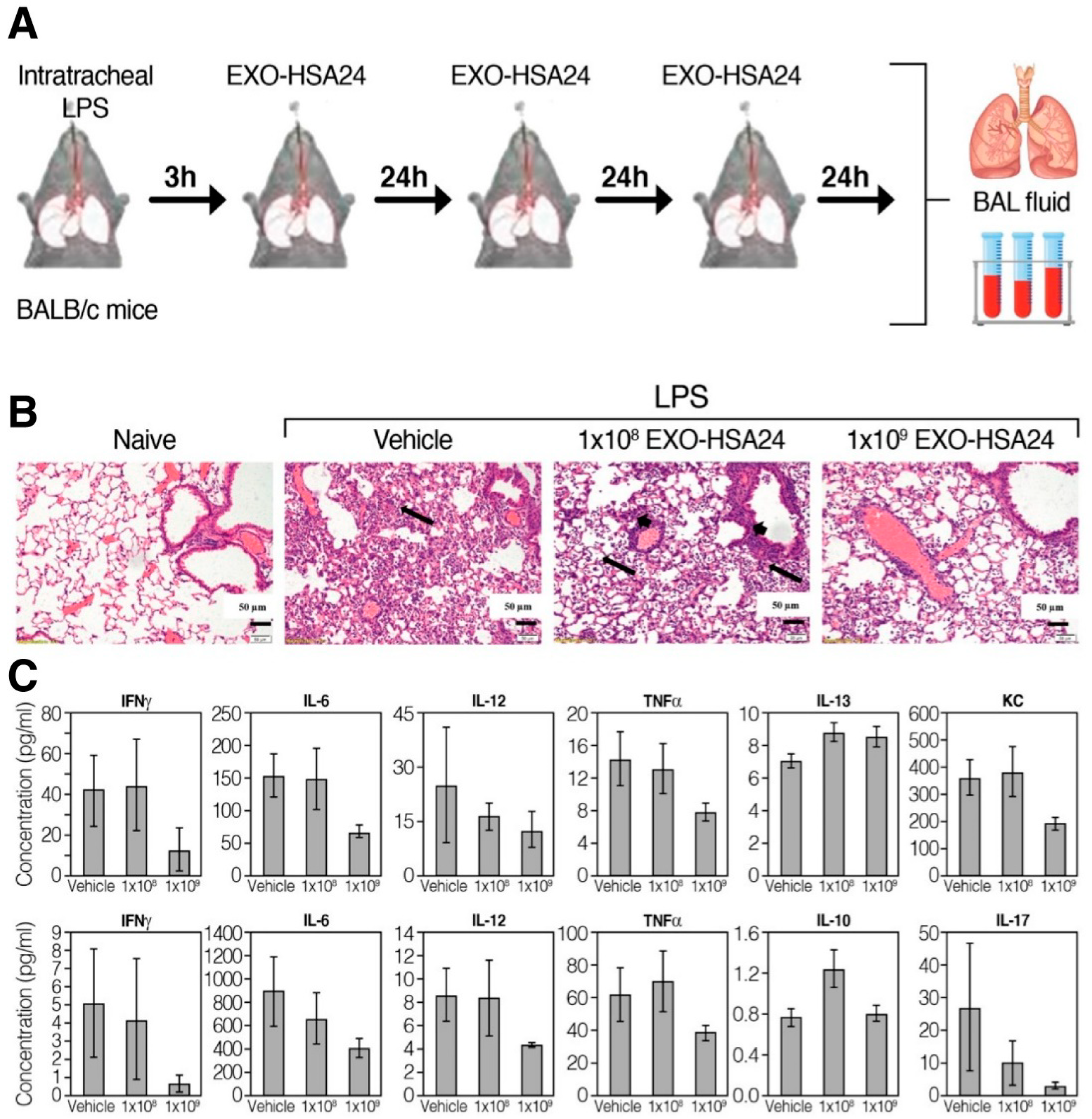
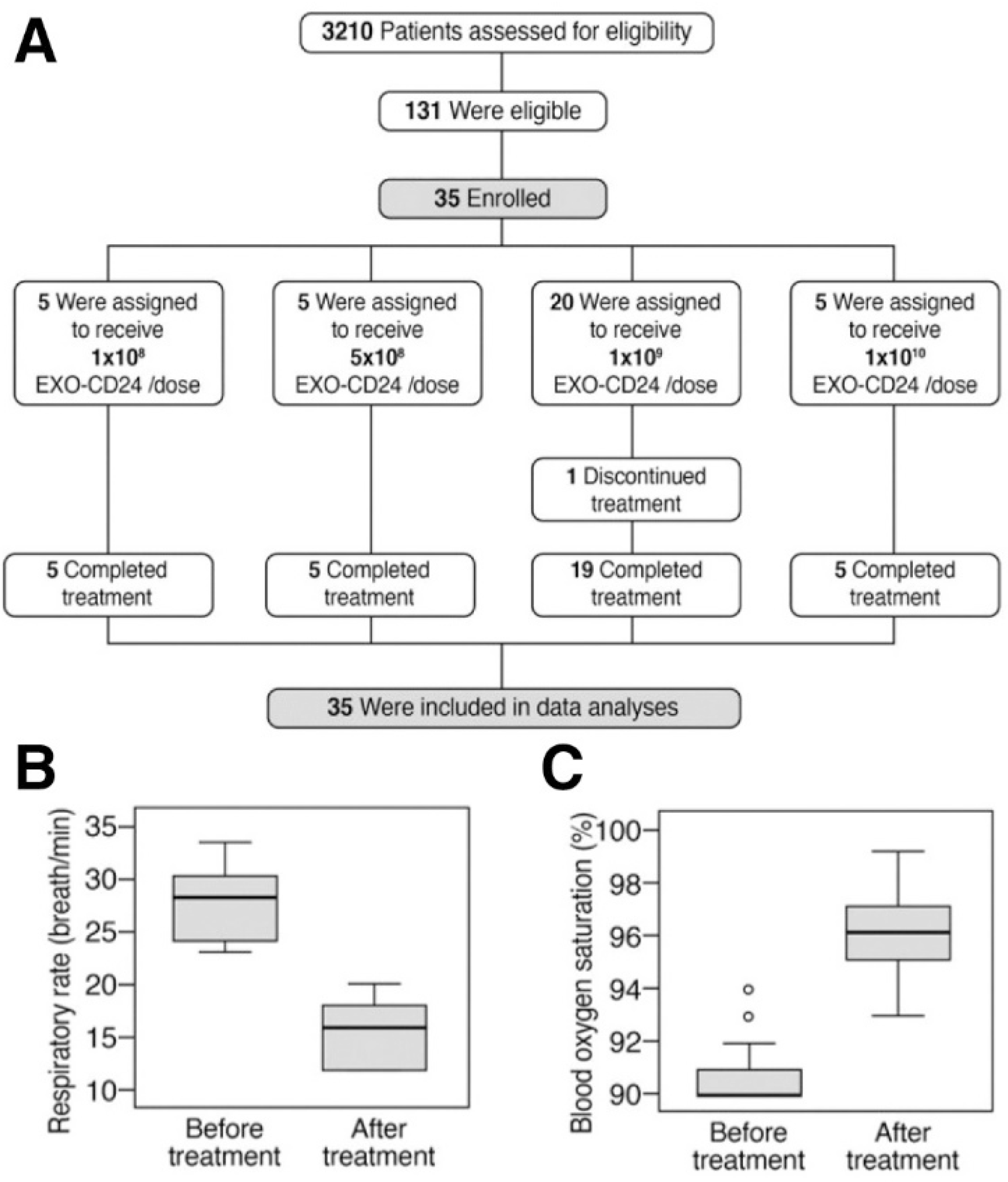
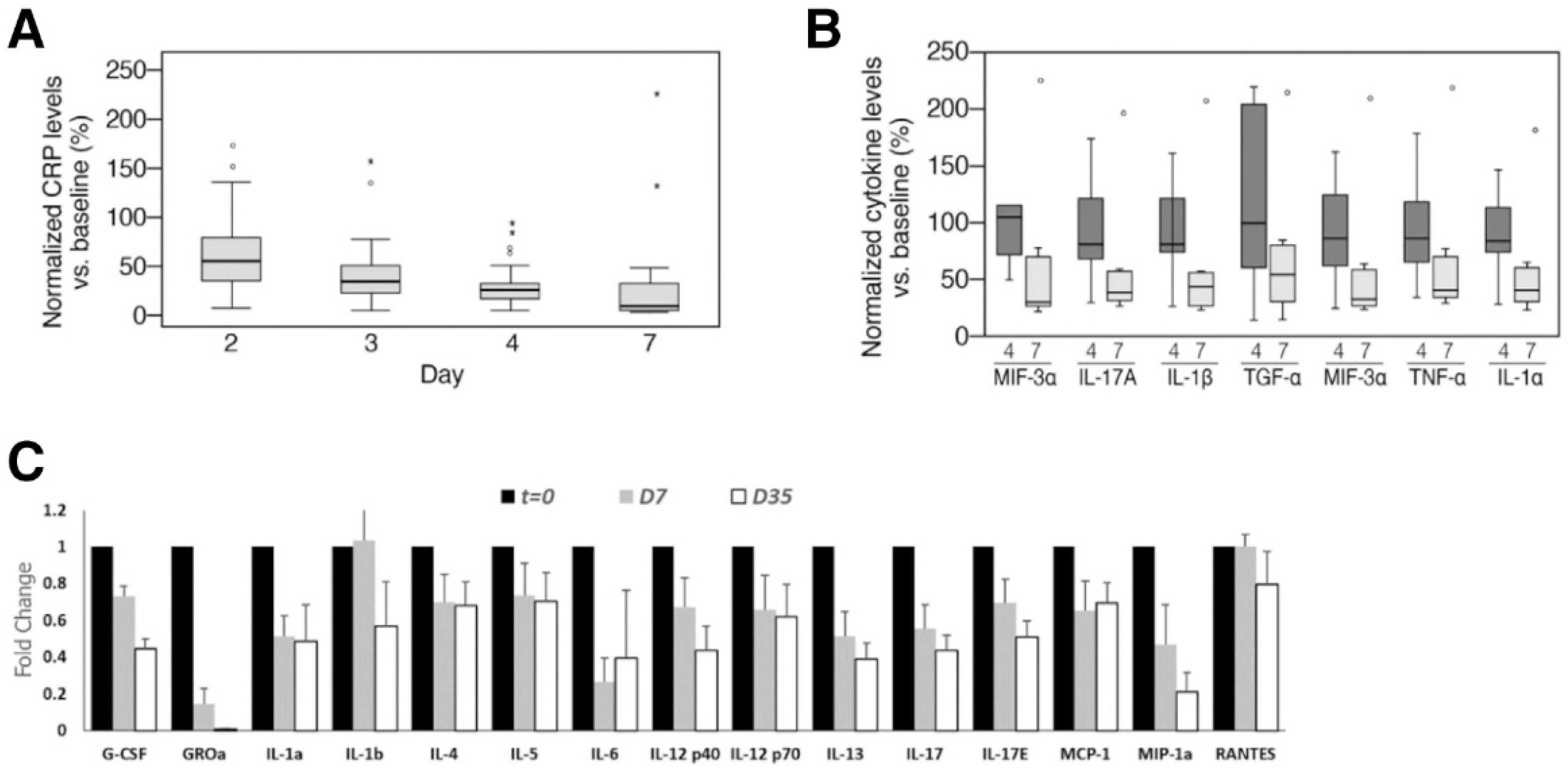
4.2. CD-24 Exosomes
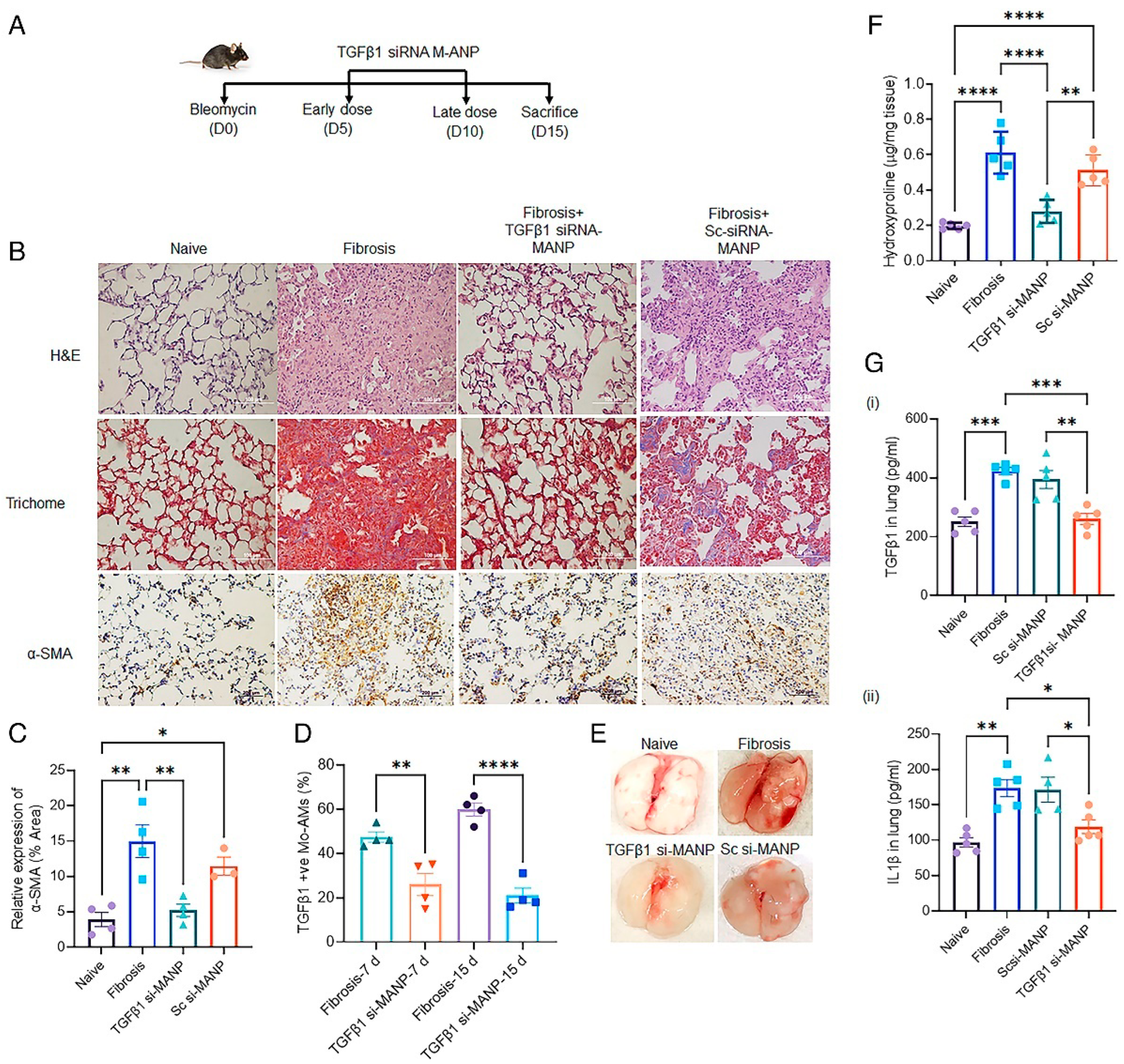
4.3. Mannosylated Albumin-siRNA NPs
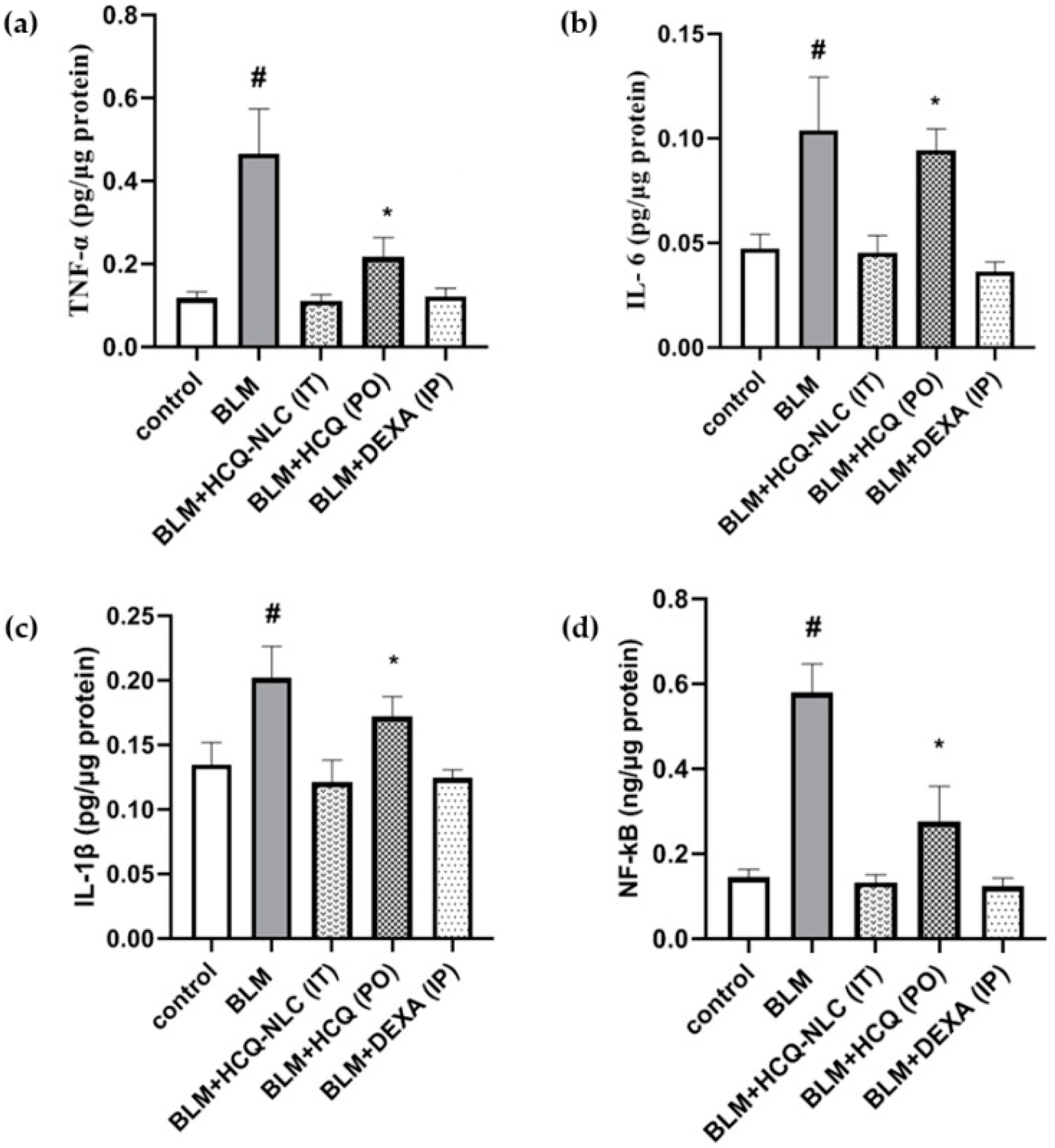
4.4. Nanostructured Hydroxychloroquine
4.5. PLGA-PEG-G0-C14-IL11 siRNA NPs
5. Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- George, P.M.; Patterson, C.M.; Reed, A.K.; Thillai, M. Lung Transplantation for Idiopathic Pulmonary Fibrosis. Lancet Respir. Med. 2019, 7, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C. Pulmonary Fibrosis, Part I: Epidemiology, Pathogenesis, and Diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic Pulmonary Fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-Related Lung Fibrosis Is Diagnostically Heterogeneous but Uniformly Progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Yang, I. V Epigenomics of Idiopathic Pulmonary Fibrosis. Epigenomics 2012, 4, 195–203. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Kaminski, N. Epigenetics in Idiopathic Pulmonary Fibrosis. Biochem. Cell Biol. 2015, 93, 159–170. [Google Scholar] [CrossRef]
- Caminati, A.; Madotto, F.; Cesana, G.; Conti, S.; Harari, S. Epidemiological Studies in Idiopathic Pulmonary Fibrosis: Pitfalls in Methodologies and Data Interpretation. Eur. Respir. Rev. 2015, 24, 436–444. [Google Scholar] [CrossRef]
- Raghu, G.; Chen, S.-Y.; Yeh, W.-S.; Maroni, B.; Li, Q.; Lee, Y.-C.; Collard, H.R. Idiopathic Pulmonary Fibrosis in US Medicare Beneficiaries Aged 65 Years and Older: Incidence, Prevalence, and Survival, 2001–11. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Tanni, S.E.; Fabro, A.T.; de Albuquerque, A.; Ferreira, E.V.M.; Verrastro, C.G.Y.; Sawamura, M.V.Y.; Ribeiro, S.M.; Baldi, B.G. Pulmonary Fibrosis Secondary to COVID-19: A Narrative Review. Expert Rev. Respir. Med. 2021, 15, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.-N.; Sun, L.; Wang, B.-R.; Zou, Y.; Xu, S.; Ding, Y.-J.; Shen, L.-J.; Huang, W.-C.; Jiang, X.-J.; Chen, S.-M. The Characteristics and Evolution of Pulmonary Fibrosis in COVID-19 Patients as Assessed by AI-Assisted Chest HRCT. PLoS ONE 2021, 16, e0248957. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Nathan, S.D.; Hill, C.; Marshall, J.; Dejonckheere, F.; Thuresson, P.-O.; Maher, T.M. Predicting Life Expectancy for Pirfenidone in Idiopathic Pulmonary Fibrosis. J. Manag. Care Spec. Pharm. 2017, 23, S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Fadista, J.; Kraven, L.M.; Karjalainen, J.; Andrews, S.J.; Geller, F.; Baillie, J.K.; Wain, L.V.; Jenkins, R.G.; Feenstra, B. Shared Genetic Etiology between Idiopathic Pulmonary Fibrosis and COVID-19 Severity. eBioMedicine 2021, 65. [Google Scholar] [CrossRef]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 Infection Triggers Profibrotic Macrophage Responses and Lung Fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Parimon, T.; Espindola, M.; Marchevsky, A.; Rampolla, R.; Chen, P.; Hogaboam, C.M. Potential Mechanisms for Lung Fibrosis Associated with COVID-19 Infection. QJM An Int. J. Med. 2022, 1–6. [Google Scholar] [CrossRef]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The Spatial Landscape of Lung Pathology during COVID-19 Progression. Nature 2021, 593, 564–569. [Google Scholar] [CrossRef]
