
Pre-Clinical Research Advancements Relating to Improving the Diagnosis and Treatment of Malignant Pleural Mesothelioma: A Review


Abstract
:Simple Summary
Abstract
1. Introduction
2. Pre-Clinical Research
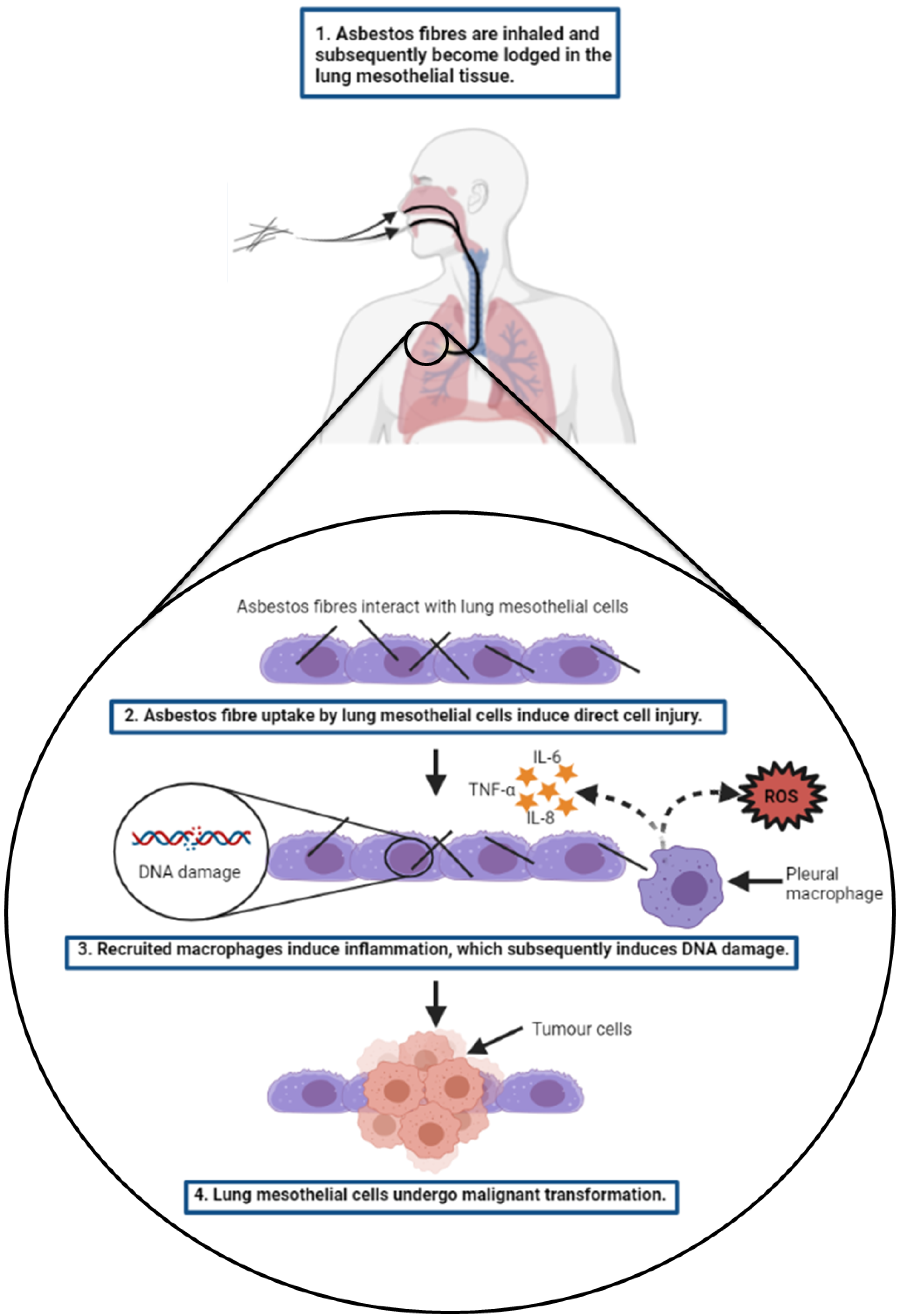
2.1. Disease Mechanism
2.1.1. Pathogenesis
2.1.2. Tumour Suppressor Genes in MPM
2.1.3. Oncogenes in MPM

{kind=link}
{kind=link}
| Gene/miRNA Name | Gene Symbol | Chromosomal Region | General Function | Specific Function | Alteration | Reference |
|---|---|---|---|---|---|---|
| BRCA1-associated protein 1 | BAP1 | 3p21.3 | Tumour suppressor gene | DNA damage repair and cell cycle regulation | Gene deletion | [26,27] |
| p16/cyclin-dependent kinase inhibitor 2A | CDKN2A | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32,39,40] |
| p15/cyclin-dependent kinase inhibitor 2B | CDKN2B | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32,33,38] |
| p19/alternate reading frame | ARF | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32,42] |
| Neurofibromin 2 | NF2 | 22q12 | Tumour suppressor gene | Regulation of cell proliferation, motility, and survival | Gene deletion | [38,43,44] |
| Tumour protein p53 | Tp53 | 17p13.1 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [45,46] |
| Epidermal Growth Factor Receptor | EGFR | 7p11.2 | Oncogene | Promotes angiogenesis | Overexpression | [68] |
| Vascular Endothelial Growth Factor | VEGF | 6p21.3 | Oncogene | Promotes angiogenesis | Overexpression | [71] |
| Notch homolog 1, translocation-associated (Drosophila) | NOTCH1 | 9q34.3 | Oncogene | Promotes cell proliferation, differentiation and survival | Overexpression | [81] |
| Notch homolog 2 (Drosophila) | NOTCH2 | 1p12 | Tumour suppressor | Suppresses cell proliferation, motility and survival | Underexpression | [81] |
| miR-182-5p, miR-183-5p and miR-24-3p | N/A | N/A | OncomiR’s | Promote cell proliferation, adhesion and invasion | Overexpression | [74,75] |
| miR-320a, miR-34a and miR-200a | N/A | N/A | OncomiR’s | Suppress the expression of PDL-1 | Underexpression | [76] |
2.2. Emerging Biomarkers for the Diagnosis of MPM
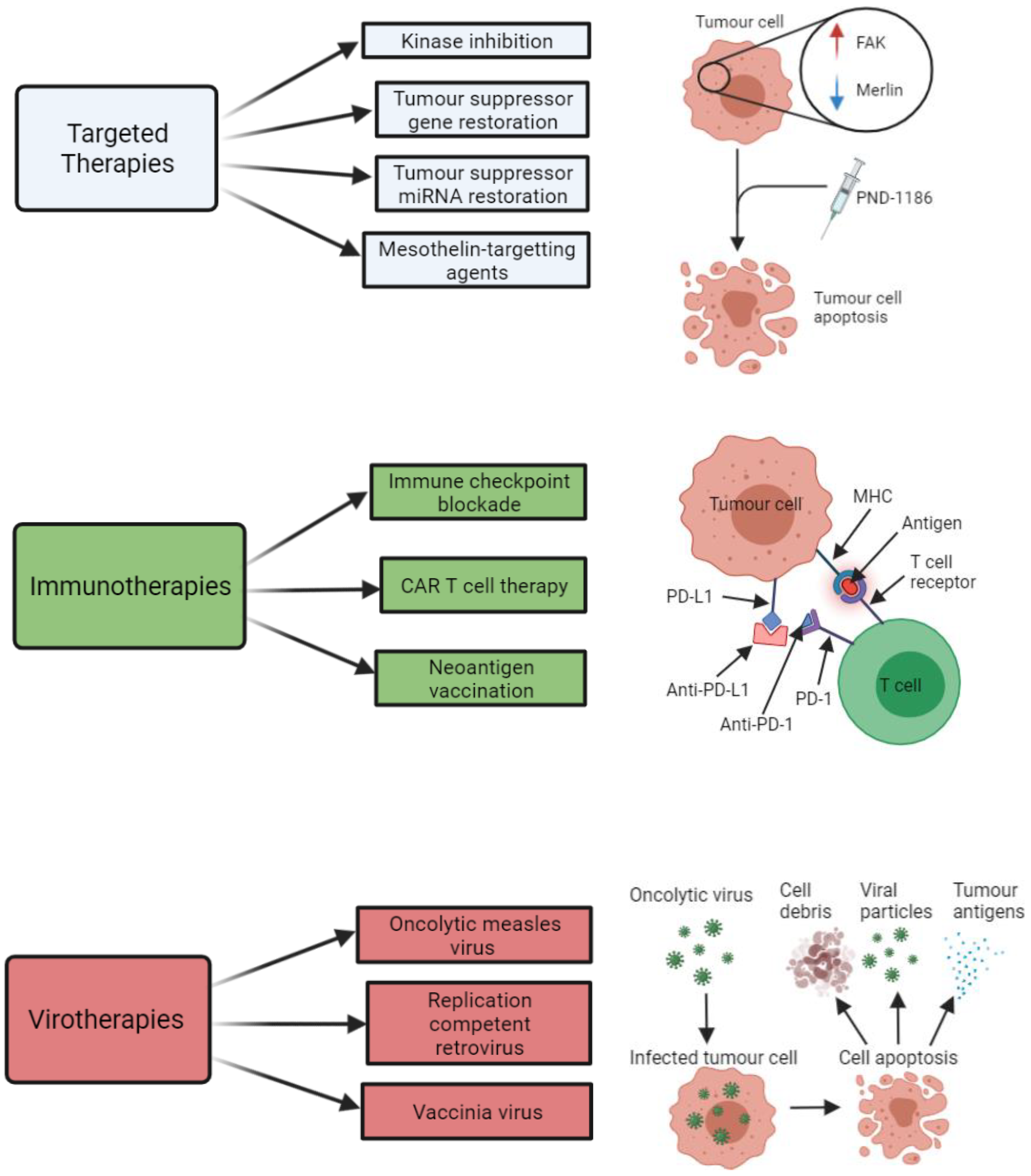
2.3. Treatment Development
2.3.1. Current Standard of Care
2.3.2. Targeted Therapies
2.3.3. Immunotherapies
2.3.4. Virotherapies
3. Expert Commentary and Recommendations for Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, R. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man Volume 2 Some Inorganic and Organometallic Compounds. J. Clin. Pathol. 1974, 27, 171–172. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, metals, fibres, and dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11–465. [Google Scholar]
