
Role of NK Cells in Cancer and Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
2. NK-Cell-Based Cancer Immunotherapy
3. Adoptive NK Cell Therapy
4. Use of Ex Vivo-Activated NK Cells
5. NK Cells and CARs
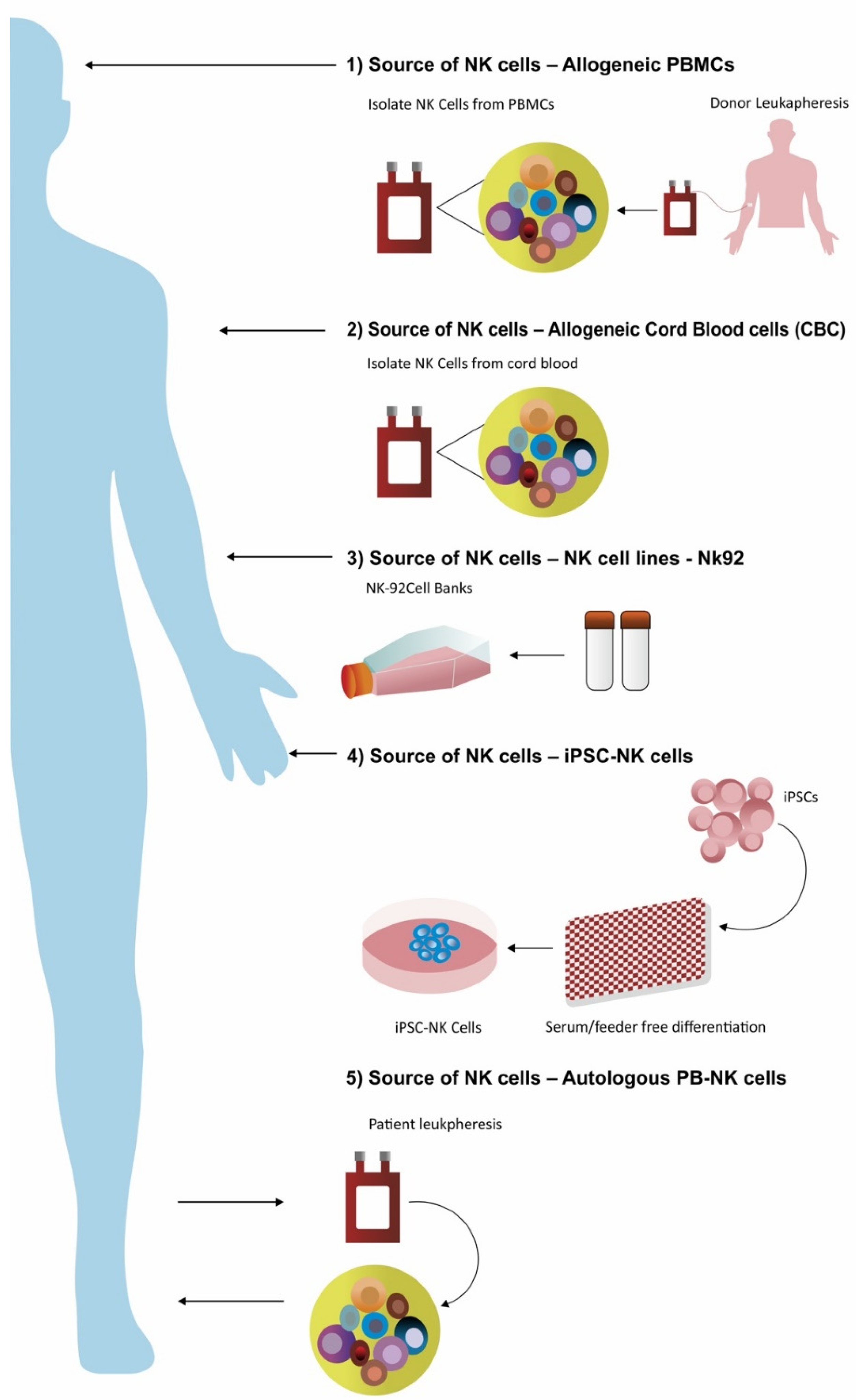
5.1. NK Cells Derived from Peripheral Blood
5.2. Immortalized Cells Lines as a Source of NK Cells
5.3. NK Cells Derived from Cord Blood
5.4. NK Cells Derived from Induced Pluripotent Stem Cell
6. Novel Bispecific Antibodies or Single-Chain Variable Fragment Targeting NK Cells
6.1. Bispecific NK Cell Engager (BiKE)
6.1.1. CD16xCD33
6.1.2. CD30/CD16A
7. Limitations
8. Future and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.G. Cellular therapy of cancer with natural killer cells-where do we stand? Cytotherapy 2013, 15, 1185–1194. [Google Scholar] [CrossRef]
- A Geller, M.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56bright NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Gasser, S.; Raulet, D.H. Activation and self-tolerance of natural killer cells. Immunol. Rev. 2006, 214, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.; Lanier, L.L. Natural Killer Cell Education and Tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8+ T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef]
- O’Leary, J.G.; Goodarzi, M.; Drayton, D.L.; von Andrian, U.H. T Cell– and B Cell–Independent Adaptive Immunity Mediated by Natural Killer Cells. Nat. Immunol. 2006, 7, 507–516. Available online: https://www.nature.com/articles/ni1332 (accessed on 26 September 2021). [CrossRef]
- Paust, S.; Gill, H.S.; Wang, B.Z.; Flynn, M.P.; Moseman, E.A.; Senman, B.; Szczepanik, M.; Telenti, A.; Askenase, P.W.; Compans, R.W.; et al. Critical Role for the Chemokine Receptor CXCR6 in NK Cell–Mediated antigen-Specific Memory of Haptens and Viruses. Nat. Immunol. 2010, 11, 1127–1135. Available online: https://www.nature.com/articles/ni.1953 (accessed on 26 September 2021). [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Rokni, M.; Marofi, F.; Panahi, Y.; Yousefi, M. Natural killer cell-based immunotherapy: From transplantation toward targeting cancer stem cells. J. Cell Physiol. 2018, 234, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Mancusi, A.; Urbani, E.; Perruccio, K.; Burchielli, E.; Tosti, A.; Topini, F.; Aversa, F.; Martelli, M.F.; et al. Alloreactive natural killer cells in mismatched hematopoietic stem cell transplantation. Blood Cells Mol. Dis. 2004, 33, 216–221. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Fogel, L.A.; Sun, M.M.; Geurs, T.L.; Carayannopoulos, L.N.; French, A.R. Markers of Nonselective and Specific NK Cell Activation. J. Immunol. 2013, 190, 6269–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Anfossi, N. Inhibitory NK-cell receptors on T cells: Witness of the past, actors of the future. Nat. Rev. Immunol. 