


Inverse Problems in Pump–Probe Spectroscopy


Abstract
:1. Introduction
2. Inverse Problem of Experimental Data Analysis
2.1. Formulation of the Problem
2.2. Least-Squares Formulation of the Inverse Problem
- Using one of the minimization algorithms, such as the Conjugate Gradient Method [21] or Powell’s algorithm [22], we first get iteration trial parameter values . Depending on the chosen minimization algorithm, we may require either calculation of the LSQ function’s gradient (), Hessian (), or evaluate the LSQ function’s values in a few neighboring points around .
- Then, we again compute the and find second (), third (), fourth (), and so on, values of the parameters, trying to minimize the value of the LSQ function (Equation (6)).
- We halt this iterative procedure when we reach a pre-defined convergence criterion. For instance, if the change of the parameter value from iteration to iteration is smaller than some small value (), e.g., as , then the convergence criterion can be said to satisfy. In this case, we take the last value obtained in the procedure to be our solution, and then we estimate the uncertainties of the parameters and correlations between them using an approximate normal distribution computed from the second derivatives of the LSQ function (see Equation (10)).
2.3. Regularization of the Least-Squares Inverse Problem
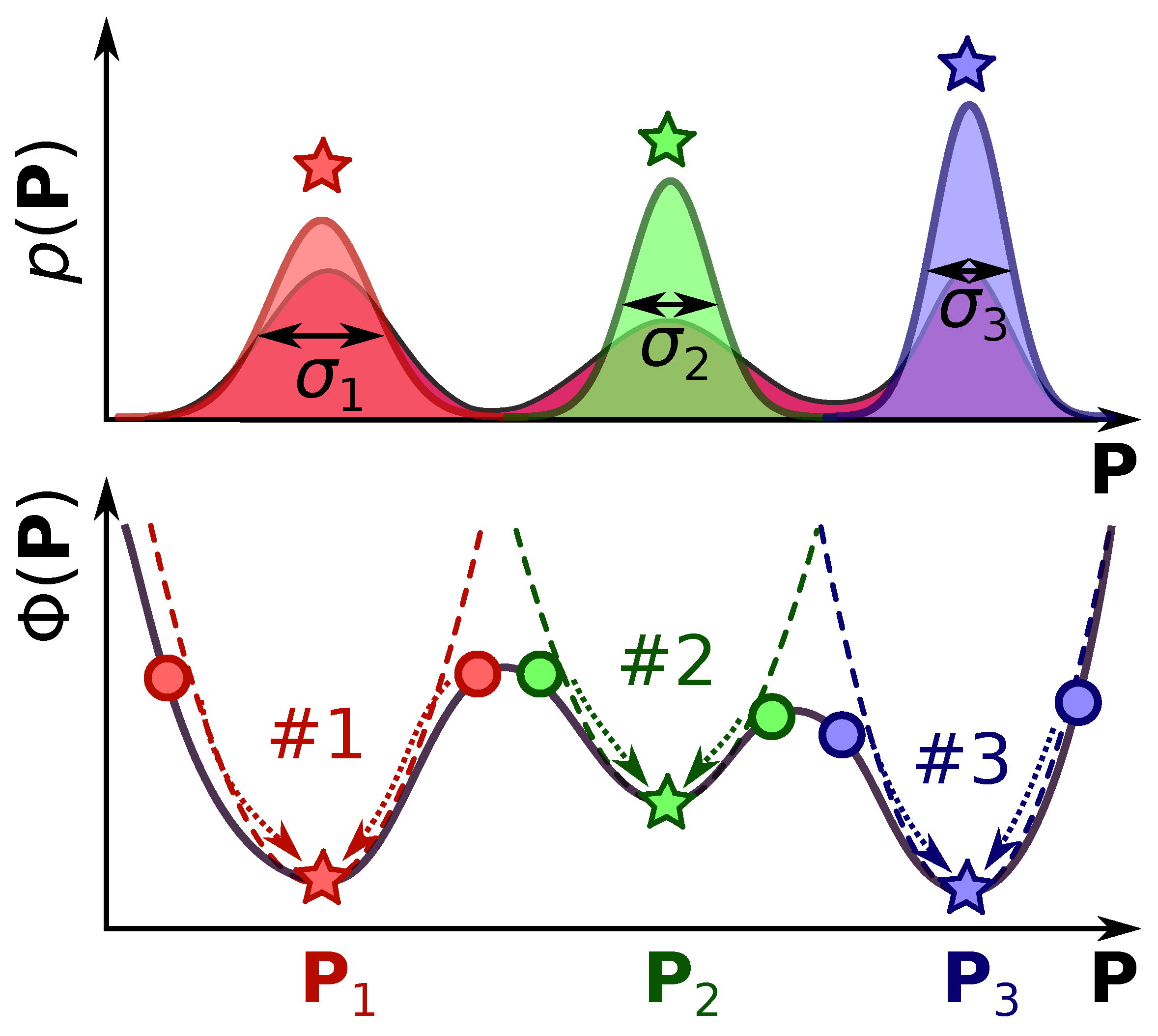
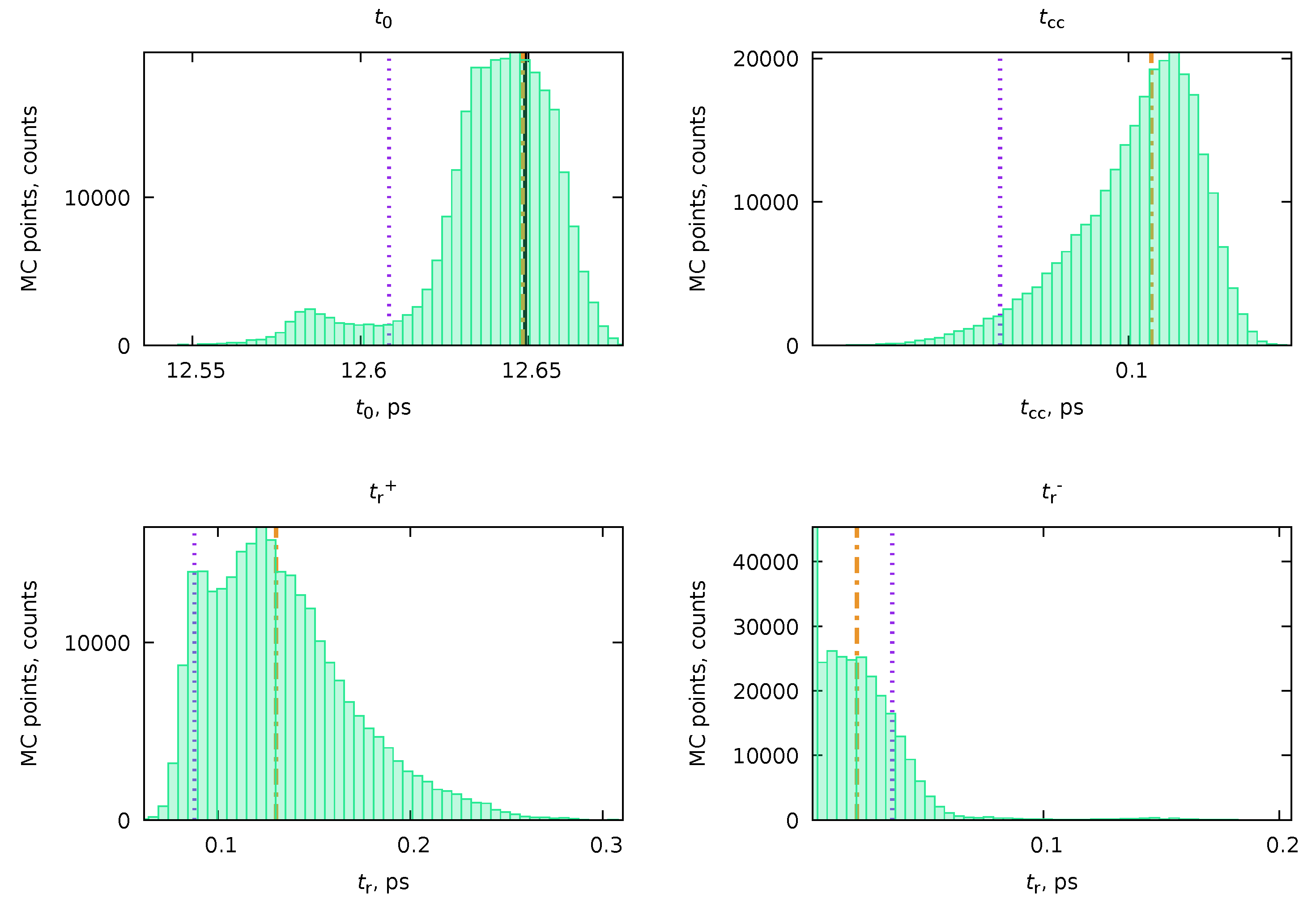
2.4. Monte-Carlo Importance Sampling of the Parameter Space
- We start from the initial set of parameters , which we assign to be the current state of the simulation (i.e., we set ). Generally, we can use any set of parameters for the initial guess, but a faster simulation convergence is reached if we provide initial values in the desired solution region, e.g., as the solution of the LSQ fitting problem (Equation (9)).
- From the current state, we generate a new trial set of parameters , and then we compute the transition probability . This value describes a chance of changing our current state to a new state (i.e., reassigning ). The probability should be related to the probabilities given in Equation (5), and we will discuss it in detail further in the text (see Equation (21)).
- Then, we draw a random value from a uniform distribution between 0 and 1, and compare the with .
- If , then becomes the new state of the system, i.e., we reassign . This state we will call an accepted step.
- If , this means that the transition does not happen (we disregard the ). The new state of the system becomes the same old value . We will call this state a declined step.
- By repeating steps #2 and #3 for a sufficient amount of iterations (N), we generate a trajectory of states , where index denotes the state at the n-th iteration of the algorithm. Naturally, some sets of parameters will be repeated multiple times throughout the trajectory. Furthermore, this trajectory will encode inside the desired distribution given in Equation (5). In practice, the initial part of the trajectory (e.g., first 10% of steps) is disregarded as an equilibration phase. The acceptance ratio refers to the accepted steps in algorithm step #3 () to the total number of steps (, where is the number of rejected steps). A general requirement for the simulation to be reasonably good is that this ratio should not be too big or too small. A simple rule of thumb can be that the acceptance rate should be in the range .