- Carbone, M.; Ly, B.H.; Dodson, R.F.; Pagano, I.; Morris, P.T.; Dogan, U.A.; Gazdar, A.F.; Pass, H.I.; Yang, H. Malignant mesothelioma: Facts, myths, and hypotheses. J. Cell. Physiol. 2012, 227, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzati, M.; Lopez, A.D.; Rodgers, A.A.; Murray, C.J.L. Comparative Quantification of Health Risks: Global and Regional Burden of Disease Attributable to Selected Major Risk Factors; Ezzati, M., Lopez, A.D., Rodgers, A., Murray, C.J.L., Eds.; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Driscoll, T.; Nelson, D.I.; Steenland, K.; Leigh, J.; Concha-Barrientos, M.; Fingerhut, M.; Prüss-Üstün, A. The global burden of disease due to occupational carcinogens. Am. J. Ind. Med. 2005, 48, 419–431. [Google Scholar] [CrossRef]
- Chimed-Ochir, O.; Takahashi, K.; Sorahan, T.; Driscoll, T.; Fitzmaurice, C.; Yoko-O, M.; Sawanyawisuth, K.; Furuya, S.; Tanaka, F.; Horie, S.; et al. 1554 Estimation of the global burden of mesothelioma deaths from incomplete national mortality data. Occup. Environ. Med. 2018, 75, A131. [Google Scholar] [CrossRef]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2015, 387, 1405–1414. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin Versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Bianchi, C.; Giarelli, L.; Grandi, G.; Brollo, A.; Ramani, L.; Zuch, C. Latency periods in asbestos-related mesothelioma of the pleura. Eur. J. Cancer Prev. 1997, 6, 162–166. [Google Scholar]
- Lanphear, B.P.; Buncher, C.R. Latent period for malignant mesothelioma of occupational origin. J. Occup. Med. 1992, 34, 718–721. [Google Scholar]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Kadariya, Y.; Menges, C.W.; Talarchek, J.; Cai, K.Q.; Klein-Szanto, A.J.; Pietrofesa, R.A.; Christofidou-Solomidou, M.; Cheung, M.; Mossman, B.T.; Shukla, A.; et al. Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev. Res. 2016, 9, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekido, Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Choe, N.; Tanaka, S.; Kagan, E. Asbestos Fibers and Interleukin-1 Upregulate the Formation of Reactive Nitrogen Species in Rat Pleural Mesothelial Cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.; Laczko, R.; DeMay, R.M.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous Exposure to Chrysotile Asbestos Can Cause Transformation of Human Mesothelial Cells via HMGB1 and TNF-α Signaling. Am. J. Pathol. 