Torsional Disorder in Tetraphenyl [3]-Cumulenes: Insight into Excited State Quenching


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Spectroscopy
2.3. Calculations
3. Results
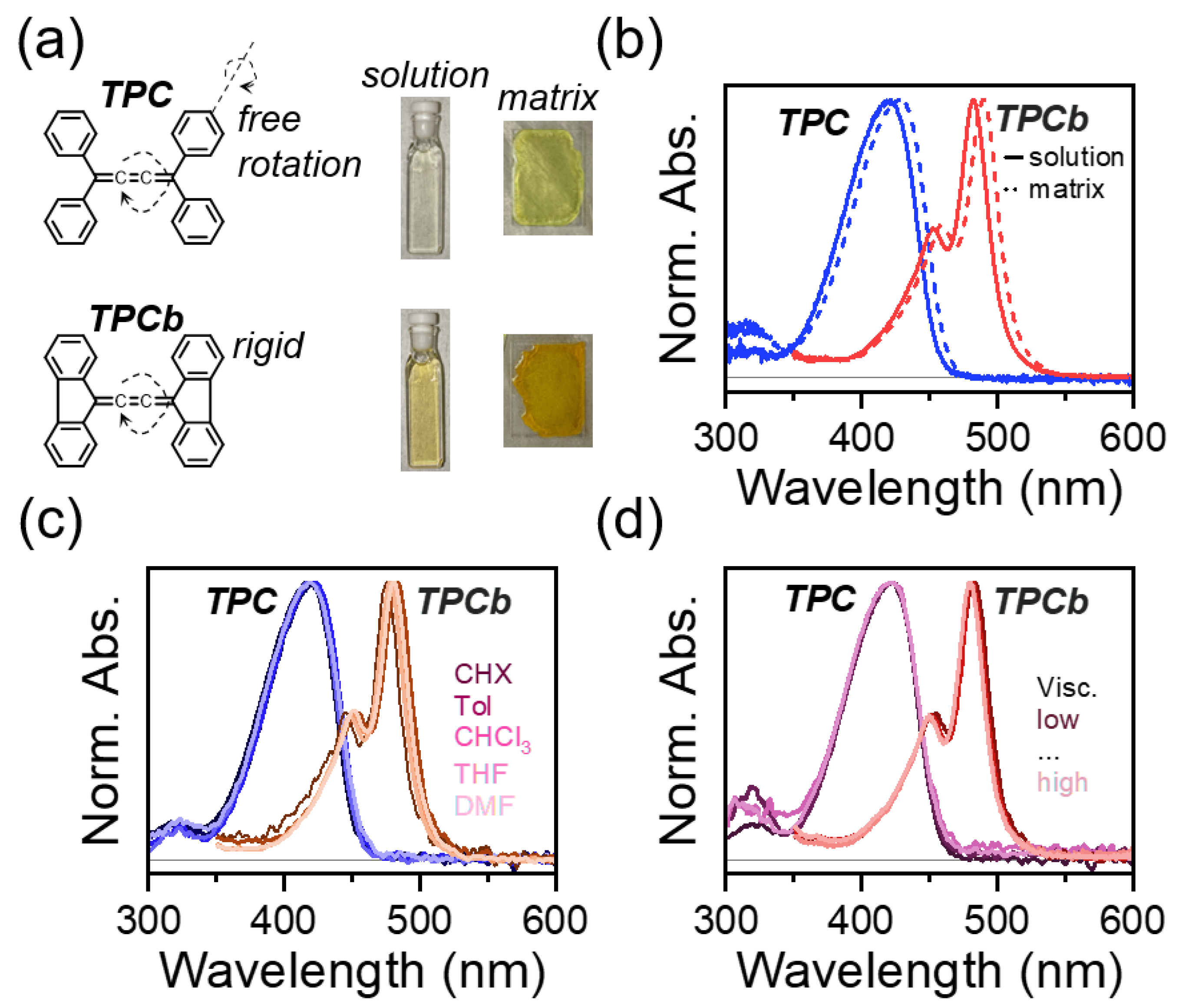
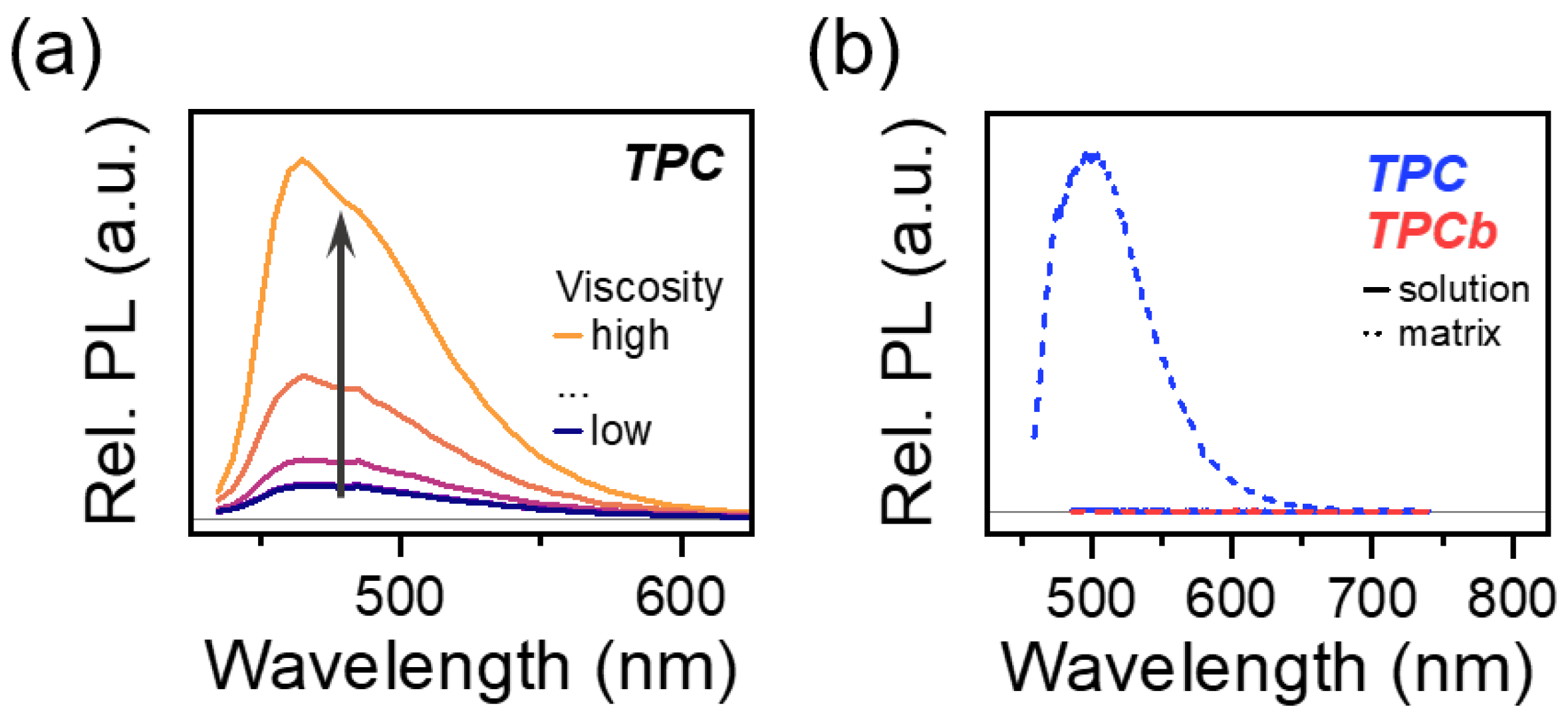
3.1. Steady-State Characterization
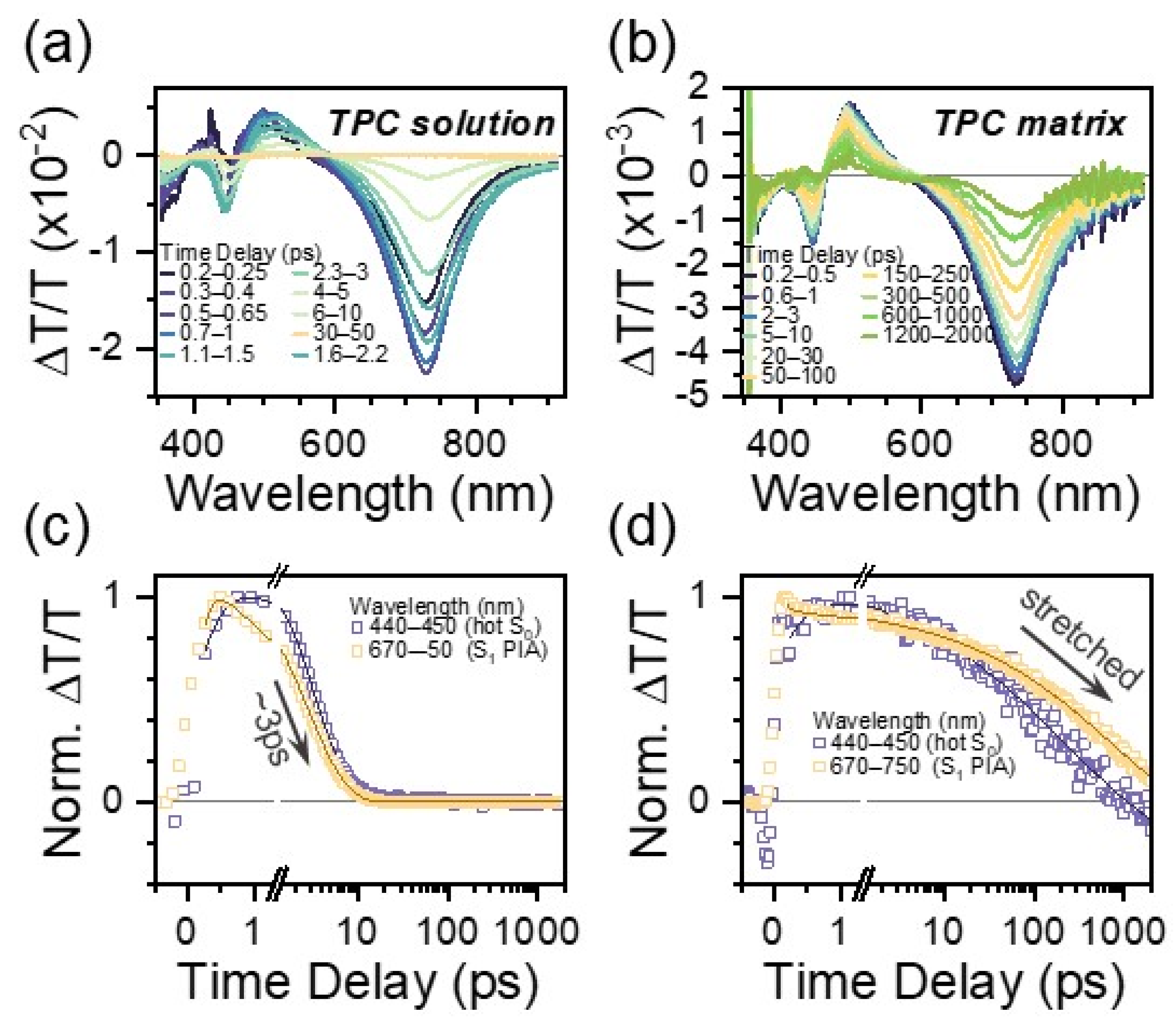
3.2. TPC Excited State Characterization
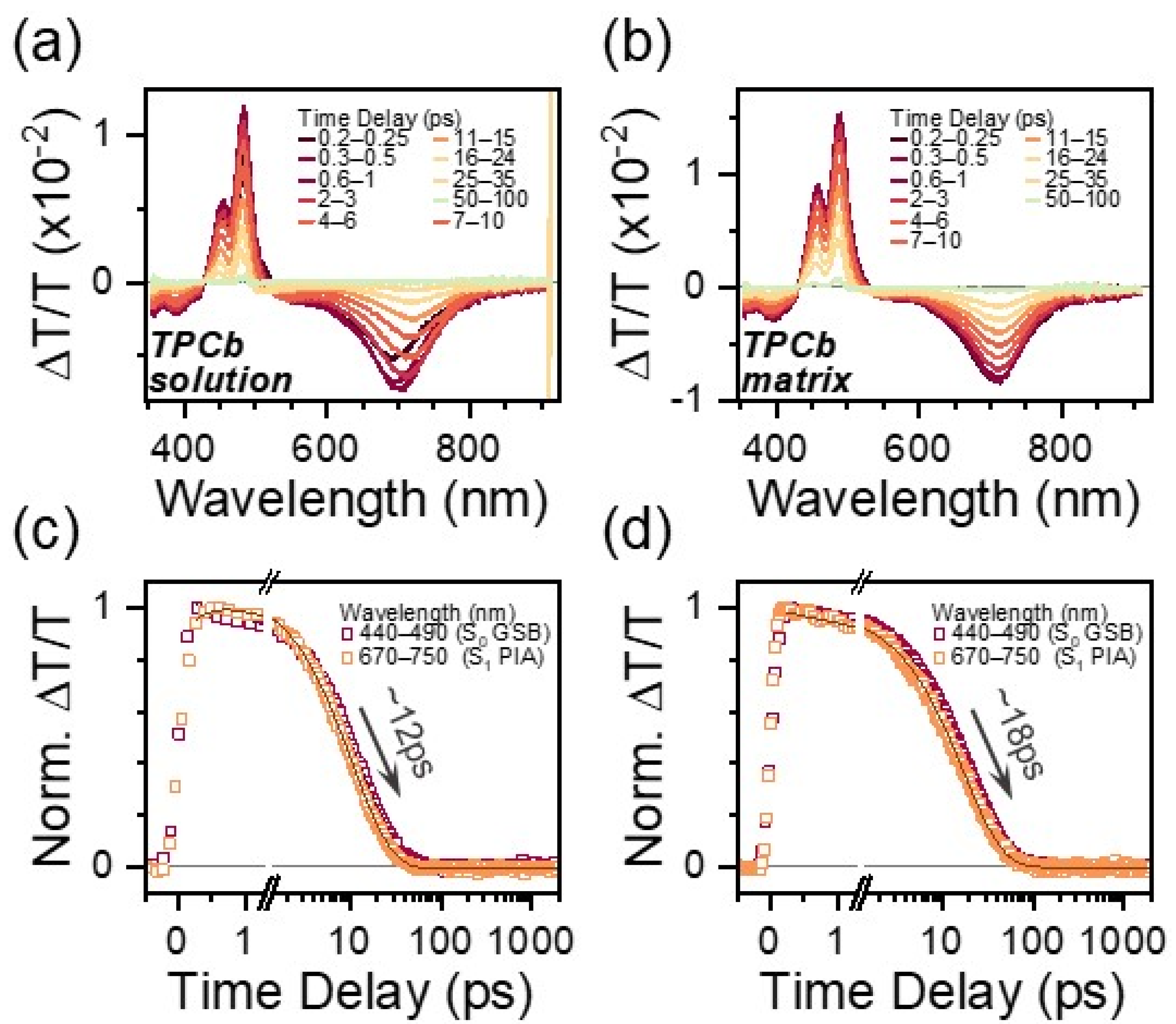
3.3. TPCb Excited State Characterization
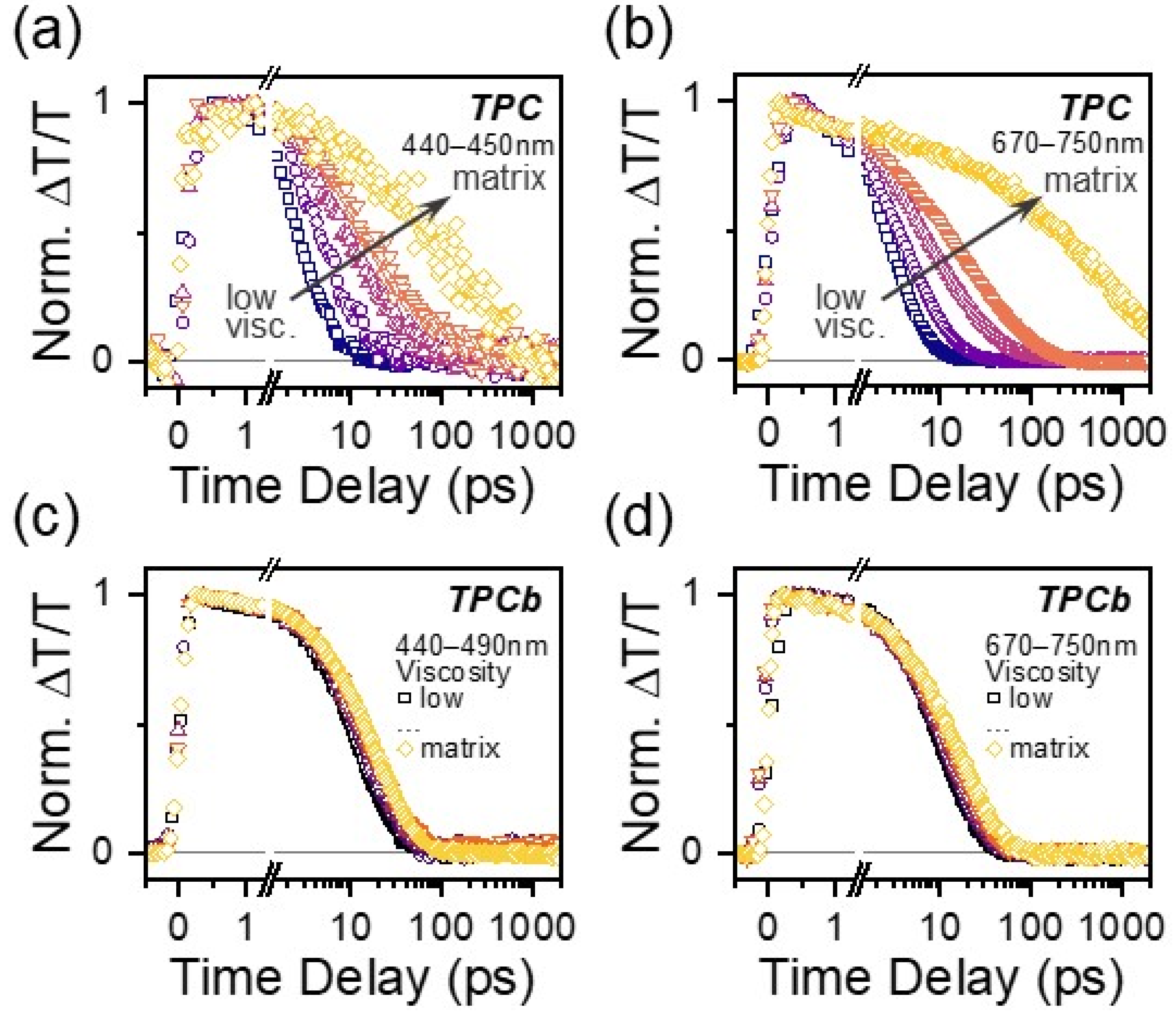
3.4. Environmental Effects on Excited State Relaxation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brand, K. Über Untersuchungen in der Tetraarylbutan-Reihe und Über Das 1.1 4.4-Tetraphenyl-Butatrien. (4. Mitteilung Über die Reduktion Organischer Halogen-Verbindungen.). Berichte Dtsch. Chem. Ges. A B Ser. 1921, 54, 1987–2006. [Google Scholar] [CrossRef]
- Tammann, G. Über Kohlenstoff, der Bei der Einwirkung von Quecksilber Auf CCl4, CBr4 Und CJ4 Entsteht. Z. Anorg. Allg. Chem. 1921, 115, 145–158. [Google Scholar] [CrossRef]
- Januszewski, J.A.; Tykwinski, R.R. Synthesis and Properties of Long [n]Cumulenes (n ≥ 5). Chem. Soc. Rev. 2014, 43, 3184–3203. [Google Scholar] [CrossRef] [PubMed]