- Flaifel, A.; Kwok, B.; Ko, J.; Chang, S.; Smith, D.; Zhou, F.; Chiriboga, L.A.; Zeck, B.; Theise, N.; Rudym, D.; et al. Pulmonary Pathology of End-Stage COVID-19 Disease in Explanted Lungs and Outcomes After Lung Transplantation. Am. J. Clin. Pathol. 2022, 157, 908–926. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative Stress and Pulmonary Fibrosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Ksajikian, A.; Overbey, J.; Li, P.; Li, Y. Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review. Cells 2022, 11. [Google Scholar] [CrossRef]
- Sehlmeyer, K.; Ruwisch, J.; Roldan, N.; Lopez-Rodriguez, E. Corrigendum: Alveolar Dynamics and Beyond—The Importance of Surfactant Protein C and Cholesterol in Lung Homeostasis and Fibrosis. Front. Physiol. 2020, 11, 943. [Google Scholar] [CrossRef]
- Seibold, M.A.; Smith, R.W.; Urbanek, C.; Groshong, S.D.; Cosgrove, G.P.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A.; Reynolds, S.D. The Idiopathic Pulmonary Fibrosis Honeycomb Cyst Contains A Mucocilary Pseudostratified Epithelium. PLoS ONE 2013, 8, e58658. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Ridley, R.; Brereton, C.; Alzetani, A.; Johnson, B.; Marshall, B.G.; Fletcher, S.V.; Ottensmeier, C.H.; Richeldi, L.; Skipp, P.; et al. Paracrine SPARC Signaling Dysregulates Alveolar Epithelial Barrier Integrity and Function in Lung Fibrosis. Cell Death Discov. 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Van Winkle, L.S.; Fanucchi, M.V.; Plopper, C.G. The Attenuated Fibroblast Sheath of the Respiratory Tract Epithelial–Mesenchymal Trophic Unit. Am. J. Respir. Cell Mol. Biol. 1999, 21, 655–657. [Google Scholar] [CrossRef]
- Ghazavi, A.; Ganji, A.; Keshavarzian, N.; Rabiemajd, S.; Mosayebi, G. Cytokine Profile and Disease Severity in Patients with COVID-19. Cytokine 2021, 137, 155323. [Google Scholar] [CrossRef]
- Colarusso, C.; Maglio, A.; Terlizzi, M.; Vitale, C.; Molino, A.; Pinto, A.; Vatrella, A.; Sorrentino, R. Post-COVID-19 Patients Who Develop Lung Fibrotic-like Changes Have Lower Circulating Levels of IFN-β but Higher Levels of IL-1α and TGF-β. Biomedicines 2021, 9, 1931. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A Molecular Single-Cell Lung Atlas of Lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Yao, C.; Bora, S.A.; Parimon, T.; Zaman, T.; Friedman, O.A.; Palatinus, J.A.; Surapaneni, N.S.; Matusov, Y.P.; Cerro Chiang, G.; Kassar, A.G.; et al. Cell-Type-Specific Immune Dysregulation in Severely Ill COVID-19 Patients. Cell Rep. 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung Transplantation for Patients with Severe COVID-19. Sci. Transl. Med. 2020, 12, eabe4282. [Google Scholar] [CrossRef] [PubMed]