- From the obtained trajectory , we can compute all the required parameters. For instance, the mean value of parameter (from the set of parameters ) can be computed as:where is the value of in the parameter set . Similarly, we can compute the standard deviation of parameter from the mean value as:The covariance between the parameters and will be given similarly, as:
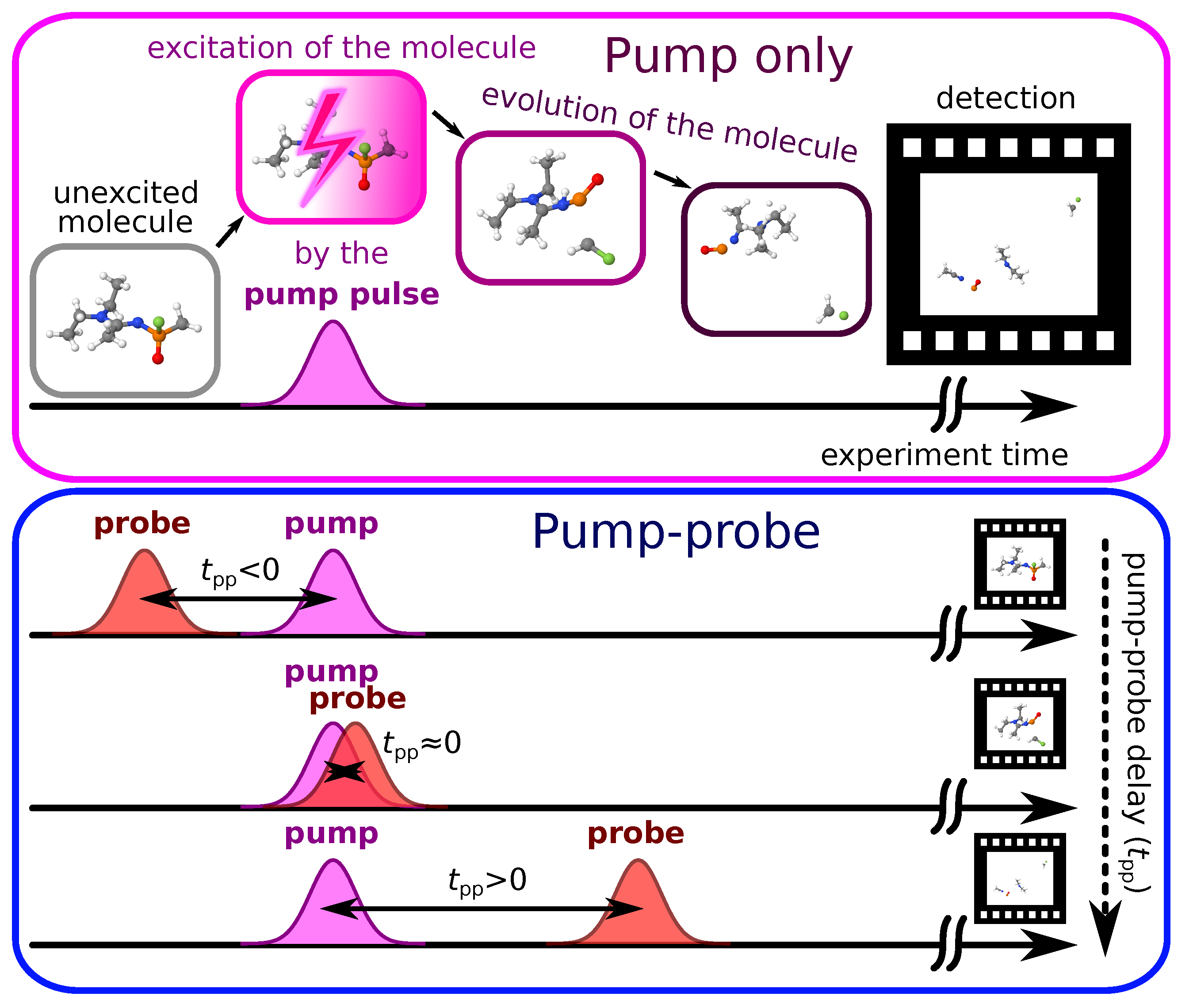
3. Fitting Model of Pump–Probe Spectroscopy
3.1. General Considerations
- If both the pump and the probe pulses act on the molecule simultaneously, then (this temporal overlap of the pump and probe pulses is also called );
- If the probe pulse interacts with the molecule before the pump pulse, then ;
- If the probe pulse interacts with the molecule after the pump pulse, then .

3.2. Delta-Shaped Pump–Probe Model
3.2.1. Assumptions of the Model
3.2.2. Step Function Dynamics
3.2.3. Instant Dynamics
3.2.4. Transient Pump–Probe Signatures of Metastable Species
3.2.5. Coherent Oscillations without Decay
3.2.6. Coherent Oscillations with Decay
3.2.7. More Complicated Dynamics Models
- The first type is simply the constant (“c”) defined as:This function (with coefficient in Equation (59)) has no parameters and describes the background of the pump–probe experiment.
- The second type is the step or “switch” function (“s”):This basis function (with coefficient in Equation (59)) describes the switching of the background between and regimes.
- The third type is the transient (“t”) function:This type of basis function (with coefficients in Equation (59)) describes the pump-induced decay dynamics, and it depends on a parameter , which is an effective decay time.
- This type of dynamics describes unresolvably fast relaxation dynamics.
- The second additional function, describing nondecaying coherent oscillation (“o”), can be taken from Equation (51):This basis function has two parameters: the oscillation frequency and the initial phase .
- The last additional function, describing a transient coherent oscillation (“to”), can be taken from Equation (57):This basis function has three parameters: the oscillation frequency , the initial phase , and the decay time .
3.3. Accounting for Finite Duration of the Pulses and Experiment Jitters
- The real pulses are not delta-shaped but have a finite duration.
- Real experimental setups have fluctuations (jitters) of the pump–probe delay, arising from different physical processes.
- Pump pulse duration .
- Probe pulse duration .
- Instrument jitter magnitude. .
- The first function is the constant (“c”) function:
- The second type is the step function (“s”):where is the error function.
- The third type is the transient (“t”) function:
- The fourth type is the instant (“i”) dynamics function:
- The fifth type is the nondecaying coherent oscillation (“o”) function:
- Furthermore, the sixth type is the decaying (transient) coherent oscillation (“to”) function:

4. Estimation Procedure for the Parameters and Their Uncertainties
4.1. Single Dataset Case
- The first ones are the linear coefficients before basis functions. These parameters depict effective cross-sections and quantum yields for a given dynamics. We will represent these parameters as an N-dimensional vector .
- The second set of parameters defines each basis function . There are several types of actual parameters.
- The first type is , representing the temporal overlap of the pump and the probe pulses on the molecular sample. This parameter is not always known in advance from the experimental setup (e.g., in the cases of experiments at the FELs with conventional lasers [6]); it might be needed to be fit. In this case, the parameter is provided to a given basis function by replacing it with . In most cases, is a shared parameter for all the basis functions and datasets. However, in some cases, some of the basis functions can have a different parameter to account for Wigner time delay in photoionization [52].
- The second type of parameter is the cross-correlation time . This parameter might differ for various basis functions since some processes can require different numbers of photons to be pumped/probed.
- The third type is the decay time . Various decay processes usually have different parameters.
- The fourth type is the coherent oscillation frequency .
- Furthermore, the last, fifth, parameter type is the oscillation phase .
These values are required to fully describe the model of the observable. We will denote all of these parameters with a vector .
4.2. Multiple Dataset Case
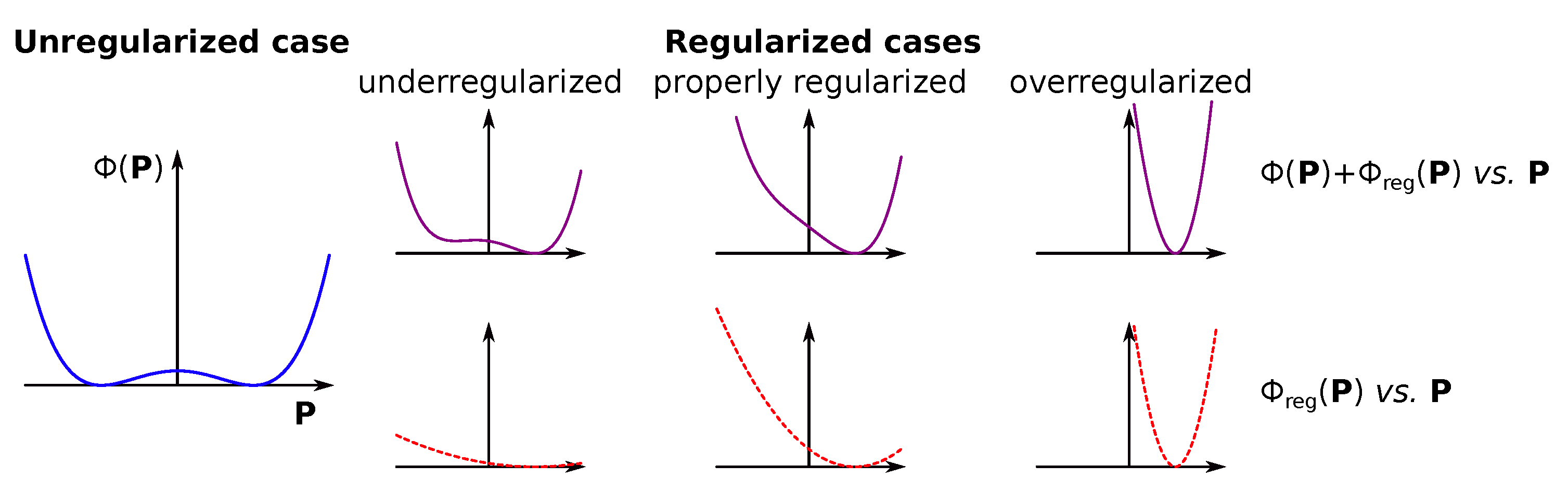
4.3. Inverse Problem Regularization
4.4. Inverse Problem Solution Algorithm
- Construct a regularization functional for parameters . Two types are available.The total regularization function can be either:
- , if both regularization cases are applicable;
- or , if only one regularization case in demand;
- , if no regularization is required.
- Define an effective function (Equation (89)) as a sum of experimental and regularization functions.
- Find a solution of the LSQ problem as using local or global fitting.
- Start a MC sampling procedure (see Section 2.4) with probability to sample nonlinear () and linear () parameters.
- In addition to the values and uncertainties, Pearson correlation coefficients (Equation (16)), histograms of parameter distributions, and higher distribution moments can also be calculated from the MC trajectory.