- Banhart, F. Chains of Carbon Atoms: A Vision or a New Nanomaterial? Beilstein J. Nanotechnol. 2015, 6, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Bryce, M.R. A Review of Functional Linear Carbon Chains (Oligoynes, Polyynes, Cumulenes) and Their Applications as Molecular Wires in Molecular Electronics and Optoelectronics. J. Mater. Chem. C 2021, 9, 10524–10546. [Google Scholar] [CrossRef]
- Casari, C.S.; Milani, A. Carbyne: From the Elusive Allotrope to Stable Carbon Atom Wires. MRS Commun. 2018, 8, 207–219. [Google Scholar] [CrossRef]
- Casari, C.S.; Tommasini, M.; Tykwinski, R.R.; Milani, A. Carbon-Atom Wires: 1-D Systems with Tunable Properties. Nanoscale 2016, 8, 4414–4435. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lan, H.; Peng, L.; Suenaga, K.; Iijima, S. Deriving Carbon Atomic Chains from Graphene. Phys. Rev. Lett. 2009, 102, 205501. [Google Scholar] [CrossRef]
- La Torre, A.; Botello-Mendez, A.; Baaziz, W.; Charlier, J.-C.; Banhart, F. Strain-Induced Metal–Semiconductor Transition Observed in Atomic Carbon Chains. Nat. Commun. 2015, 6, 6636. [Google Scholar] [CrossRef]
- Zang, Y.; Fu, T.; Zou, Q.; Ng, F.; Li, H.; Steigerwald, M.L.; Nuckolls, C.; Venkataraman, L. Cumulene Wires Display Increasing Conductance with Increasing Length. Nano Lett. 2020, 20, 8415–8419. [Google Scholar] [CrossRef]
- Wang, M.; Lin, S. Ballistic Thermal Transport in Carbyne and Cumulene with Micron-Scale Spectral Acoustic Phonon Mean Free Path. Sci. Rep. 2015, 5, 18122. [Google Scholar] [CrossRef] [PubMed]