2004, 4, 190–198. [Google Scholar] [CrossRef]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell–cancer cycle: Advances and new challenges in NK cell–based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Corat, M.A.F.; Schlums, H.; Wu, C.; Theorell, J.; Espinoza, D.A.; Sellers, S.E.; Townsley, D.M.; Young, N.S.; Bryceson, Y.T.; Dunbar, C.E.; et al. Acquired somatic mutations in PNH reveal long-term maintenance of adaptive NK cells independent of HSPCs. Blood 2017, 129, 1940–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wallace, D.L.; De Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsis-Mortari, A.; Keever-Taylor, C.A.; Zhang, M.J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow Transplant. 2003, 32, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walewski, J.; Paietta, E.; Dutcher, J.; Wiernik, P.H. Evaluation of natural killer and lymphokine-activated killer (LAK) cell activity in vivo in patients treated with high-dose interleukin-2 and adoptive transfer of autologous LAK cells. J. Cancer Res. Clin. Oncol. 1989, 115, 170–174. [Google Scholar] [CrossRef]
- Benyunes, M.C.; Higuchi, C.; York, A.; Lindgren, C.; A Thompson, J.; Buckner, C.D.; Fefer, A. Immunotherapy with interleukin 2 with or without lymphokine-activated killer cells after autologous bone marrow transplantation for malignant lymphoma: A feasibility trial. Bone Marrow Transplant. 1995, 16, 283–288. [Google Scholar]
- Ni, J.; Miller, M.; Stojanovic, A.; Cerwenka, A. Toward the next generation of NK cell-based adoptive cancer immunotherapy. Oncoimmunology 2013, 2, e23811. [Google Scholar] [CrossRef] [Green Version]
- Stern, M.; Passweg, J.R.; Meyer-Monard, S.; Esser, R.; Tonn, T.; Soerensen, J.; Paulussen, M.; Gratwohl, A.; Klingebiel, T.; Bader, P.; et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: A prospective phase II study in two centers. Bone Marrow Transplant 2013, 48, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Sun, H.; Zhang, C.; Tian, Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell. Mol. Immunol. 2014, 12, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Kono, K.; Kawaguchi, Y.; Mizukami, Y.; Mimura, K.; Maruyama, T.; Izawa, S.; Fujii, H. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis. Esophagus 2010, 23, 675–681. [Google Scholar] [CrossRef]
- Izawa, S.; Kono, K.; Mimura, K.; Kawaguchi, Y.; Watanabe, M.; Maruyama, T.; Fujii, H. H2O2; production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: Possible mechanisms of NK cell dysfunction. Cancer Immunol. Immunother. 2011, 60, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Rego, R.L.; Ansell, S.M.; Knutson, K.L.; Foster, N.R.; Sargent, D.J. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology 2009, 137, 1270–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartigan-O’Connor, D.J.; Poon, C.; Sinclair, E.; McCune, J.M. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J. Immunol. Methods 2007, 319, 41–52. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Moretta, L.; Moretta, A. Unravelling natural killer cell function: Triggering and inhibitory human NK receptors. EMBO J. 2003, 23, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Navarro, F.; Bellón, T.; Llano, M.; García, P.; Samaridis, J.; Angman, L.; Cella, M.; López-Botet, M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997, 186, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Lundqvist, A.; McCoy, P.; Samsel, L.; Fan, Y.; Tawab, A.; Childs, R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy 2009, 11, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of Highly Cytotoxic Human Natural Killer Cells for Cancer Cell Therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, J.W.; Chase, J.; Romee, R.; Schneider, S.E.; Sullivan, R.P.; Cooper, M.; Fehniger, T.A. Preactivation with IL-12, IL-15, and IL-18 Induces CD25 and a Functional High-Affinity IL-2 Receptor on Human Cytokine-Induced Memory-like Natural Killer Cells. Biol. Blood Marrow Transplant. 2014, 20, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Miller, M.; Stojanovic, A.; Garbi, N.; Cerwenka, A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J. Exp. Med. 2012, 209, 2351–2365. [Google Scholar] [CrossRef]
- Imamura, M.; Shook, D.; Kamiya, T.; Shimasaki, N.; Chai, S.M.H.; Coustan-Smith, E.; Imai, C.; Campana, D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood 2014, 124, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Barrett, D.M.; Singh, N.; Porter, D.L.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor Therapy for Cancer. Annu. Rev. Med. 2014, 65, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Brentjens, R.J. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov. Med. 2010, 9, 277–288. [Google Scholar]
- Hernández-López, A.; Téllez-González, M.A.; Mondragón-Terán, P.; Meneses-Acosta, A. Chimeric Antigen Receptor-T Cells: A Pharmaceutical Scope. Front. Pharmacol. 2021, 12, 720692. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8417740/ (accessed on 26 September 2021). [PubMed]
- Shimasaki, N.; Coustan-Smith, E.; Kamiya, T.; Campana, D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy 2016, 18, 1422–1434. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Glienke, W.; Esser, R.; Priesner, C.; Suerth, J.D.; Schambach, A.; Wels, W.S.; Grez, M.; Kloess, S.; Arseniev, L.; Koehl, U. Advantages and applications of CAR-expressing natural killer cells. Front. Pharmacol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Betancur, M.; Lu, W.; Wels, W.S.; Marino, T.; Van Etten, R.A.; Klingemann, H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk. Lymphoma 2012, 53, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [Green Version]
- ImmunityBio, Inc. Phase I Study of Adoptive Immunotherapy Using the Natural Killer Cell Line, Neukoplast™ (NK-92), for the Treatment of Refractory or Relapsed Acute Myeloid Leukemia (Internet). Report No.: NCT00900809. Available online: https://clinicaltrials.gov/ct2/show/NCT00900809 (accessed on 7 October 2021).