- Aesif, S.W.; Bribriesco, A.C.; Yadav, R.; Nugent, S.L.; Zubkus, D.; Tan, C.D.; Mehta, A.C.; Mukhopadhyay, S. Pulmonary Pathology of COVID-19 Following 8 Weeks to 4 Months of Severe Disease: A Report of Three Cases, Including One With Bilateral Lung Transplantation. Am. J. Clin. Pathol. 2021, 155, 506–514. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Thannickal, V.J. Epithelial-Mesenchymal Interactions in Pulmonary Fibrosis. Semin. Respir. Crit. Care Med. 2006, 27, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic Pulmonary Fibrosis: Multiple Causes and Multiple Mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Uhal, B.D.; Joshi, I.; Hughes, W.F.; Ramos, C.; Pardo, A.; Selman, M. Alveolar Epithelial Cell Death Adjacent to Underlying Myofibroblasts in Advanced Fibrotic Human Lung. Am. J. Physiol. Cell. Mol. Physiol. 1998, 275, L1192–L1199. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine Signalling during ZEB1-Mediated Epithelial–Mesenchymal Transition Augments Local Myofibroblast Differentiation in Lung Fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2–to–Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef]
- Cassandras, M.; Wang, C.; Kathiriya, J.; Tsukui, T.; Matatia, P.; Matthay, M.; Wolters, P.; Molofsky, A.; Sheppard, D.; Chapman, H.; et al. Gli1+ Mesenchymal Stromal Cells Form a Pathological Niche to Promote Airway Progenitor Metaplasia in the Fibrotic Lung. Nat. Cell Biol. 2020, 22, 1295–1306. [Google Scholar] [CrossRef]
- Prasse, A.; Binder, H.; Schupp, J.C.; Kayser, G.; Bargagli, E.; Jaeger, B.; Hess, M.; Rittinghausen, S.; Vuga, L.; Lynn, H.; et al. BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 199, 622–630. [Google Scholar] [CrossRef]
- Blackwell, T.S.; Tager, A.M.; Borok, Z.; Moore, B.B.; Schwartz, D.A.; Anstrom, K.J.; Bar-Joseph, Z.; Bitterman, P.; Blackburn, M.R.; Bradford, W.; et al. Future Directions in Idiopathic Pulmonary Fibrosis Research. An NHLBI Workshop Report. Am. J. Respir. Crit. Care Med. 2013, 189, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Koli, K.; Myllärniemi, M.; Vuorinen, K.; Salmenkivi, K.; Ryynänen, M.J.; Kinnula, V.L.; Keski-Oja, J. Bone Morphogenetic Protein-4 Inhibitor Gremlin Is Overexpressed in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2006, 169, 61–71. [Google Scholar] [CrossRef]
- Keski-Oja, J.; Koli, K.; Lohi, J.; Laiho, M. Growth Factors in the Regulation of Plasminogen-Plasmin System in Tumor Cells. Semin. Thromb. Hemost. 1991, 17, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Raghow, R.; Postlethwaite, A.E.; Keski-Oja, J.; Moses, H.L.; Kang, A.H. Transforming Growth Factor-Beta Increases Steady State Levels of Type I Procollagen and Fibronectin Messenger RNAs Posttranscriptionally in Cultured Human Dermal Fibroblasts. J. Clin. Investig. 1987, 79, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Shannon, J.M.; Hyatt, B.A. Epithelial-Mesenchymal Interactions in the Developing Lung. Annu. Rev. Physiol. 2004, 66, 625–645. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary Fibrosis, Part II: State-of-the-Art Patient Management. Expert Rev. Respir. Med. 2017, 11, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M. Pirfenidone: An Update on Clinical Trial Data and Insights from Everyday Practice. Eur. Respir. Rev. 2014, 23, 111–117. [Google Scholar] [CrossRef]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Spagnolo, P.; Maher, T.M.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Recent Advances on Pharmacological Therapy. Pharmacol. Ther. 2015, 152, 18–27. [Google Scholar] [CrossRef]