2013, 183, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bocchetta, M.; Kroczynska, B.; Elmishad, A.G.; Chen, Y.; Liu, Z.; Bubici, C.; Mossman, B.T.; Pass, H.I.; Testa, J.R.; et al. TNF-α inhibits asbestos-induced cytotoxicity via a NF-κB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 10397–10402. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Gasparri, F.; Galvani, A.; Nici, L.; Darnowski, J.W.; Barbone, D.; Fennell, D.A.; Gaudino, G.; Porta, C.; Mutti, L. Bortezomib Inhibits Nuclear Factor-κB–Dependent Survival and Has Potent In vivo Activity in Mesothelioma. Clin. Cancer Res. 2007, 13, 5942–5951. [Google Scholar] [CrossRef] [Green Version]
- Rossini, M.; Rizzo, P.; Bononi, I.; Clementz, A.; Ferrari, R.; Martini, F.; Tognon, M.G. New Perspectives on Diagnosis and Therapy of Malignant Pleural Mesothelioma. Front. Oncol. 2018, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.W.; Lake, R.A. Advances in malignant mesothelioma. N. Engl. J. Med. 2005, 353, 1591–1603. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 111, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Kobrinski, D.A.; Yang, H.; Kittaneh, M. BAP1: Role in carcinogenesis and clinical implications. Transl. Lung Cancer Res. 2020, 9, S60–S66. [Google Scholar] [CrossRef]
- Ismail, I.H.; Davidson, R.; Gagné, J.-P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasu, M.; Emi, M.; Pastorino, S.; Tanji, M.; Powers, A.; Luk, H.; Baumann, F.; Zhang, Y.; Gazdar, A.; Kanodia, S.; et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J. Thorac. Oncol. 2015, 10, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2015, 75, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, M.; Flores, E.G.; Emi, M.; Johnson, T.A.; Tsunoda, T.; Behner, D.; Hoffman, H.; Hesdorffer, M.; Nasu, M.; Napolitano, A.; et al. Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet. 2015, 11, e1005633. [Google Scholar] [CrossRef]
- Napolitano, A.; Pellegrini, L.; Dey, A.; Larson, D.; Tanji, M.; Flores, E.G.; Kendrick, B.; Lapid, D.; Powers, A.C.; Kanodia, S.; et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 2015, 35, 1996–2002. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Jhanwar, S.C.; Klein, W.M.; Bell, D.W.; Lee, W.C.; Altomare, D.A.; Nobori, T.; Olopade, O.I.; Buckler, A.J.; Testa, J.R. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994, 54, 5547–5551. [Google Scholar]
- Andujar, P.; LeComte, C.; Renier, A.; Fleury-Feith, J.; Kheuang, L.; Daubriac, J.; Janin, A.; Jaurand, M.-C. Clinico-pathological features and somatic gene alterations in refractory ceramic fibre-induced murine mesothelioma reveal mineral fibre-induced mesothelioma identities. Carcinogenesis 2007, 28, 1599–1605. [Google Scholar] [CrossRef] [Green Version]
- Ouelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeComte, C.; Andujar, P.; Renier, A.; Kheuang, L.; Abramowski, V.; Mellotte, L.; Fleury-Feith, J.; Zucman-Rossi, J.; Giovanni, M.; Jaurand, M.-C. Similar Tumor Suppressor Gene Alteration Profiles in Asbestos-Induced Murine and Human Mesothelioma. Cell Cycle 2005, 4, 1862–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomare, D.A.; Vaslet, C.A.; Skele, K.L.; De Rienzo, A.; Devarajan, K.; Jhanwar, S.C.; McClatchey, A.I.; Kane, A.B.; Testa, J.R. A Mouse Model Recapitulating Molecular Features of Human Mesothelioma. Cancer Res. 2005, 65, 8090–8095. [Google Scholar] [CrossRef] [Green Version]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illei, P.B.; Rusch, V.W.; Zakowski, M.F.; Ladanyi, M. Homozygous Deletion of CDKN2A and Codeletion of the Methylthioadenosine Phosphorylase Gene in the Majority of Pleural Mesotheliomas. Clin. Cancer Res. 2003, 9, 2108–2113. [Google Scholar]
- Bononi, A.; Napolitano, A.; Pass, H.; Yang, H.; Carbone, M. Latest developments in our understanding of the pathogenesis of mesothelioma and the design of targeted therapies. Expert Rev. Respir. Med. 2015, 9, 633–654. [Google Scholar] [CrossRef] [Green Version]
- Altomare, D.A.; Menges, C.W.; Xu, J.; Pei, J.; Zhang, L.; Tadevosyan, A.; Neumann-Domer, E.; Liu, Z.; Carbone, M.; Chudoba, I.; et al. Losses of Both Products of the Cdkn2a/Arf Locus Contribute to Asbestos-Induced Mesothelioma Development and Cooperate to Accelerate Tumorigenesis. PLoS ONE 2011, 6, e18828. [Google Scholar] [CrossRef]
- Andujar, P.; Pairon, J.-C.; Renier, A.; Descatha, A.; Hysi, I.; Abd-Alsamad, I.; Billon-Galland, M.-A.; Blons, H.; Clin, B.; Danel, C.; et al. Differential mutation profiles and similar intronic TP53 polymorphisms in asbestos-related lung cancer and pleural mesothelioma. Mutagenesis 2013, 28, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.Q.; Lee, W.C.; Klein, M.A.; Cheng, G.Z.; Jhanwar, S.C.; Testa, J.R. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: Evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosom. Cancer 1999, 24, 238–242. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, D.; Hong, Y.S.; Kim, K.-P.; Yoon, Y.K.; Lee, D.H.; Kim, S.-W.; Chun, S.-M.; Jang, S.J.; Kim, T.W. Mutational Profiling of Malignant Mesothelioma Revealed Potential Therapeutic Targets in EGFR and NRAS. Transl. Oncol. 