- Scaccabarozzi, A.D.; Milani, A.; Peggiani, S.; Pecorario, S.; Sun, B.; Tykwinski, R.R.; Caironi, M.; Casari, C.S. A Field-Effect Transistor Based on Cumulenic Sp-Carbon Atomic Wires. J. Phys. Chem. Lett. 2020, 11, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Pecorario, S.; Scaccabarozzi, A.D.; Fazzi, D.; Gutiérrez-Fernández, E.; Vurro, V.; Maserati, L.; Jiang, M.; Losi, T.; Sun, B.; Tykwinski, R.R.; et al. Stable and Solution-Processable Cumulenic Sp-Carbon Wires: A New Paradigm for Organic Electronics. Adv. Mater. 2022, 34, 2110468. [Google Scholar] [CrossRef] [PubMed]
- Hendon, C.H.; Tiana, D.; Murray, A.T.; Carbery, D.R.; Walsh, A. Helical Frontier Orbitals of Conjugated Linear Molecules. Chem. Sci. 2013, 4, 4278–4284. [Google Scholar] [CrossRef]
- Garner, M.H.; Hoffmann, R.; Rettrup, S.; Solomon, G.C. Coarctate and Möbius: The Helical Orbitals of Allene and Other Cumulenes. ACS Cent. Sci. 2018, 4, 688–700. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Venkataraman, L. Tight-Binding Analysis of Helical States in Carbyne. J. Chem. Phys. 2020, 153, 124304. [Google Scholar] [CrossRef]
- Lai, Y.; Halder, A.; Kim, J.; Hicks, T.J.; Milner, P.J. Electroreductive Radical Borylation of Unactivated (Hetero)Aryl Chlorides Without Light by Using Cumulene-Based Redox Mediators. Angew. Chem. Int. Ed. 2023, 62, e202310246. [Google Scholar] [CrossRef]