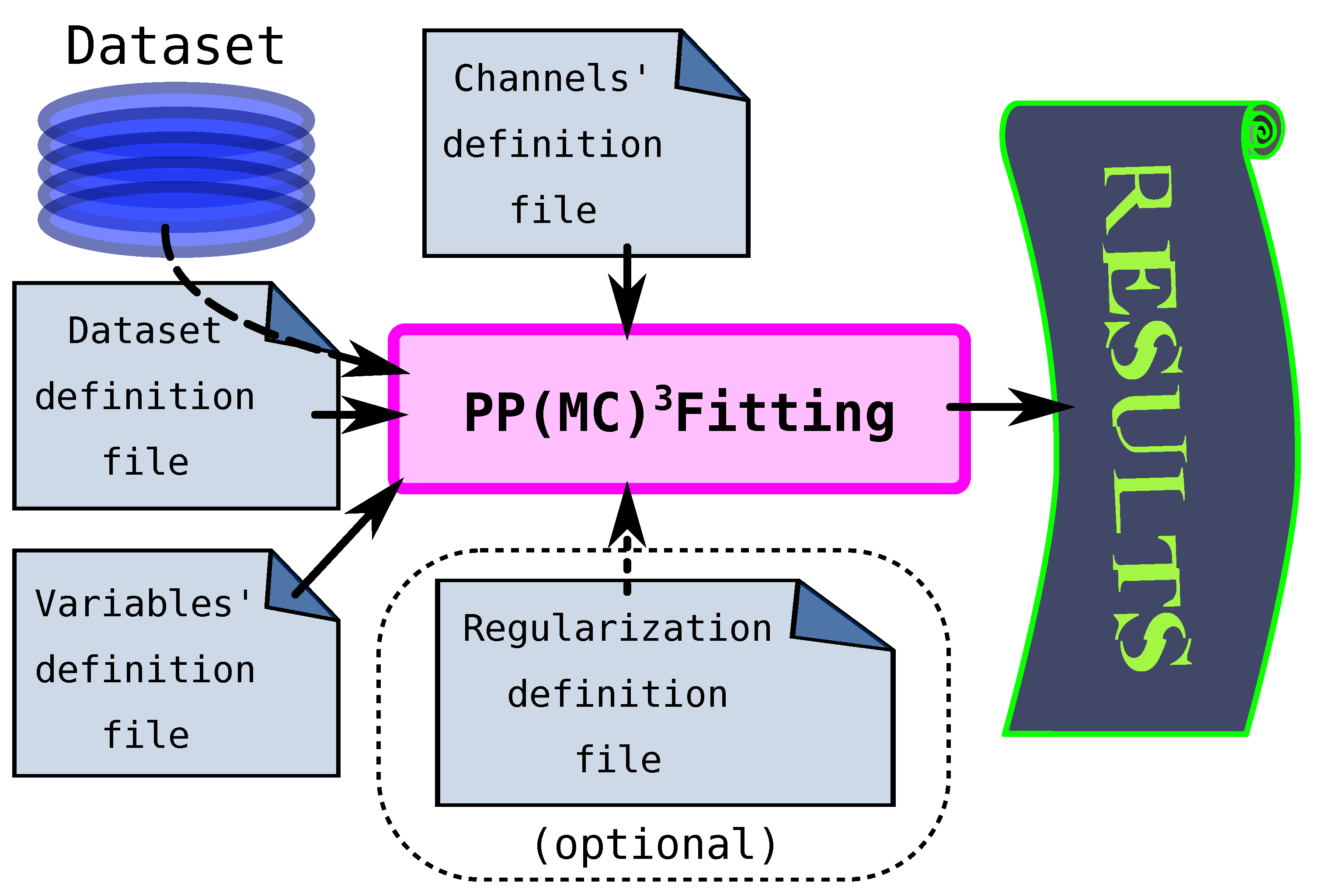
5. PP(MCFitting Software
6. Numerical Examples
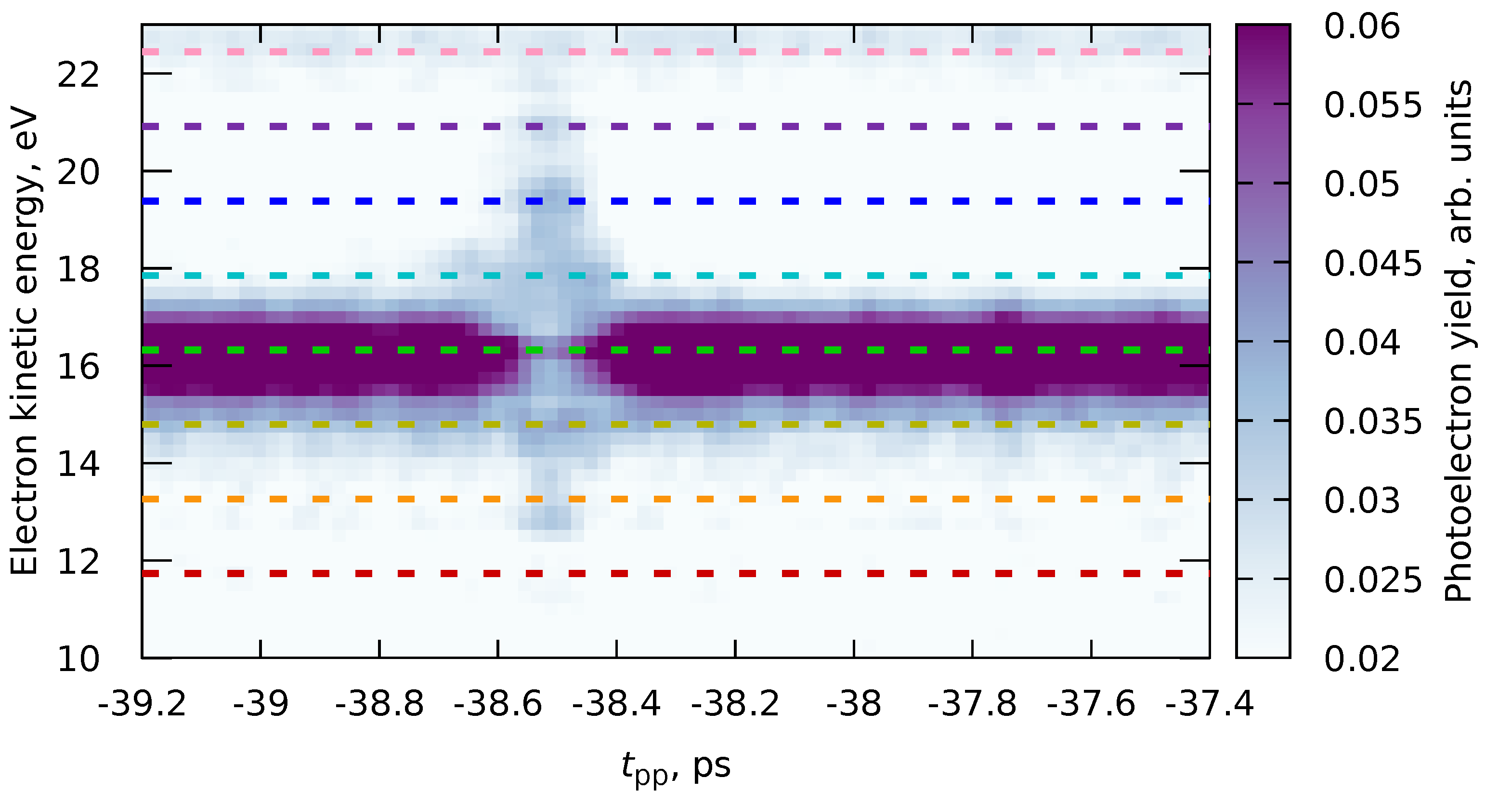
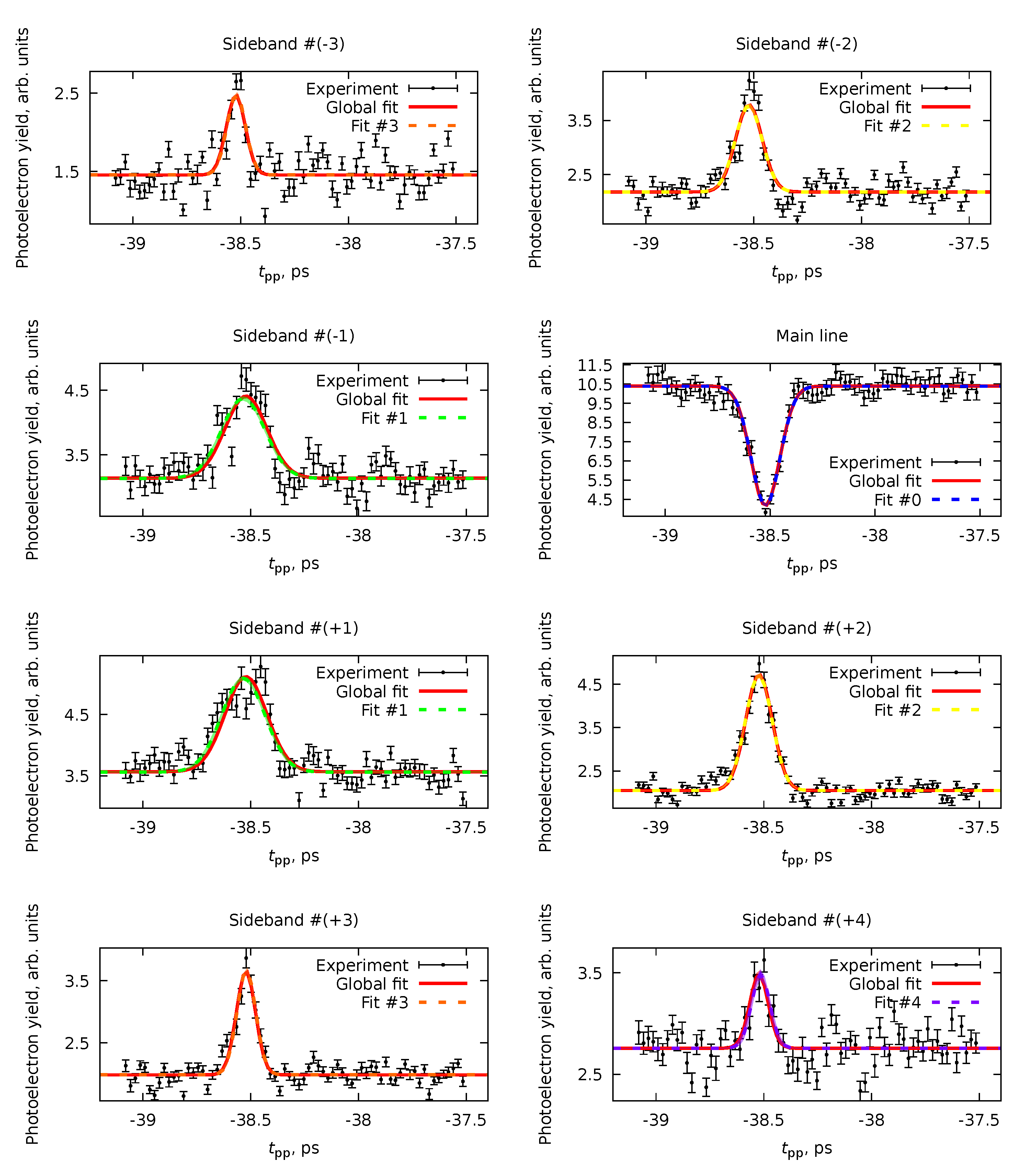
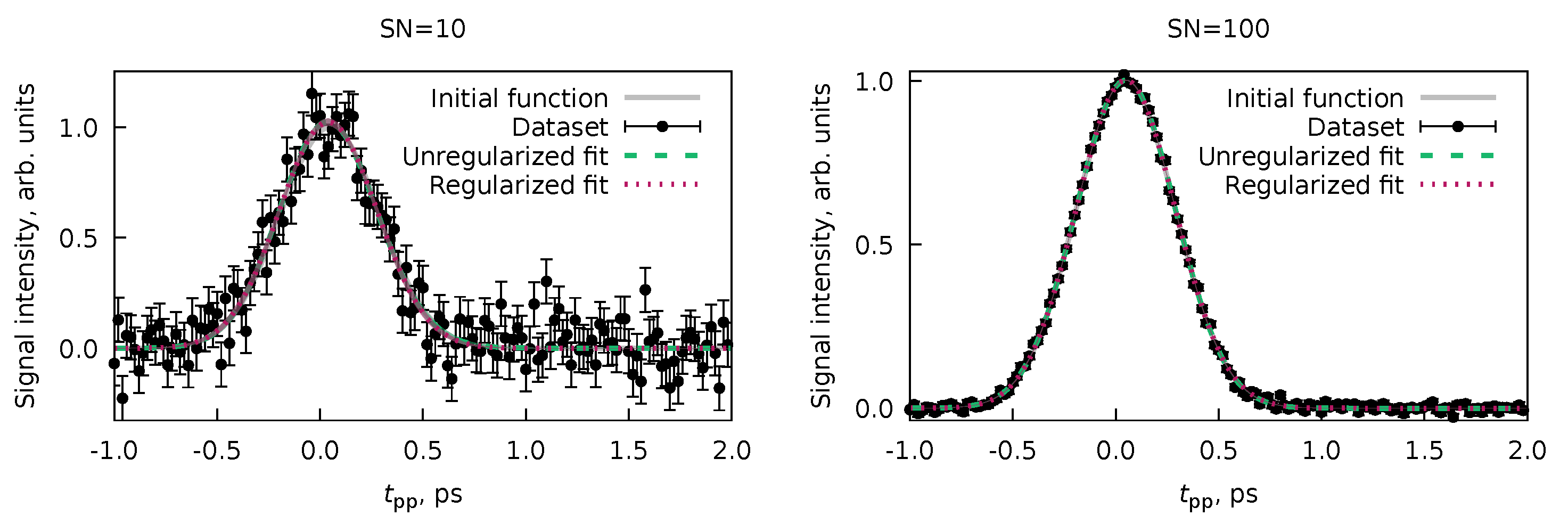
6.1. Multiple Datasets with Shared Parameters
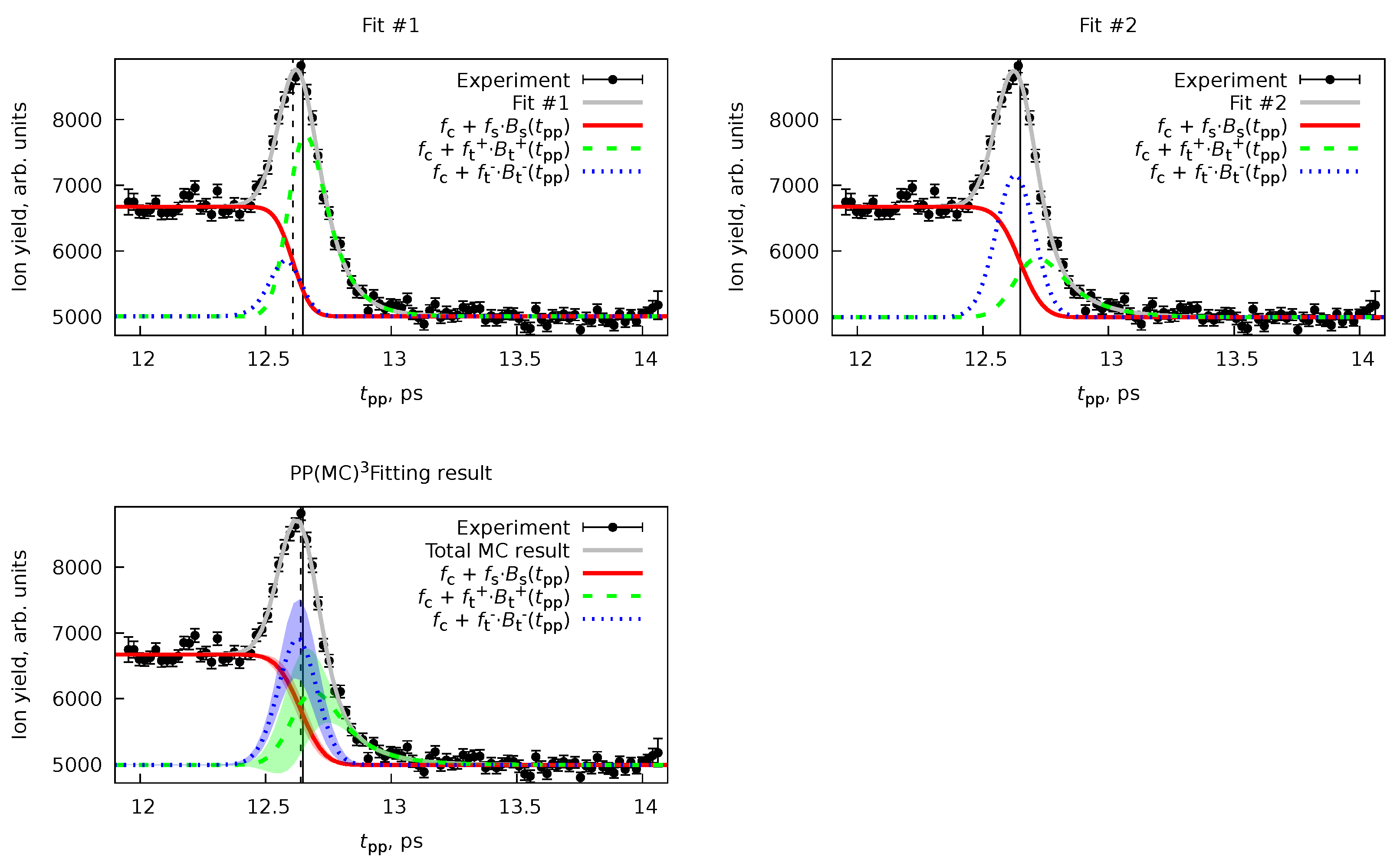
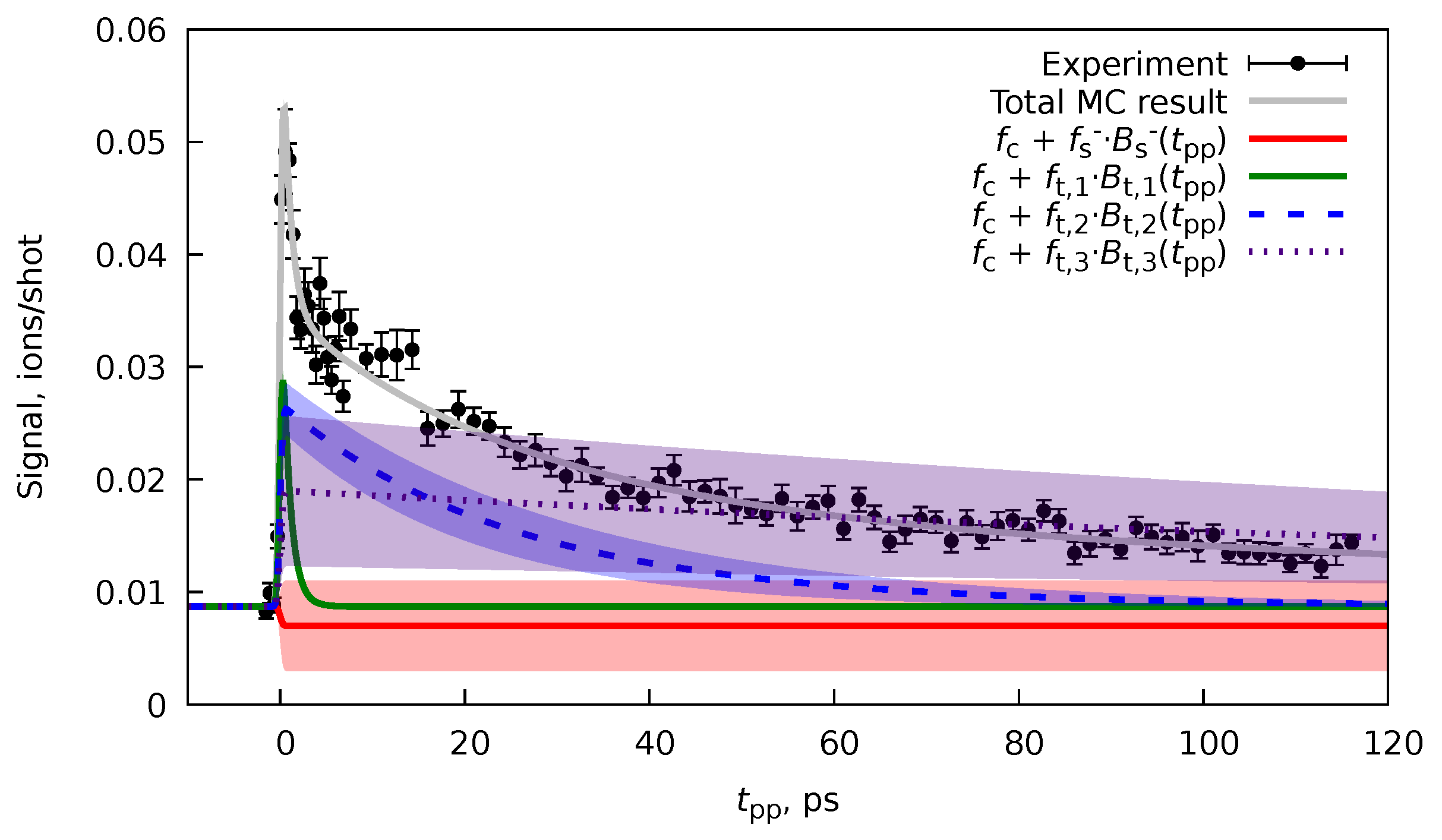
6.2. Forward-Backward Channel Dataset
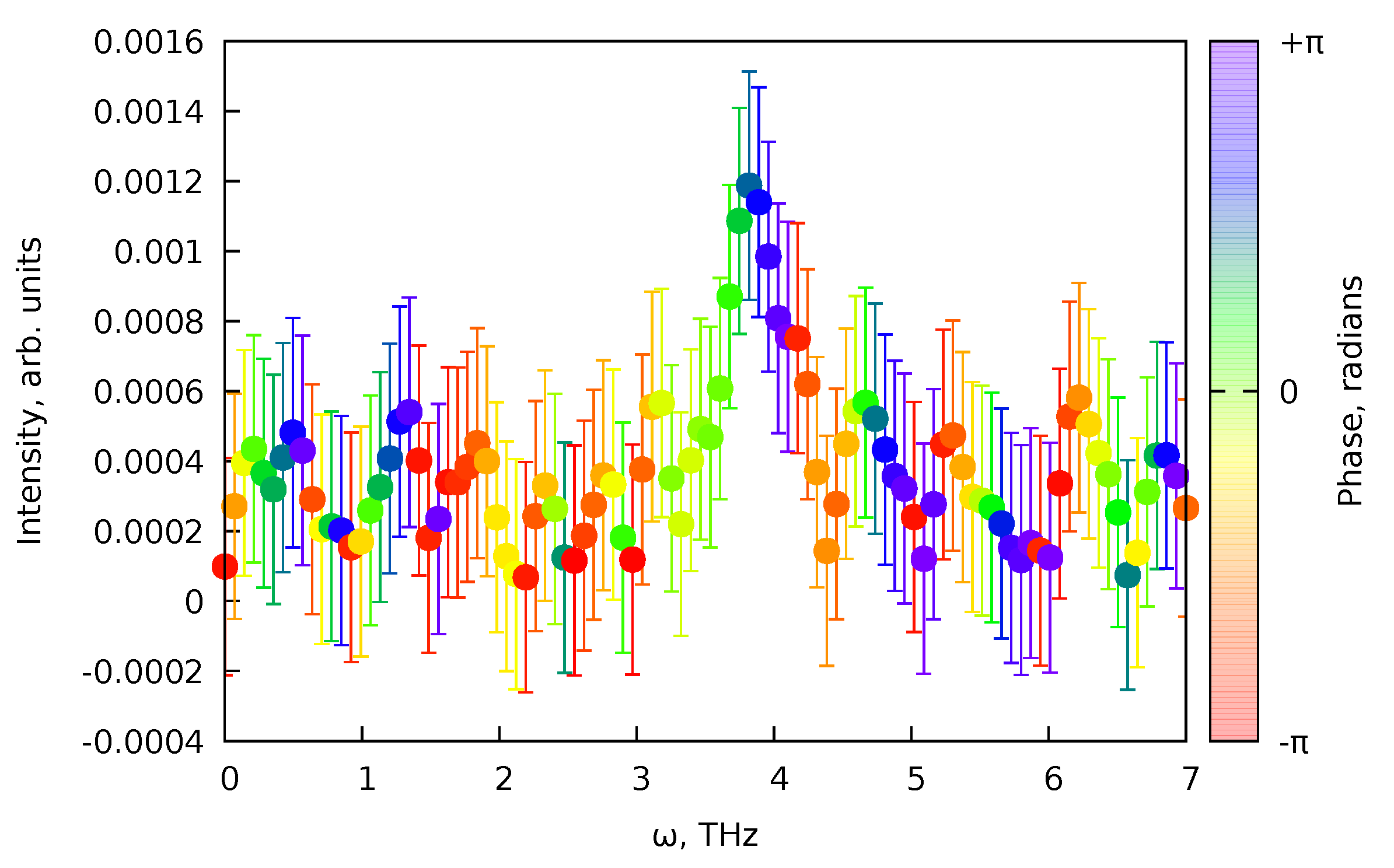
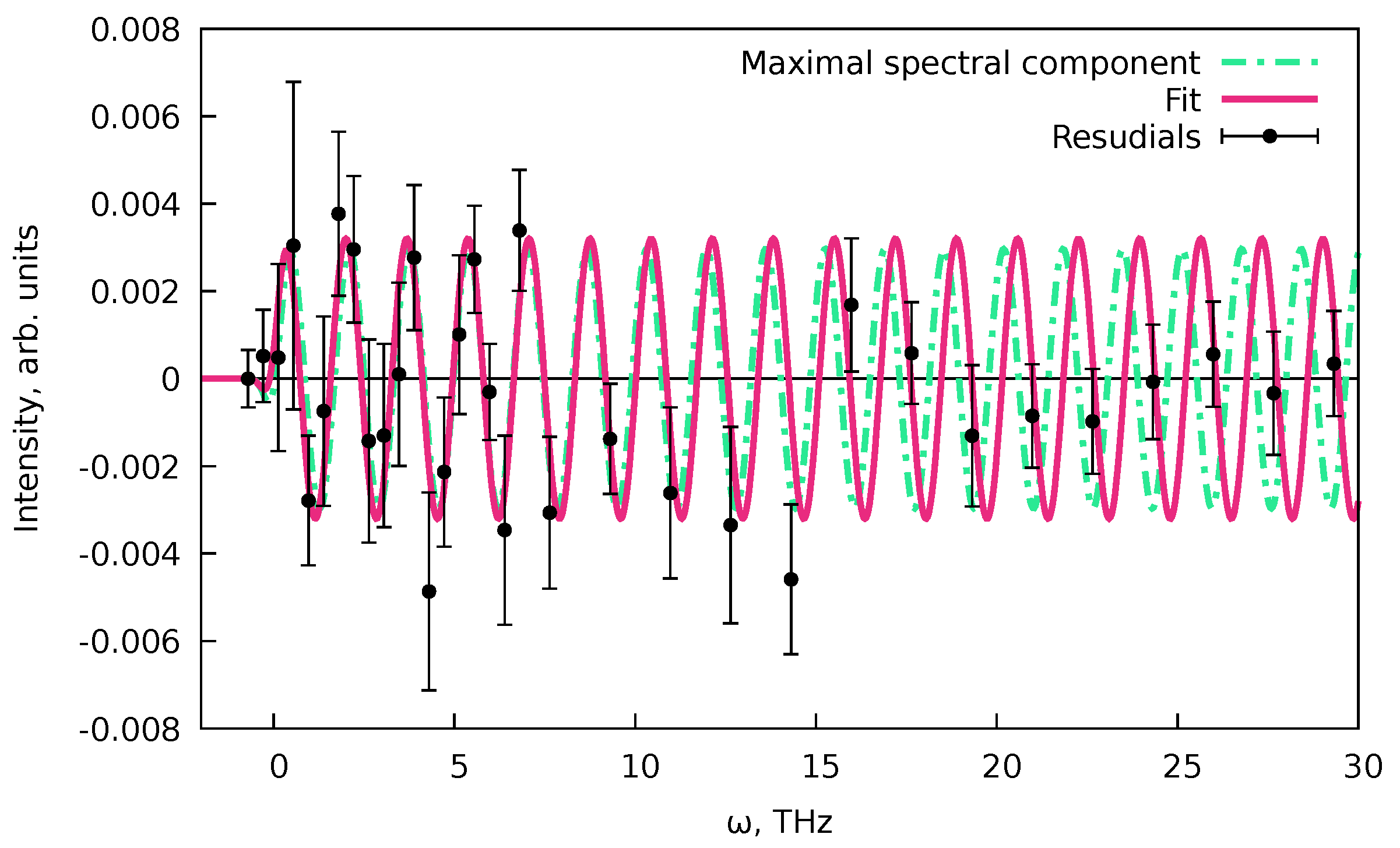
6.3. Treatment of the Data with Coherent Oscillations
- First, with the algorithm from Section 4.4 implemented in PP(MCFitting, we fit the nonoscillating part of the dynamics.
- Then, we perform an rwLSSA analysis [68] (see Appendix C) for the residuals of the fit that are also printed by the PP(MCFitting. This analysis will allow us to check whether the signal contains any systematic oscillations. Note that the oscillations should be present only in or in parts of the pump–probe data (see Equations (82) and (83)).
- If the rwLSSA spectrum shows a presence of statistically meaningful oscillations in a reasonable range of frequencies, then the frequencies and the phases of the maximal amplitude signals from rwLSSA spectra can be used as initial guesses to fit the residuals of the PP(MCFitting result with expression Equation (76) and basis functions (Equation (82)) and (Equation (83)). Since the coherent oscillations should correspond to the incoherent processes, the cross-correlations and decay times from the PP(MCFitting results can be used.