- King, C.S.; Nathan, S.D. Practical Considerations in the Pharmacologic Treatment of Idiopathic Pulmonary Fibrosis. Curr. Opin. Pulm. Med. 2015, 21, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.; Savithra, G.H.L.; Abyad, A.; Kanvinde, S.; Li, J.; Brock, S.; Oupický, D. Surface PEGylation of Mesoporous Silica Nanorods (MSNR): Effect on Loading, Release, and Delivery of Mitoxantrone in Hypoxic Cancer Cells. Sci. Rep. 2017, 7, 2274. [Google Scholar] [CrossRef] [PubMed]
- Roggers, R.; Kanvinde, S.; Boonsith, S.; Oupický, D. The Practicality of Mesoporous Silica Nanoparticles as Drug Delivery Devices and Progress toward This Goal. AAPS PharmSciTech 2014, 15, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, J.; Kanvinde, S.; Lin, Z.; Hazeldine, S.; Singh, R.K.; Oupický, D. Self-Immolative Polycations as Gene Delivery Vectors and Prodrugs Targeting Polyamine Metabolism in Cancer. Mol. Pharm. 2015, 12, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, S.; Sillman, B.; Bade, A.N.; Avedissian, S.N.; Podany, A.T.; McMillan, J.M.; Gautam, N.; Hanson, B.; Dyavar Shetty, B.L.; Szlachetka, A.; et al. Transformation of Dolutegravir into an Ultra-Long-Acting Parenteral Prodrug Formulation. Nat. Commun. 2022, 13, 3226. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.M.; Siccardi, M. Optimizing Nanomedicine Pharmacokinetics Using Physiologically Based Pharmacokinetics Modelling. Br. J. Pharmacol. 2014, 171, 3963–3979. [Google Scholar] [CrossRef]
- Gautam, N.; McMillan, J.M.; Kumar, D.; Bade, A.N.; Pan, Q.; Kulkarni, T.A.; Li, W.; Sillman, B.; Smith, N.A.; Shetty, B.L.D.; et al. Lipophilic Nanocrystal Prodrug-Release Defines the Extended Pharmacokinetic Profiles of a Year-Long Cabotegravir. Nat. Commun. 2021, 12, 3453. [Google Scholar] [CrossRef]
- Kulkarni, T.A.; Bade, A.N.; Sillman, B.; Shetty, B.L.D.; Wojtkiewicz, M.S.; Gautam, N.; Hilaire, J.R.; Sravanam, S.; Szlachetka, A.; Lamberty, B.G.; et al. A Year-Long Extended Release Nanoformulated Cabotegravir Prodrug. Nat. Mater. 2020, 19, 910–920. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-Converting Enzyme 2 (ACE2) Expression and Tissue Susceptibility to SARS-CoV-2 Infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular Interaction and Inhibition of SARS-CoV-2 Binding to the ACE2 Receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Z.; Dinh, P.-U.C.; Zhu, D.; Popowski, K.D.; Lutz, H.; Hu, S.; Lewis, M.G.; Cook, A.; Andersen, H.; et al. Cell-Mimicking Nanodecoys Neutralize SARS-CoV-2 and Mitigate Lung Injury in a Non-Human Primate Model of COVID-19. Nat. Nanotechnol. 2021, 16, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.; Ben Shimon, M.; Hay-Levi, M.; Shenberg, G.; Choshen, G.; Bannon, L.; Tepper, M.; Kazanov, D.; Seni, J.; Lev-Ari, S.; et al. A Novel Platform for Attenuating Immune Hyperactivity Using EXO-CD24 in COVID-19 and Beyond. EMBO Mol. Med. 2022, 14, e15997. [Google Scholar] [CrossRef]
- Fang, X.; Zheng, P.; Tang, J.; Liu, Y. CD24: From A to Z. Cell. Mol. Immunol. 2010, 7, 100–103. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Zeng, X.; Schwarz, H.; Nanda, H.S.; Peng, X.; Zhou, Y. Exosomes, a New Star for Targeted Delivery. Front. Cell Dev. Biol. 2021, 9, 2827. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular Vesicles as a Next-Generation Drug Delivery Platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Dinh, P.-U.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C.; et al. Inhalation of Lung Spheroid Cell Secretome and Exosomes Promotes Lung Repair in Pulmonary Fibrosis. Nat. Commun. 2020, 11, 1064. [Google Scholar] [CrossRef]