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [Green Version]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-Scale Derivation of Natural Killer Cells from Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci. Rep. 2019, 9, 18729. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S.; et al. Specific growth inhibition of ErbB2-expressing human breast cancer cells by genetically modified NK-92 cells. Oncol. Rep. 2014, 33, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell–mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Chu, Y.; Yahr, A.; Huang, B.; Ayello, J.; Barth, M.; Cairo, M.S. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. OncoImmunology 2017, 6, e1341031. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Kerbauy, L.N.; Marin, N.D.; Kaplan, M.; Banerjee, P.P.; Berrien-Elliott, M.M.; Becker-Hapak, M.; Basar, R.; Foster, M.; Garcia Melo, L.; Neal, C.C.; et al. Combining AFM13, a Bispecific CD30/CD16 Antibody, with Cytokine-Activated Blood and Cord Blood-Derived NK Cells Facilitates CAR-like Responses Against CD30+ Malignancies. Clin. Cancer Res. 2021, 27, 3744–3756. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Lu, W.; Krause, D.; Van Etten, R.; Wels, W.; Klingemann, H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. OncoImmunology 2013, 2, e26527. [Google Scholar] [CrossRef] [Green Version]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- University Health Network, Toronto. A Dose Escalation Study of NK-92 Cell Infusions in Patients with Hematological Malignancies in Relapse After Autologous Stem Cell Transplantation. (Internet). Report No.: NCT00990717. Available online: https://clinicaltrials.gov/ct2/show/NCT00990717 (accessed on 7 October 2021).
- St. Jude Children’s Research Hospital. Pilot Study of Genetically Modified Haploidentical Natural Killer Cell Infusions for B-Lineage Acute Lymphoblastic Leukemia (Internet). Report No.: NCT00995137. Available online: https://clinicaltrials.gov/ct2/show/NCT00995137 (accessed on 7 October 2021).
- Lin, T.P. Pilot Study of Redirected Haploidentical Natural Killer Cell Infusions for B-Lineage Acute Lymphoblastic Leukemia (Internet). Report No.: NCT01974479. Available online: https://clinicaltrials.gov/ct2/show/NCT01974479 (accessed on 7 October 2021).