2018, 11, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Krontira, A.; Marazioti, A.; Vreka, M.; Iliopoulou, M.; Lilis, I.; Giotopoulou, G.; Spella, M.; Petrou, H.; Stathopoulos, G. LSC-2017-KRAS & TP53 mutations cause malignant mesothelioma. Eur. Respir. J. 2017, 50, OA293. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Ordaz, J.; Lo, C.L.; Damayanti, N.P.; Zhou, F.; Irudayaraj, J. From the Cover: Zinc oxide Nanoparticles-Induced Reactive Oxygen Species Promotes Multimodal Cyto- and Epigenetic Toxicity. Toxicol. Sci. 2017, 156, 261–274. [Google Scholar] [PubMed] [Green Version]
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of miRNA Expression in Malignant Mesothelioma: MiRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 1293. [Google Scholar] [CrossRef]
- Chatterjee, R.; Vinson, C. CpG methylation recruits sequence specific transcription factors essential for tissue specific gene expression. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1819, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Toyooka, S.; Soh, J.; Tsukuda, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Maki, Y.; Ueno, T.; Yamamoto, H.; et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol. Rep. 2013, 29, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Toyooka, S.; Tsukuda, K.; Sakaguchi, M.; Fukazawa, T.; Soh, J.; Asano, H.; Ueno, T.; Muraoka, T.; Yamamoto, H.; et al. Epigenetic Silencing of MicroRNA-34b/c Plays an Important Role in the Pathogenesis of Malignant Pleural Mesothelioma. Clin. Cancer Res. 2011, 17, 4965–4974. [Google Scholar] [CrossRef] [Green Version]
- Cioce, M.; Ganci, F.; Canu, V.; Sacconi, A.; Mori, F.; Canino, C.; Korita, E.; Casini, B.; Alessandrini, G.; Cambria, A.; et al. Protumorigenic effects of mir-145 loss in malignant pleural mesothelioma. Oncogene 2013, 33, 5319–5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.; Trapani, D.; Ravn, J.; Sørensen, J.B.; Andersen, C.B.; Grauslund, M.; Santoni-Rugiu, E. Methylation-associated Silencing of microRNA-126 and its Host Gene EGFL7 in Malignant Pleural Mesothelioma. Anticancer. Res. 2015, 35, 6223–6229. [Google Scholar]
- Jiang, R.; Zhang, C.; Liu, G.; Gu, R.; Wu, H. Retracted: MicroRNA-126 Inhibits Proliferation, Migration, Invasion, and EMT in Osteosarcoma by Targeting ZEB1. J. Cell. Biochem. 