- Hirao, Y.; Ihara, K.; Ishibashi, Y.; Tiu, E.G.; Asahi, T.; Kubo, T. Mechanism and Kinetics of Fluorescence Quenching of Fluorene-Endcapped Butatriene: A Microspectroscopic Study of the Discrete State Constructed in Microcrystals. J. Phys. Chem. C 2022, 126, 1196–1203. [Google Scholar] [CrossRef]
- Hirao, Y.; Daifuku, Y.; Ihara, K.; Kubo, T. Spin–Spin Interactions in One-Dimensional Assemblies of a Cumulene-Based Singlet Biradical. Angew. Chem. 2021, 133, 21489–21496. [Google Scholar] [CrossRef]
- Messelberger, J.; Grünwald, A.; Pinter, P.; Hansmann, M.M.; Munz, D. Carbene Derived Diradicaloids—Building Blocks for Singlet Fission? Chem. Sci. 2018, 9, 6107–6117. [Google Scholar] [CrossRef]
- Morris, V.R.; Pollack, S.K. Singlet−Triplet Gap in 1,2,3-Butatriene and Its Consequences on the Mechanism of Its Spontaneous Polymerization. J. Phys. Chem. B 1998, 102, 5042–5046. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Zhu, R. Conjugated [5]Cumulene Polymers Enabled by Condensation Polymerization of Propargylic Electrophiles. J. Am. Chem. Soc. 2023, 145, 23755–23763. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Kaiser, R.I.; Mebel, A.M. Chemistry of Energetically Activated Cumulenes—From Allene (H2CCCH2) to Hexapentaene (H2CCCCCCH2). ChemPhysChem 2008, 9, 350–369. [Google Scholar] [CrossRef] [PubMed]