- Williams, S.M.; Sumstad, D.; Kadidlo, D.; Curtsinger, J.; Luo, X.; Miller, J.S.; McKenna, D.H., Jr. Clinical-scale production of cGMP compliant CD3/CD19 cell-depleted NK cells in the evolution of NK cell immunotherapy at a single institution. Transfusion 2018, 58, 1458–1467. [Google Scholar] [CrossRef]
- Li, H.W.; Vishwasrao, P.; Hölzl, M.A.; Chen, S.; Choi, G.; Zhao, G.; Sykes, M. Impact of Mixed Xenogeneic Porcine Hematopoietic Chimerism on Human NK Cell Recognition in a Humanized Mouse Model. Arab. Archaeol. Epigr. 2016, 17, 353–364. [Google Scholar] [CrossRef] [Green Version]
- A Grimm, E.; Robb, R.J.; A Roth, J.; Neckers, L.M.; Lachman, L.B.; Wilson, D.J.; A Rosenberg, S. Lymphokine-activated killer cell phenomenon. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J. Exp. Med. 1983, 158, 1356–1361. [Google Scholar] [CrossRef]
- Phillips, J.H.; Lanier, L.L. Dissection of the lymphokine-activated killer phenomenon. Relative contribution of peripheral blood natural killer cells and T lymphocytes to cytolysis. J. Exp. Med. 1986, 164, 814–825. [Google Scholar] [CrossRef]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018, 3. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Esser, R.; Müller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schönfeld, K.; et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Spanholtz, J.; Preijers, F.; Tordoir, M.; Trilsbeek, C.; Paardekooper, J.; De Witte, T.; Schaap, N.; Dolstra, H. Clinical-Grade Generation of Active NK Cells from Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PLoS ONE 2011, 6, e20740. [Google Scholar] [CrossRef]
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front. Immunol. 2017, 8, 329. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Voskinarian-Berse, V.; Law, E.; Reddin, T.; Bhatia, M.; Hariri, A.; Ning, Y.; Dong, D.; Maguire, T.; Yarmush, M.; et al. Characterization and ex vivo Expansion of Human Placenta-Derived Natural Killer Cells for Cancer Immunotherapy. Front. Immunol. 2013, 4, 101. [Google Scholar] [CrossRef] [Green Version]
- Bernareggi, D.; Pouyanfard, S.; Kaufman, D.S. Development of innate immune cells from human pluripotent stem cells. Exp. Hematol. 2019, 71, 13–23. [Google Scholar] [CrossRef]
- Karagiannis, P.; Kim, S.I. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 2015, 34, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Fate Therapeutics. FT500 as Monotherapy and in Combination with Immune Checkpoint Inhibitors in Subjects with Advanced Solid Tumors (Phase 1) (Internet). Report No.: NCT03841110. Available online: https://clinicaltrials.gov/ct2/show/NCT03841110 (accessed on 25 November 2021).
- Fate Therapeutics. A Phase I Study of FT516 as Monotherapy in Relapsed/Refractory Acute Myelogenous Leukemia and in Combination with Monoclonal Antibodies in Relapsed/Refractory B-Cell Lymphoma (Internet). Report No.: NCT04023071. Available online: https://clinicaltrials.gov/ct2/show/NCT04023071 (accessed on 25 November 2021).
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [Green Version]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. Available online: https://www.nature.com/articles/nature21405 (accessed on 27 November 2021). [CrossRef] [Green Version]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Daniel, A.; Vallera, D.A. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar] [PubMed] [Green Version]
- Zhao, X.; Rajasekaran, N.; Reusch, U.; Marschner, J.P.; Treder, M.; Kohrt, H. CD30/CD16A Tandab AFM13-Induced Target Cell Lysis By NK-Cells Is Enhanced by CD137 Co-Stimulation and Blocking PD-1. Blood 2015, 126, 2747. [Google Scholar] [CrossRef]