- Shapira, S.; Kazanov, D.; Weisblatt, S.; Starr, A.; Arber, N.; Kraus, S. The CD24 Protein Inducible Expression System Is an Ideal Tool to Explore the Potential of CD24 as an Oncogene and a Target for Immunotherapy in Vitro and in Vivo. J. Biol. Chem. 2011, 286, 40548–40555. [Google Scholar] [CrossRef]
- Wick, G.; Backovic, A.; Rabensteiner, E.; Plank, N.; Schwentner, C.; Sgonc, R. The Immunology of Fibrosis: Innate and Adaptive Responses. Trends Immunol. 2010, 31, 110–119. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or Foe in Idiopathic Pulmonary Fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Suzuki, Y.; Yoshimura, K.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; et al. Macrophage Mannose Receptor CD206 Predicts Prognosis in Community-Acquired Pneumonia. Sci. Rep. 2019, 9, 18750. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Castells, E.; Dixon, K.; Mason, S.; Elliott, G.; Marshall, H.; Poblocka, M.A.; Macip, S.; Richardson, M.; Khalfaoui, L.; et al. Evaluation of Pirfenidone and Nintedanib in a Human Lung Model of Fibrogenesis. Front. Pharmacol. 2021, 12, 679388. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Rackov, G.; Izquierdo, E.; Alonso, V.; Gortazar, A.R.; Escribese, M.M. Targeting Macrophages: Friends or Foes in Disease? Front. Pharmacol. 2019, 10, 1255. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Chakraborty, S.; Wong, S.W.; Hefner, N.A.; Stuart, A.; Qadir, A.S.; Mukhopadhyay, A.; Bachmaier, K.; Shin, J.-W.; Rehman, J.; et al. Nanoparticle Targeting of de Novo Profibrotic Macrophages Mitigates Lung Fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121098119. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Shahjin, F.; Cohen, J.D.; Hasan, M.; Machhi, J.; Chugh, H.; Singh, S.; Das, S.; Kulkarni, T.A.; Herskovitz, J.; et al. The Immunopathobiology of SARS-CoV-2 Infection. FEMS Microbiol. Rev. 2021, 45, fuab035. [Google Scholar] [CrossRef]
- Fox, R.I. Mechanism of Action of Hydroxychloroquine as an Antirheumatic Drug. Semin. Arthritis Rheum. 1993, 23, 82–91. [Google Scholar] [CrossRef]
- Chhonker, Y.S.; Kanvinde, S.; Ahmad, R.; Singh, A.B.; Oupický, D.; Murry, D.J. Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites 2021, 11, 106. [Google Scholar] [CrossRef]
- Kesharwani, S.S.; Ahmad, R.; Bakkari, M.A.; Rajput, M.K.S.; Dachineni, R.; Valiveti, C.K.; Kapur, S.; Jayarama Bhat, G.; Singh, A.B.; Tummala, H. Site-Directed Non-Covalent Polymer-Drug Complexes for Inflammatory Bowel Disease (IBD): Formulation Development, Characterization and Pharmacological Evaluation. J. Control. Release 2018, 290, 165–179. [Google Scholar] [CrossRef]
- Kanvinde, S.; Chhonker, Y.S.; Ahmad, R.; Yu, F.; Sleightholm, R.; Tang, W.; Jaramillo, L.; Chen, Y.; Sheinin, Y.; Li, J.; et al. Pharmacokinetics and Efficacy of Orally Administered Polymeric Chloroquine as Macromolecular Drug in the Treatment of Inflammatory Bowel Disease. Acta Biomater. 2018, 82, 158–170. [Google Scholar] [CrossRef]