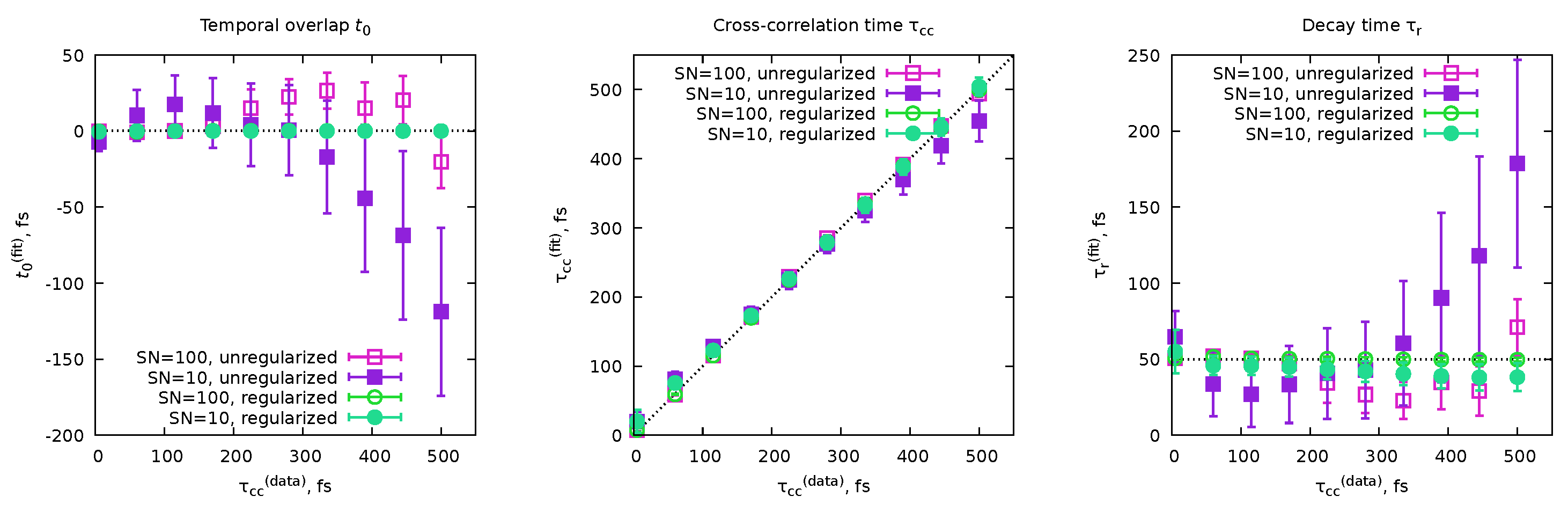
6.4. Cross-Correlation Time and Time Resolution
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FEL | Free-electron laser |
| FT | Fourier transform |
| IR | Infrared |
| LSQ | Least-squares |
| (rw)LSSA | (Regularized weighted) least-squares spectral analysis |
| MC | Monte-Carlo |
| PAH | Polycyclic aromatic hydrocarbon |
| SN | Signal-to-noise ratio |
| XUV | Extreme ultraviolet |
Appendix A. Detailed Derivations of Delta-Shaped Pump–Probe Dynamics
Appendix A.1. Solution of the First-Order Kinetics Equations
Appendix A.2. Coherent Quantum Dynamics without Decay
Appendix A.3. Relation between Quantum and Classical Regimes
Appendix A.4. Coherent Quantum Dynamics with Decay
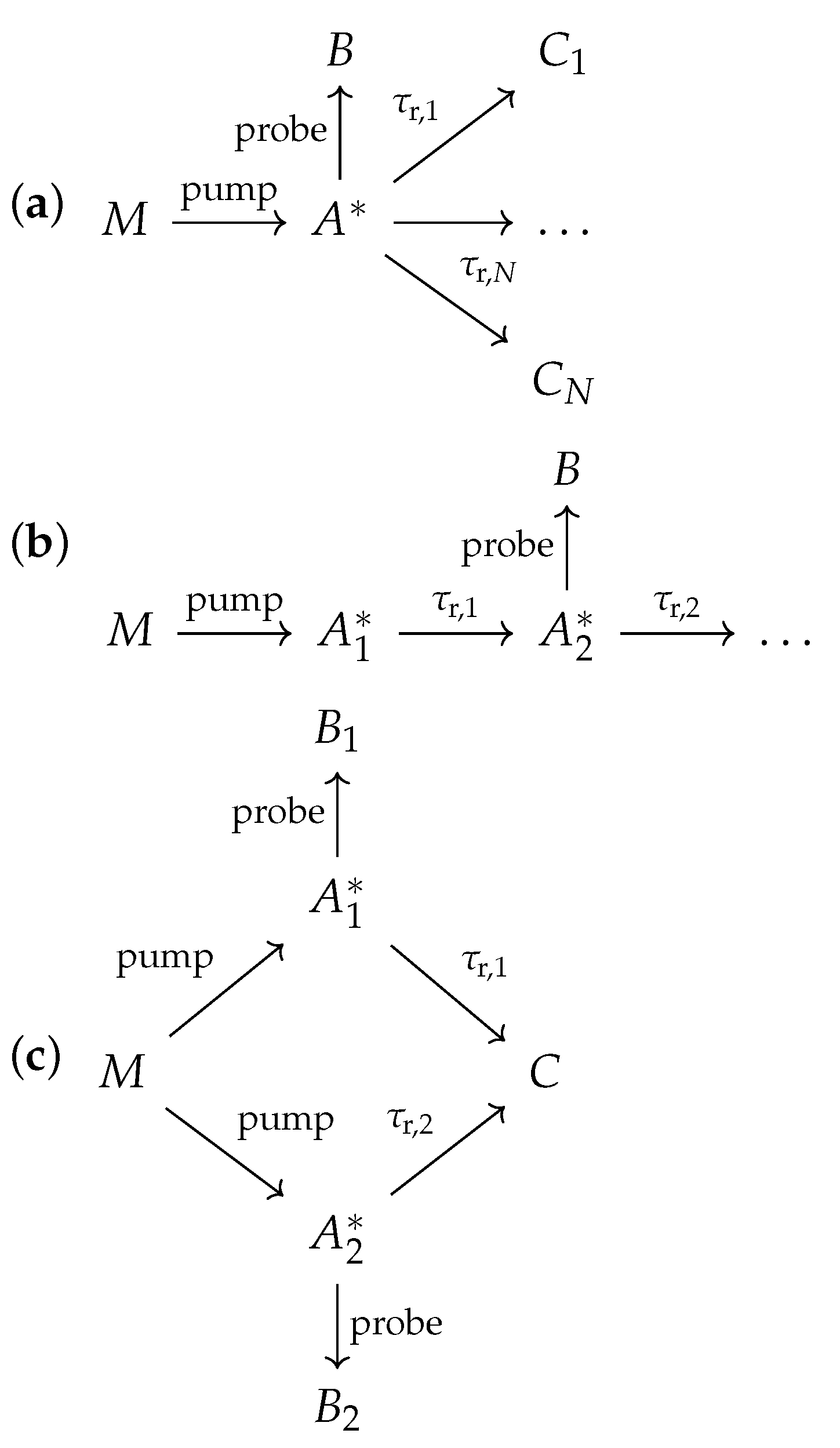
Appendix A.5. Reaction Scheme with Multiple Products
Appendix A.6. Reaction Scheme with Sequential Metastable Intermediates
Appendix A.7. Reaction Scheme with Multiple Intermediates Forming a Single Product
Appendix A.8. General form of the Decay Dynamics Pump–Probe Equations
Appendix B. Effects of the Duration of the Pulses and Experimental Setup Jitter
Appendix B.1. Sequential Convolution with Gaussian-Shaped Pulses
Appendix B.2. Basis Functions for Fitting Observables with Finite Duration Pump/Probe Pulses and Experimental Jitter
Appendix B.2.1. Constant Function
Appendix B.2.2. Step Function
Appendix B.2.3. Transient Function
Appendix B.2.4. Instant Increase Function
Appendix B.2.5. Nondecaying Coherent Oscillation Function
Appendix B.2.6. Transient Coherent Oscillation Function
Appendix C. Regularized Weighted Least-Squares Spectral Analysis (rwLSSA)
- is the N-dimensional vector of the data points;
- is the M-dimensional vector of spectral representation;
- is the matrix of size with elements ;
- is the diagonal matrix of weights;
- is the regularization parameter;
- is the covariance matrix defined as , where is the unit matrix of size .
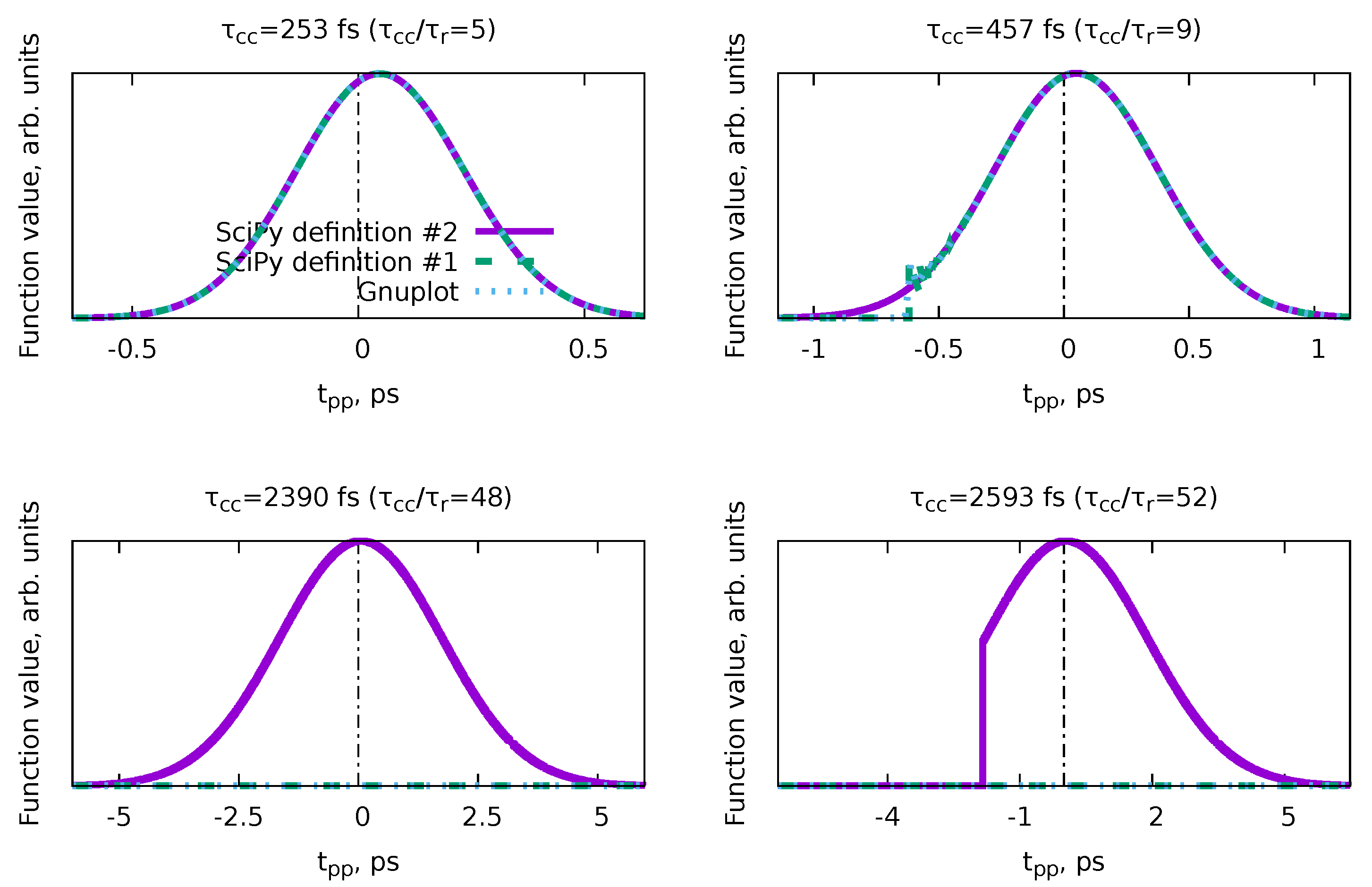
Appendix D. Issues with Numerical Implementation of the Bt(tpp) Basis Function

References
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond Using Ultrafast Lasers (Nobel Lecture). Angew. Chem. Int. Ed. 2000, 39, 2586–2631. [Google Scholar] [CrossRef]
- Young, L.; Ueda, K.; Gühr, M.; Bucksbaum, P.H.; Simon, M.; Mukamel, S.; Rohringer, N.; Prince, K.C.; Masciovecchio, C.; Meyer, M.; et al. Roadmap of ultrafast X-ray atomic and molecular physics. J. Phys. At. Mol. Opt. Phys. 2018, 51, 032003. [Google Scholar] [CrossRef]
- Zettergren, H.; Domaracka, A.; Schlathölter, T.; Bolognesi, P.; Díaz-Tendero, S.; Łabuda, M.; Tosic, S.; Maclot, S.; Johnsson, P.; Steber, A.; et al. Roadmap on dynamics of molecules and clusters in the gas phase. Eur. Phys. J. 2021, 75, 152. [Google Scholar] [CrossRef]
- Hertel, I.V.; Radloff, W. Ultrafast dynamics in isolated molecules and molecular clusters. Rep. Prog. Phys. 2006, 69, 1897. [Google Scholar] [CrossRef]
- Scutelnic, V.; Tsuru, S.; Pápai, M.; Yang, Z.; Epshtein, M.; Xue, T.; Haugen, E.; Kobayashi, Y.; Krylov, A.I.; Møller, K.B.; et al. X-ray transient absorption reveals the 1Au (nπ*) state of pyrazine in electronic relaxation. Nat. Commun. 2021, 12, 5003. [Google Scholar] [CrossRef]
- Lee, J.W.L.; Tikhonov, D.S.; Chopra, P.; Maclot, S.; Steber, A.L.; Gruet, S.; Allum, F.; Boll, R.; Cheng, X.; Düsterer, S.; et al. Time-resolved relaxation and fragmentation of polycyclic aromatic hydrocarbons investigated in the ultrafast XUV-IR regime. Nat. Commun. 2021, 12, 6107. [Google Scholar] [CrossRef] [PubMed]
- Garg, D.; Lee, J.W.L.; Tikhonov, D.S.; Chopra, P.; Steber, A.L.; Lemmens, A.K.; Erk, B.; Allum, F.; Boll, R.; Cheng, X.; et al. Fragmentation Dynamics of Fluorene Explored Using Ultrafast XUV-Vis Pump-Probe Spectroscopy. Front. Phys. 2022, 10, 880793. [Google Scholar] [CrossRef]
- Calegari, F.; Ayuso, D.; Trabattoni, A.; Belshaw, L.; Camillis, S.D.; Anumula, S.; Frassetto, F.; Poletto, L.; Palacios, A.; Decleva, P.; et al. Ultrafast electron dynamics in phenylalanine initiated by attosecond pulses. Science 2014, 346, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.J.A.; Paul, A.C.; Folkestad, S.D.; Myhre, R.H.; Cryan, J.P.; Berrah, N.; Bucksbaum, P.H.; Coriani, S.; Coslovich, G.; Feifel, R.; et al. Transient resonant Auger—Meitner spectra of photoexcited thymine. Faraday Discuss. 2021, 228, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Onvlee, J.; Trippel, S.; Küpper, J. Ultrafast light-induced dynamics in the microsolvated biomolecular indole chromophore with water. Nat. Commun. 2022, 13, 7462. [Google Scholar] [CrossRef]
- Malý, P.; Brixner, T. Fluorescence-Detected Pump–Probe Spectroscopy. Angew. Chem. Int. Ed. 2021, 60, 18867–18875. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes 3rd Edition: The Art of Scientific Computing, 3rd ed.; Cambridge University Press: New York, NY, USA, 2007. [Google Scholar]
- Mosegaard, K.; Tarantola, A. Monte Carlo sampling of solutions to inverse problems. J. Geophys. Res. Solid Earth 1995, 100, 12431–12447. [Google Scholar] [CrossRef]
- Bingham, D.; Butler, T.; Estep, D. Inverse Problems for Physics-Based Process Models. Annu. Rev. Stat. Its Appl. 2024, 11. [Google Scholar] [CrossRef]
- Tikhonov, A.N. Solution of incorrectly formulated problems and the regularization method. Soviet Math. Dokl. 1963, 4, 1035–1038. [Google Scholar]
- Tikhonov, A.; Leonov, A.; Yagola, A. Nonlinear Ill-Posed Problems; Chapman & Hall: London, UK, 1998. [Google Scholar]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Pedersen, S.; Zewail, A.H. Femtosecond real time probing of reactions XXII Kinetic description of probe absorption fluorescence depletion and mass spectrometry. Mol. Phys. 1996, 89, 1455–1502. [Google Scholar] [CrossRef]
- Hadamard, J. Sur les problèmes aux dérivés partielles et leur signification physique. Princet. Univ. Bull. 1902, 13, 49–52. [Google Scholar]
- Tikhonov, D.S.; Vishnevskiy, Y.V.; Rykov, A.N.; Grikina, O.E.; Khaikin, L.S. Semi-experimental equilibrium structure of pyrazinamide from gas-phase electron diffraction. How much experimental is it? J. Mol. Struct. 2017, 1132, 20–27. [Google Scholar] [CrossRef]
- Hestenes, M.R.; Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bur. Stand. 1952, 49, 409–436. [Google Scholar] [CrossRef]