- Cho, M.M.; Quamine, A.E.; Olsen, M.R.; Capitini, C.M. Programmed cell death protein 1 on natural killer cells: Fact or fiction? J. Clin. Investig. 2020, 130, 2816–2819. [Google Scholar] [CrossRef]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Tassev, D.V.; Cheng, M.; Cheung, N.K. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 2012, 19, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.P.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2013, 28, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Year | Researcher | Target | CAR Construct | Method | Tumor Targets | Ref |
|---|---|---|---|---|---|---|
| 2002 | Uherek et al. | ErbB2 (HER-2) | mCD8α hinge/CD3ζ | Amphotropicvirus | Tumors of epithelial origin | [61] |
| 2008 | Müller et al. | CD20 | mCD8α hinge/CD3ζ | Amphotropicvirus | Lymphoma and leukemia | [62] |
| 2009 | Boissel et al. | CD19 | CD8α TM/CD3ζ | mRNA transfection (50%) | Chronic lymphocytes Leukemia | [63] |
| 2012 | Sahm et al. | EpCAM | CD8α hinge/CD28/ CD3ζ | Lentivirus along with IL-15 | Breast carcinoma | [64] |
| 2012 | Tassev et al. | HLA-A2 EBNA3C | CD8α TM/CD137/CD3ζ | Retrovirus | EBV-positive T cells | [65] |
| 2012 | Esser et al. | GD2 | mCD8α hinge/CD3ζ | Amphotropicvirus | Neuroblastoma | [66] |
| 2013 | Zhang et al. | HLA-2 complex-associated gp100 peptide | A2 TM/CD3ζ | Transfection | Melanoma | [67] |
| 2015 | Töpfer et al. | PSCA | CD28 hinge/CD28 TM/CD3ζ DAP12 cell TM and signaling | Lentivirus in YTSNK cells and primary NK cells | Solid Tumors prostate cancer | [68] |
| 2015 | Schönfled et al. | ErbB2 (HER-2) | CD8α hinge/CD3ζ CD8α hinge/CD137/CD3ζ CD8α hinge/CD28/CD3ζ | Lentivirus | Tumors of epithelial origin | [69] |
| 2015 | Liu et al. | ErbB2 (HER-2) | CD8αhinge/CD28/CD3ζ | Transfections | Breast carcinoma | [70] |
| 2014 | Jiang et al. | CD138 | CD8α hinge/CD3ζ | Lentivirus | Multiple myeloma | [66] |
| 2014 | Chu | CS1 | CD28 TM/CD28/ CD3ζ | Lentivirus | Multiple myeloma | [16] |
| 2013 | Boissel | CD19/CD20 | CD3ζ | Lentivirus | Lymphoblastic leukemia | [44] |
| 2012 | Boissel | CD19/CD20 | CD3ζ | mRNA transfection (30–70%) and lentivirus | Lymphoblastic leukemia | [71] |
| Year | Cell Line | Phase | Disease | Clinical Trial Nos | Sponsors and Collaborator | Study Type | Gender | Study Design |
|---|---|---|---|---|---|---|---|---|
| 2009–2016 | NK-92 cells | Phase I | Acute myeloid leukemia | NCT00900809 [57] | University of Pittsburgh ZelleRX Corporation | Interventional | Both | Treatment |
| 2005–2016 | NK-92 cells | Phase I | Leukemia Lymphoma myeloma Hodgkin’s disease | NCT00990717 [72] | University Health Network, Toronto | Interventional | Both | Treatment |
| 2009–2015 | NK Cell Infusion | Phase I | Lymphoblastic leukemia Refractory B-lineage acute lymphoblastic leukemia (ALL) | NCT00995137 [73] David Shook, MD | St.Jude Children’s Research Hospital National Cancer Institute (NCI) Assisi Foundation | Interventional (NK cell infusion) | Both | Treatment |
| 2013–2017 | NK Cell Infusion | Phase I | B-cell lymphoma, non-Hodgkin, lymphoproliferative disorders, neoplasms | NCT01974479 [74] Dario Campana | National University Health System, Singapore Singapore General Hospital | Interventional (anti-CD19 redirected NK cells) | Both | Treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishwasrao, P.; Hui, S.K.; Smith, D.L.; Khairnar, V. Role of NK Cells in Cancer and Immunotherapy. Onco 2021, 1, 158-175. https://doi.org/10.3390/onco1020013
Vishwasrao P, Hui SK, Smith DL, Khairnar V. Role of NK Cells in Cancer and Immunotherapy. Onco. 2021; 1(2):158-175. https://doi.org/10.3390/onco1020013
Chicago/Turabian StyleVishwasrao, Paresh, Susanta K. Hui, D. Lynne Smith, and Vishal Khairnar. 2021. "Role of NK Cells in Cancer and Immunotherapy" Onco 1, no. 2: 158-175. https://doi.org/10.3390/onco1020013
APA StyleVishwasrao, P., Hui, S. K., Smith, D. L., & Khairnar, V. (2021). Role of NK Cells in Cancer and Immunotherapy. Onco, 1(2), 158-175. https://doi.org/10.3390/onco1020013