- Kabaciński, P.; Marabotti, P.; Fazzi, D.; Petropoulos, V.; Iudica, A.; Serafini, P.; Cerullo, G.; Casari, C.S.; Zavelani-Rossi, M. Disclosing Early Excited State Relaxation Events in Prototypical Linear Carbon Chains. J. Am. Chem. Soc. 2023, 145, 18382–18390. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, R.; Sander, W.; Cremer, D.; Kraka, E. Photochemistry of Butatriene—Spectroscopic Evidence for the Existence of Allenylcarbene. J. Phys. Chem. A 2000, 104, 3819–3825. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E.; Joo, H.; A. Stearns, J.; S. Zwier, T. Exploration of the Potential Energy Surface of C4H4 for Rearrangement and Decomposition Reactions of Vinylacetylene: A Computational Study. Part I. Phys. Chem. Chem. Phys. 2006, 8, 5304–5316. [Google Scholar] [CrossRef] [PubMed]
- Connors, R.E.; Mochel, J.; Chynwat, V. Electronic Absorption Spectrum of 1,1,4,4-Tetraphenylbutatriene. J. Phys. Chem. 1988, 92, 1792–1793. [Google Scholar] [CrossRef]
- Ermer, S.P.; Lovejoy, S.M.; Leung, D.S.; Altman, J.C.; Aron, K.P.; Spitzer, R.C.; Hansen, G.A. Synthesis and Evaluation of Cumulenes: Novel Rigid Nonlinear Optical Molecules. In Proceedings of SPIE 1337, Nonlinear Optical Properties of Organic Materials III. Proceedings of the 34th Annual International Technical Symposium on Optical and Optoelectronic Applied Science and Engineering, San Diego, CA, USA, 8–13 July 1990; Khanarian, G., Ed.; SPIE: Bellingham, WA, USA, 1990; p. 89. [Google Scholar]