- Powell, M.J.D. An efficient method for finding the minimum of a function of several variables without calculating derivatives. Comput. J. 1964, 7, 155–162. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Santosa, F.; Symes, W.W. Linear Inversion of Band-Limited Reflection Seismograms. SIAM J. Sci. Stat. Comput. 1986, 7, 1307–1330. [Google Scholar] [CrossRef]
- Bauer, F.; Lukas, M.A. Comparing parameter choice methods for regularization of ill-posed problems. Math. Comput. Simul. 2011, 81, 1795–1841. [Google Scholar] [CrossRef]
- Chiu, N.; Ewbank, J.; Askari, M.; Schäfer, L. Molecular orbital constrained gas electron diffraction studies: Part I. Internal rotation in 3-chlorobenzaldehyde. J. Mol. Struct. 1979, 54, 185–195. [Google Scholar] [CrossRef]
- Baše, T.; Holub, J.; Fanfrlík, J.; Hnyk, D.; Lane, P.D.; Wann, D.A.; Vishnevskiy, Y.V.; Tikhonov, D.; Reuter, C.G.; Mitzel, N.W. Icosahedral Carbaboranes with Peripheral Hydrogen—Chalcogenide Groups: Structures from Gas Electron Diffraction and Chemical Shielding in Solution. Chem.—Eur. J. 2019, 25, 2313–2321. [Google Scholar] [CrossRef]
- Vishnevskiy, Y.V.; Tikhonov, D.S.; Reuter, C.G.; Mitzel, N.W.; Hnyk, D.; Holub, J.; Wann, D.A.; Lane, P.D.; Berger, R.J.F.; Hayes, S.A. Influence of Antipodally Coupled Iodine and Carbon Atoms on the Cage Structure of 9,12-I2-closo-1,2-C2B10H10: An Electron Diffraction and Computational Study. Inorg. Chem. 2015, 54, 11868–11874. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 2004, 21, 1087–1092. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Rosenbluth, M.N. Genesis of the Monte Carlo Algorithm for Statistical Mechanics. AIP Conf. Proc. 2003, 690, 22–30. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.; Seger, R.; Mullen, K.M.; van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Müller, C.; Pascher, T.; Eriksson, A.; Chabera, P.; Uhlig, J. KiMoPack: A python Package for Kinetic Modeling of the Chemical Mechanism. J. Phys. Chem. 2022, 126, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Arecchi, F.; Bonifacio, R. Theory of optical maser amplifiers. IEEE J. Quantum Electron. 1965, 1, 169–178. [Google Scholar] [CrossRef]
- Lindh, L.; Pascher, T.; Persson, S.; Goriya, Y.; Wärnmark, K.; Uhlig, J.; Chábera, P.; Persson, P.; Yartsev, A. Multifaceted Deactivation Dynamics of Fe(II) N-Heterocyclic Carbene Photosensitizers. J. Phys. Chem. 2023, 127, 10210–10222. [Google Scholar] [CrossRef] [PubMed]
- Brückmann, J.; Müller, C.; Friedländer, I.; Mengele, A.K.; Peneva, K.; Dietzek-Ivanšić, B.; Rau, S. Photocatalytic Reduction of Nicotinamide Co-factor by Perylene Sensitized RhIII Complexes**. Chem.—Eur. J. 2022, 28, e202201931. [Google Scholar] [CrossRef] [PubMed]
- Shchatsinin, I.; Laarmann, T.; Zhavoronkov, N.; Schulz, C.P.; Hertel, I.V. Ultrafast energy redistribution in C60 fullerenes: A real time study by two-color femtosecond spectroscopy. J. Chem. Phys. 2008, 129, 204308. [Google Scholar] [CrossRef] [PubMed]
- Van Stokkum, I.H.; Larsen, D.S.; van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1657, 82–104. [Google Scholar] [CrossRef]
- Connors, K. Chemical Kinetics: The Study of Reaction Rates in Solution; VCH Publishers, Inc.: New York, NY, USA, 1990. [Google Scholar]
- Atkins, P.; Paula, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Rivas, D.E.; Serkez, S.; Baumann, T.M.; Boll, R.; Czwalinna, M.K.; Dold, S.; de Fanis, A.; Gerasimova, N.; Grychtol, P.; Lautenschlager, B.; et al. High-temporal-resolution X-ray spectroscopy with free-electron and optical lasers. Optica 2022, 9, 429–430. [Google Scholar] [CrossRef]
- Debnath, T.; Mohd Yusof, M.S.B.; Low, P.J.; Loh, Z.H. Ultrafast structural rearrangement dynamics induced by the photodetachment of phenoxide in aqueous solution. Nat. Commun. 2019, 10, 2944. [Google Scholar] [CrossRef]
- Atkins, P.; Friedman, R. Molecular Quantum Mechanics; OUP Oxford: Oxford, UK, 2011. [Google Scholar]
- Manzano, D. A short introduction to the Lindblad master equation. AIP Adv. 2020, 10, 025106. [Google Scholar] [CrossRef]
- Tikhonov, D.S.; Blech, A.; Leibscher, M.; Greenman, L.; Schnell, M.; Koch, C.P. Pump-probe spectroscopy of chiral vibrational dynamics. Sci. Adv. 2022, 8, eade0311. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tikhonov, D.S.; Singh, H.; Steber, A.L.; Pérez, C.; Schnell, M. Inducing transient enantiomeric excess in a molecular quantum racemic mixture with microwave fields. Nat. Commun. 2023, 14, 934. [Google Scholar] [CrossRef] [PubMed]
- Hatano, N. Exceptional points of the Lindblad operator of a two-level system. Mol. Phys. 2019, 117, 2121–2127. [Google Scholar] [CrossRef]
- Schulz, S.; Grguraš, I.; Behrens, C.; Bromberger, H.; Costello, J.T.; Czwalinna, M.K.; Felber, M.; Hoffmann, M.C.; Ilchen, M.; Liu, H.Y.; et al. Femtosecond all-optical synchronization of an X-ray free-electron laser. Nat. Commun. 2015, 6, 5938. [Google Scholar] [CrossRef]
- Savelyev, E.; Boll, R.; Bomme, C.; Schirmel, N.; Redlin, H.; Erk, B.; Düsterer, S.; Müller, E.; Höppner, H.; Toleikis, S.; et al. Jitter-correction for IR/UV-XUV pump-probe experiments at the FLASH free-electron laser. New J. Phys. 2017, 19, 043009. [Google Scholar] [CrossRef]
- Schirmel, N.; Alisauskas, S.; Huülsenbusch, T.; Manschwetus, B.; Mohr, C.; Winkelmann, L.; Große-Wortmann, U.; Zheng, J.; Lang, T.; Hartl, I. Long-Term Stabilization of Temporal and Spectral Drifts of a Burst-Mode OPCPA System. In Proceedings of the Conference on Lasers and Electro-Optics, San Jose, CA, USA, 5–10 May 2019; p. STu4E.4. [Google Scholar] [CrossRef]
- Lambropoulos, P. Topics on Multiphoton Processes in Atoms**Work supported by a grant from the National Science Foundation No. MPS74-17553. Adv. At. Mol. Phys. 1976, 12, 87–164. [Google Scholar] [CrossRef]
- Kheifets, A.S. Wigner time delay in atomic photoionization. J. Phys. At. Mol. Opt. Phys. 2023, 56, 022001. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Chopra, P. Astrochemically Relevant Polycyclic Aromatic Hydrocarbons Investigated Using Ultrafast Pump-Probe Spectroscopy and Near-Edge X-ray Absorption Fine Structure Spectroscopy. Ph.D. Thesis, Christian-Albrechts-Universität zu Kiel, Kiel, Germany, 2022. [Google Scholar]