- Ali, A.S.; Alrashedi, M.G.; Ahmed, O.A.; Ibrahim, I.M. Pulmonary Delivery of Hydroxychloroquine Nanostructured Lipid Carrier as a Potential Treatment of COVID-19. Polymers (Basel) 2022, 14, 2616. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhao, G.; Chen, Q.; Li, Z.; Gao, M.; Ho, W.; Xu, X.; Zhang, X.-Q. Inhaled SiRNA Nanoparticles Targeting IL11 Inhibit Lung Fibrosis and Improve Pulmonary Function Post-Bleomycin Challenge. Sci. Adv. 2023, 8, eabn7162. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.; Louis, C.; Metcalfe, R.D.; Kosasih, C.C.; Wicks, I.P.; Griffin, M.D.W.; Putoczki, T.L. Emerging Roles for IL-11 in Inflammatory Diseases. Cytokine 2022, 149, 155750. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Kleeberger, S.R. Mouse Models of Bleomycin-Induced Pulmonary Fibrosis. Curr. Protoc. Pharmacol. 2008, 40, 5.46.1–5.46.17. [Google Scholar] [CrossRef]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An Overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Levine, R.L. Addressing the Long-Term Effects of COVID-19. JAMA 2022, 328, 823–824. [Google Scholar] [CrossRef]
- Suran, M. Autopsies Reveal Lung Damage Patterns From COVID-19. JAMA 2021, 326, 2463. [Google Scholar] [CrossRef]
- McGroder, C.F.; Zhang, D.; Choudhury, M.A.; Salvatore, M.M.; D’Souza, B.M.; Hoffman, E.A.; Wei, Y.; Baldwin, M.R.; Garcia, C.K. Pulmonary Fibrosis 4 Months after COVID-19 Is Associated with Severity of Illness and Blood Leucocyte Telomere Length. Thorax 2021, 76, 1242–1245. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef]
- Hama Amin, B.J.; Kakamad, F.H.; Ahmed, G.S.; Ahmed, S.F.; Abdulla, B.A.; Mohammed, S.H.; Mikael, T.M.; Salih, R.Q.; Ali, R.K.; Salh, A.M.; et al. Post COVID-19 Pulmonary Fibrosis; a Meta-Analysis Study. Ann. Med. Surg. 2022, 77, 103590. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Balan, I.; Yadav, S.; Matos, W.F.; Kharawala, A.; Gaddam, M.; Sarabia, N.; Koneru, S.C.; Suddapalli, S.K.; Marzban, S. Post-COVID-19 Pulmonary Fibrosis. Cureus 2022, 14, e22770. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating Mechanisms of Pulmonary Fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Kanvinde, S.; Kulkarni, T.; Deodhar, S.; Bhattacharya, D.; Dasgupta, A. Non-Viral Vectors for Delivery of Nucleic Acid Therapies for Cancer. BioTech 2022, 11, 6. [Google Scholar] [CrossRef]
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, Properties, and Regulatory Issues. Front. Chem. 2018, 6, 360. [Google Scholar] [CrossRef]
- Ren, J.; Cai, R.; Wang, J.; Daniyal, M.; Baimanov, D.; Liu, Y.; Yin, D.; Liu, Y.; Miao, Q.; Zhao, Y.; et al. Precision Nanomedicine Development Based on Specific Opsonization of Human Cancer Patient-Personalized Protein Coronas. Nano Lett. 2019, 19, 4692–4701. [Google Scholar] [CrossRef]
- Velino, C.; Carella, F.; Adamiano, A.; Sanguinetti, M.; Vitali, A.; Catalucci, D.; Bugli, F.; Iafisco, M. Nanomedicine Approaches for the Pulmonary Treatment of Cystic Fibrosis. Front. Bioeng. Biotechnol. 2019, 7, 406. [Google Scholar] [CrossRef]
- Skibba, M.; Drelich, A.; Poellmann, M.; Hong, S.; Brasier, A.R. Nanoapproaches to Modifying Epigenetics of Epithelial Mesenchymal Transition for Treatment of Pulmonary Fibrosis. Front. Pharmacol. 2020, 11, 607689. [Google Scholar] [CrossRef]