- Chynwat, V.; Coffin, T.L.; Wang, H.; Connors, R.E. Excited Electronic States of Arylbutatrienes. J. Phys. Chem. 1996, 100, 5217–5223. [Google Scholar] [CrossRef]
- Connors, R.E.; Chynwat, V.; Clifton, C.H.; Coffin, T.L. Temperature Dependence of Aryl Butatriene Fluorescence: Barrier to Twisting on S 1 for 1,1,4,4-Tetraphenylbutatriene. J. Mol. Struct. 1998, 443, 107–113. [Google Scholar] [CrossRef]
- Jiménez, V.G.; Tapia, R.; Medel, M.A.; Mariz, I.F.A.; Ribeiro, T.; Blanco, V.; Cuerva, J.M.; Maçôas, E.; Campaña, A.G. Aggregation-Induced Emission of [3]Cumulenes Functionalized with Heptagon-Containing Polyphenylenes. Chem. Commun. 2018, 54, 3359–3362. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Lehtola, S.; Steigemann, C.; Oliveira, M.J.T.; Marques, M.A.L. Recent Developments in Libxc—A Comprehensive Library of Functionals for Density Functional Theory. SoftwareX 2018, 7, 1–5. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. A Simplified Tamm-Dancoff Density Functional Approach for the Electronic Excitation Spectra of Very Large Molecules. J. Chem. Phys. 2013, 138, 244104. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Lenzer, T.; Ehlers, F.; Scholz, M.; Oswald, R.; Oum, K. Assignment of Carotene S* State Features to the Vibrationally Hot Ground Electronic State. Phys. Chem. Chem. Phys. 2010, 12, 8832–8839. [Google Scholar] [CrossRef] [PubMed]
- Grilj, J.; Laricheva, E.N.; Olivucci, M.; Vauthey, E. Fluorescence of Radical Ions in Liquid Solution: Wurster’s Blue as a Case Study. Angew. Chem. Int. Ed. 2011, 50, 4496–4498. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.C. Stretched Exponential Relaxation Arising from a Continuous Sum of Exponential Decays. Phys. Rev. B 2006, 74, 184430. [Google Scholar] [CrossRef]
- Zang, Y.; Zou, Q.; Fu, T.; Ng, F.; Fowler, B.; Yang, J.; Li, H.; Steigerwald, M.L.; Nuckolls, C.; Venkataraman, L. Directing Isomerization Reactions of Cumulenes with Electric Fields. Nat. Commun. 2019, 10, 4482. [Google Scholar] [CrossRef]
- Bühringer, M.U.; Padberg, K.; Phleps, M.D.; Maid, H.; Placht, C.; Neiss, C.; Ferguson, M.J.; Görling, A.; Tykwinski, R.R. Double Bonds? Studies on the Barrier to Rotation about the Cumulenic C=C Bonds of Tetraaryl[n]Cumulenes (N = 3, 5, 7, 9). Angew. Chem. Int. Ed. 2018, 57, 8321–8325. [Google Scholar] [CrossRef]
- Kerisit, N.; Gawel, P.; Levandowski, B.; Yang, Y.-F.; García-López, V.; Trapp, N.; Ruhlmann, L.; Boudon, C.; Houk, K.N.; Diederich, F. A Four-Step Synthesis of Substituted 5,11-Dicyano-6,12-Diaryltetracenes with Enhanced Stability and High Fluorescence Emission. Chem. Eur. J. 2018, 24, 159–168. [Google Scholar] [CrossRef]