- Garg, D. Electronic Structure and Ultrafast Fragmentation Dynamics of Polycyclic Aromatic Hydrocarbons. Ph.D. Thesis, Universität Hamburg, Hamburg, Germany, 2023. [Google Scholar]
- Douguet, N.; Grum-Grzhimailo, A.N.; Bartschat, K. Above-threshold ionization in neon produced by combining optical and bichromatic XUV femtosecond laser pulses. Phys. Rev. A 2017, 95, 013407. [Google Scholar] [CrossRef]
- Strüder, L.; Epp, S.; Rolles, D.; Hartmann, R.; Holl, P.; Lutz, G.; Soltau, H.; Eckart, R.; Reich, C.; Heinzinger, K.; et al. Large-format, high-speed, X-ray pnCCDs combined with electron and ion imaging spectrometers in a multipurpose chamber for experiments at 4th generation light sources. Nucl. Instruments Methods Phys. Res. Sect. Accel. Spectrom. Detect. Assoc. Equip. 2010, 614, 483–496. [Google Scholar] [CrossRef]
- Erk, B.; Müller, J.P.; Bomme, C.; Boll, R.; Brenner, G.; Chapman, H.N.; Correa, J.; Düsterer, S.; Dziarzhytski, S.; Eisebitt, S.; et al. CAMP@FLASH: An end-station for imaging, electron- and ion-spectroscopy, and pump–probe experiments at the FLASH free-electron laser. J. Synchrotron. Radiat. 2018, 25, 1529–1540. [Google Scholar] [CrossRef]
- Rossbach, J. FLASH: The First Superconducting X-ray Free-Electron Laser. In Synchrotron Light Sources and Free-Electron Lasers: Accelerator Physics, Instrumentation and Science Applications; Jaeschke, E., Khan, S., Schneider, J.R., Hastings, J.B., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–22. [Google Scholar] [CrossRef]
- Beye, M.; Gühr, M.; Hartl, I.; Plönjes, E.; Schaper, L.; Schreiber, S.; Tiedtke, K.; Treusch, R. FLASH and the FLASH2020+ project—Current status and upgrades for the free-electron laser in Hamburg at DESY. Eur. Phys. J. Plus 2023, 138, 193. [Google Scholar] [CrossRef]
- Garcia, J.D.; Mack, J.E. Energy Level and Line Tables for One-Electron Atomic Spectra*. J. Opt. Soc. Am. 1965, 55, 654–685. [Google Scholar] [CrossRef]
- Martin, W.C. Improved 40ex0exi 1snl ionization energy, energy levels, and Lamb shifts for 1sns and 1snp terms. Phys. Rev. A 1987, 36, 3575–3589. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Kelley, C. Gnuplot 5.4: An Interactive Plotting Program. 2021. Available online: http://www.gnuplot.info (accessed on 20 January 2020).
- LEVENBERG, K. A Method for the Solution of Certain Non-Linear Problems in Least Squares. Q. Appl. Math. 1944, 2, 164–168. [Google Scholar] [CrossRef]
- Marquardt, D.W. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Vaníček, P. Approximate spectral analysis by least-squares fit. Astrophys. Space Sci. 1969, 4, 387–391. [Google Scholar] [CrossRef]
- Tikhonov, D.S. Regularized weighted sine least-squares spectral analysis for gas electron diffraction data. J. Chem. Phys. 2023, 159, 174101. [Google Scholar] [CrossRef] [PubMed]
- Ippen, E.P.; Shank, C.V. Techniques for Measurement. In Ultrashort Light Pulses: Picosecond Techniques and Applications; Shapiro, S.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1977; pp. 83–122. [Google Scholar] [CrossRef]
- Vardeny, Z.; Tauc, J. Picosecond coherence coupling in the pump and probe technique. Opt. Commun. 1981, 39, 396–400. [Google Scholar] [CrossRef]
- Lebedev, M.V.; Misochko, O.V.; Dekorsy, T.; Georgiev, N. On the nature of “coherent artifact”. J. Exp. Theor. Phys. 2005, 100, 272–282. [Google Scholar] [CrossRef]
- Rhodes, M.; Steinmeyer, G.; Ratner, J.; Trebino, R. Pulse-shape instabilities and their measurement. Laser Photonics Rev. 2013, 7, 557–565. [Google Scholar] [CrossRef]
- SciPy: Open Source Scientific Tools for Python. Available online: http://www.scipy.org/ (accessed on 18 January 2024).
- Van Stokkum, I.H.M.; Weißenborn, J.; Weigand, S.; Snellenburg, J.J. Pyglotaran: A lego-like Python framework for global and target analysis of time-resolved spectra. Photochem. Photobiol. Sci. 2023, 22, 2413–2431. [Google Scholar] [CrossRef]
- Müller, M.G.; Niklas, J.; Lubitz, W.; Holzwarth, A.R. Ultrafast Transient Absorption Studies on Photosystem I Reaction Centers from Chlamydomonas reinhardtii. 1. A New Interpretation of the Energy Trapping and Early Electron Transfer Steps in Photosystem I. Biophys. J. 2003, 85, 3899–3922. [Google Scholar] [CrossRef]
- Croce, R.; Müller, M.G.; Bassi, R.; Holzwarth, A.R. Carotenoid-to-Chlorophyll Energy Transfer in Recombinant Major Light-Harvesting Complex (LHCII) of Higher Plants. I. Femtosecond Transient Absorption Measurements. Biophys. J. 2001, 80, 901–915. [Google Scholar] [CrossRef]
- Holzwarth, A.R.; Müller, M.G.; Niklas, J.; Lubitz, W. Ultrafast Transient Absorption Studies on Photosystem I Reaction Centers from Chlamydomonas reinhardtii. 2: Mutations near the P700 Reaction Center Chlorophylls Provide New Insight into the Nature of the Primary Electron Donor. Biophys. J. 2006, 90, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Zurek, W.H. Decoherence and the Transition from Quantum to Classical. Phys. Today 1991, 44, 36–44. [Google Scholar] [CrossRef]
- Zurek, W.H. Decoherence and the Transition from Quantum to Classical–Revisited. Los Alamos Sci. 2002, 27, 86–109. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value, fs | |||||
|---|---|---|---|---|---|---|
| Global | Fit #0 | Fit #1 | Fit #2 | Fit #3 | Fit #4 | |
| ps | ||||||
| — | — | — | — | |||
| — | — | — | — | |||
| — | — | — | — | |||
| — | — | — | — | |||
| — | — | — | — | |||
| Parameter | Fit #1 | Fit #2 | PP(MC Fitting | ||
|---|---|---|---|---|---|
| Ini. | Fin. | Ini. | Fin. | ||
| ps | 0 | 0 | |||
| 97 | 97 | ||||
| 50 | 150 | ||||
| 50 | 150 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhonov, D.S.; Garg, D.; Schnell, M. Inverse Problems in Pump–Probe Spectroscopy. Photochem 2024, 4, 57-110. https://doi.org/10.3390/photochem4010005
Tikhonov DS, Garg D, Schnell M. Inverse Problems in Pump–Probe Spectroscopy. Photochem. 2024; 4(1):57-110. https://doi.org/10.3390/photochem4010005
Chicago/Turabian StyleTikhonov, Denis S., Diksha Garg, and Melanie Schnell. 2024. "Inverse Problems in Pump–Probe Spectroscopy" Photochem 4, no. 1: 57-110. https://doi.org/10.3390/photochem4010005
APA StyleTikhonov, D. S., Garg, D., & Schnell, M. (2024). Inverse Problems in Pump–Probe Spectroscopy. Photochem, 4(1), 57-110. https://doi.org/10.3390/photochem4010005