- Loo, C.-Y.; Lee, W.-H. Nanotechnology-Based Therapeutics for Targeting Inflammatory Lung Diseases. Nanomedicine 2022, 17, 865–879. [Google Scholar] [CrossRef]
- White, E.S.; Thomas, M.; Stowasser, S.; Tetzlaff, K. Challenges for Clinical Drug Development in Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 90. [Google Scholar] [CrossRef]
- Kaner, R.J.; Bajwa, E.K.; El-Amine, M.; Gorina, E.; Gupta, R.; Lazarus, H.M.; Luckhardt, T.R.; Mouded, M.; Posada, K.; Richeldi, L.; et al. Design of Idiopathic Pulmonary Fibrosis Clinical Trials in the Era of Approved Therapies. Am. J. Respir. Crit. Care Med. 2019, 200, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Harari, S.; Caminati, A. Idiopathic Pulmonary Fibrosis: From Clinical Trials to Real-Life Experiences. Eur. Respir. Rev. 2015, 24, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Crestani, B.; Hernandez, P.; Inoue, Y.; Wachtlin, D.; Loaiza, L.; Quaresma, M.; Stowasser, S.; Richeldi, L. Safety and Survival Data in Patients with Idiopathic Pulmonary Fibrosis Treated with Nintedanib: Pooled Data from Six Clinical Trials. BMJ Open Respir. Res. 2019, 6, e000397. [Google Scholar] [CrossRef] [PubMed]
- Jogdeo, C.M.; Panja, S.; Kanvinde, S.; Kapoor, E.; Siddhanta, K.; Oupický, D. Advances in Lipid-Based Codelivery Systems for Cancer and Inflammatory Diseases. Adv. Healthc. Mater. 2023, 12, 2202400. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Kumar, R.; Srivastava, V.; Baindara, P.; Ahmad, A. Thermostable Vaccines: An Innovative Concept in Vaccine Development. Expert Rev. Vaccines 2022, 21, 811–824. [Google Scholar] [CrossRef]
- Đorđević, S.; Gonzalez, M.M.; Conejos-Sánchez, I.; Carreira, B.; Pozzi, S.; Acúrcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current Hurdles to the Translation of Nanomedicines from Bench to the Clinic. Drug Deliv. Transl. Res. 2022, 12, 500–525. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trial ID | Title | Intervention | Sponsor |
|---|---|---|---|
| NCT04607928 | Phase-II Randomized Clinical Trial to Evaluate the Effect of Pirfenidone Compared to Placebo in Post-COVID19 | Drug: Pirfenidone Drug: Placebo | Institut d’Investigació Biomèdica de Bellvitge |
| NCT04818489 | Impact of Colchicine on the Clinical Outcome of COVID-19 and the Development of Post-COVID-19 Pulmonary Fibrosis: Randomized Controlled Clinical Trial | Drug: Colchicine 0.5 mg Other: the standard protocol only | ClinAmygate |
| NCT04551781 | Short Term Low Dose Corticosteroids for Management of Post Covid-19 Pulmonary Fibrosis | Drug: 20 mg Prednisone for 14 days Drug: control | South Valley University |
| NCT05648734 | Impact of Anti-Inflammatory and Anti-Fibrotic Drugs on Post-acute COVID-19 Pulmonary Fibrosis | Drug: Corticosteroids alone Drug: Corticosteroids + Colchicine Drug: Corticosteroids + Pirfenidone Drug: Corticosteroids + Colchicine + Pirfenidone for ≥14 day | Mansoura University |
| NCT04279197 | Efficacy and Safety of Fuzheng Huayu Tablets in Post-COVID-19 Patients with Pulmonary Inflammation and Fibrosis: A Multicenter Double-blind Randomized Controlled Trial | Drug: Fuzheng Huayu Tablet Drug: Vitamin C tablets Drug: Placebo Other: respiratory function rehabilitation training | ShuGuang Hospital |
| NCT04541680 | Nintedanib for the Treatment of SARS-Cov-2 Induced Pulmonary Fibrosis | Drug: Nintedanib 150 mg Other: Placebo | Assistance Publique–Hôpitaux de Paris |