- Dahl, B.J.; Mills, N.S. Antiaromatic Spacer-Bridged Bisfluorenyl Dications Generated by Superacid Induced Ionization. J. Am. Chem. Soc. 2008, 130, 10179–10186. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bain, D.; Chang, J.; Lai, Y.; Khazanov, T.; Milner, P.J.; Musser, A.J. Torsional Disorder in Tetraphenyl [3]-Cumulenes: Insight into Excited State Quenching. Photochem 2024, 4, 138-150. https://doi.org/10.3390/photochem4010008
Bain D, Chang J, Lai Y, Khazanov T, Milner PJ, Musser AJ. Torsional Disorder in Tetraphenyl [3]-Cumulenes: Insight into Excited State Quenching. Photochem. 2024; 4(1):138-150. https://doi.org/10.3390/photochem4010008
Chicago/Turabian StyleBain, David, Julia Chang, Yihuan Lai, Thomas Khazanov, Phillip J. Milner, and Andrew J. Musser. 2024. "Torsional Disorder in Tetraphenyl [3]-Cumulenes: Insight into Excited State Quenching" Photochem 4, no. 1: 138-150. https://doi.org/10.3390/photochem4010008
APA StyleBain, D., Chang, J., Lai, Y., Khazanov, T., Milner, P. J., & Musser, A. J. (2024). Torsional Disorder in Tetraphenyl [3]-Cumulenes: Insight into Excited State Quenching. Photochem, 4(1), 138-150. https://doi.org/10.3390/photochem4010008