| NCT04856111 | A Study of the Efficacy and Safety of Pirfenidone vs. Nintedanib in the Treatment of Fibrotic Lung Disease After Coronavirus Disease-19 | Drug: Pirfenidone Drug: Nintedanib | Postgraduate Institute of Medical Education and Research |
| NCT04948203 | SECOVID: A Multi-center, Randomized, Dose-ranging Parallel-group Trial Assessing the Efficacy of Sirolimus in Hospitalized Patients With COVID-19 Pneumonia for the Prevention of Post-COVID Fibrosis | Drug: Sirolimus | University of Chicago |
| NCT05387239 | Safety and Effectiveness of EV-Pure + WJ-Pure Treatment on Pulmonary Fibrosis Secondary to Covid-19 | Drug: EV-Pure™ and WJ-Pure™ plus standard care Drug: Placebo (Saline plus standard care) | Vitti Labs, LLC |
| NCT04645368 | Multicenter, Open-label Prospective Cohort Study of the Efficacy and Safety of the Inclusion of Longidaze in the Prevention and Treatment of Post-inflammatory Pulmonary Fibrosis and Interstitial Lung Diseases Caused by COVID-19 | Drug: bovhyaluronidase azoxymer | NPO Petrovax |
| NCT04805086 | Phase I/II MONACO Cell Therapy Study: Monocytes as an Anti-fibrotic Treatment After COVID-19 | Biological: MON002 | Guy’s and St Thomas’ NHS Foundation Trust |
| NCT04912011 | The Use of a Mineralocorticoid Receptor Antagonist (Spironolactone) in the Treatment of Pulmonary Fibrosis Associated With SARS-CoV-2 Infection | Drug: Canrenoate Potassium Drug: Normal Saline | Pomeranian Medical University Szczecin |
| NCT04338802 | Efficacy and Safety of Nintedanib Ethanesulfonate Soft Capsule in the Treatment of Pulmonary Fibrosis in Patients with Moderate to Severe COVID-9 (COVID 19): a Single-center, Randomized, Placebo-controlled Study | Drug: Nintedanib 150 mg Other: Placebo | Tongji Hospital |
| NCT04619680 | Early Nintedanib Deployment in COVID-19 Interstitial Lung Disease | Drug: Nintedanib Drug: Placebo | Icahn School of Medicine at Mount Sinai |
| NCT04482595 | A Phase 2 Study of BIO 300 Oral Suspension in Discharged COVID-19 Patients | Drug: BIO 300 Oral Suspension Drug: Placebo | Humanetics Corporation |
| NCT04537130 | Phase Ib Controlled Exploratory Trial for Treatment of Fibrosing Interstitial Lung Disease Patients Secondary to SARS-CoV-2 Infection with IN01 Vaccine (COVINVAC) | Biological: IN01 vaccine | Instituto Oncológico |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanvinde, S.; Deodhar, S.; Kulkarni, T.A.; Jogdeo, C.M. Nanotherapeutic Approaches to Treat COVID-19-Induced Pulmonary Fibrosis. BioTech 2023, 12, 34. https://doi.org/10.3390/biotech12020034
Kanvinde S, Deodhar S, Kulkarni TA, Jogdeo CM. Nanotherapeutic Approaches to Treat COVID-19-Induced Pulmonary Fibrosis. BioTech. 2023; 12(2):34. https://doi.org/10.3390/biotech12020034
Chicago/Turabian StyleKanvinde, Shrey, Suyash Deodhar, Tanmay A. Kulkarni, and Chinmay M. Jogdeo. 2023. "Nanotherapeutic Approaches to Treat COVID-19-Induced Pulmonary Fibrosis" BioTech 12, no. 2: 34. https://doi.org/10.3390/biotech12020034
APA StyleKanvinde, S., Deodhar, S., Kulkarni, T. A., & Jogdeo, C. M. (2023). Nanotherapeutic Approaches to Treat COVID-19-Induced Pulmonary Fibrosis. BioTech, 12(2), 34. https://doi.org/10.3390/biotech12020034