2017, 118, 3765–3774. [Google Scholar] [CrossRef]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 Suppresses Mesothelioma Malignancy by Targeting IRS1 and Interfering with the Mitochondrial Function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, M.; Monaco, F.; Manzella, N.; Rohlena, J.; Rohlenova, K.; Staffolani, S.; Gaetani, S.; Ciarapica, V.; Amati, M.; Bracci, M.; et al. MicroRNA-126 induces autophagy by altering cell metabolism in malignant mesothelioma. Oncotarget 2016, 7, 36338–36352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, X.; Xu, B.; Wang, B.; Wang, Z.; Liang, Y.; Zhou, J.; Hu, J.; Jiang, B. Epigenetic silencing of miR-126 contributes to tumor invasion and angiogenesis in colorectal cancer. Oncol. Rep. 2013, 30, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Friedman, J.M.; Chihara, Y.; Egger, G.; Chuang, J.C.; Liang, G. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem. Biophys. Res. Commun. 2009, 379, 726–731. [Google Scholar] [CrossRef]
- Pinton, G.; Manente, A.G.; Murer, B.; De Marino, E.; Mutti, L.; Moro, L. PARP1 inhibition affects pleural mesothelioma cell viability and uncouples AKT/mTOR axis via SIRT1. J. Cell. Mol. Med. 2013, 17, 233–241. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Paul, S. Risk of occupational exposure to asbestos, silicon and arsenic on pulmonary disorders: Understanding the genetic-epigenetic interplay and future prospects. Environ. Res. 2016, 147, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Amati, M.; Nocchi, L.; Saccucci, F.; Strafella, E.; Staffolani, S.; Tarquini, L.M.; Carbonari, D.; Alleva, R.; Borghi, B.; et al. Asbestos exposure affects poly(ADP-ribose) polymerase-1 activity: Role in asbestos-induced carcinogenesis. Mutagenesis 2011, 26, 585–591. [Google Scholar] [CrossRef]
- Gaetani, S.; Monaco, F.; Alessandrini, F.; Tagliabracci, A.; Sabbatini, A.; Bracci, M.; Valentino, M.; Neuzil, J.; Amati, M.; Santarelli, L.; et al. Mechanism of miR-222 and miR-126 regulation and its role in asbestos-induced malignancy. Int. J. Biochem. Cell Biol. 2020, 121, 105700. [Google Scholar] [CrossRef]
- Kirschner, M.B.; Cheng, Y.Y.; Badrian, B.; Kao, S.C.; Creaney, J.; Edelman, J.J.; Armstrong, N.J.; Vallely, M.P.; Musk, A.W.; Robinson, B.W.S.; et al. Increased circulating miR-625-3p: A potential biomarker for patients with malignant pleural mesothelioma. J. Thorac. Oncol. 2012, 7, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef]
- Williams, M.; Kirschner, M.B.; Cheng, Y.Y.; Hanh, J.; Weiss, J.; Mugridge, N.; Wright, C.; Linton, A.; Kao, S.C.; Edelman, J.J.B.; et al. miR-193a-3p is a potential tumor suppressor in malignant pleural mesothelioma. Oncotarget 2015, 6, 23480–23495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Taffaro, M.L.; Salgia, R.; Johnson, B.E. Inhibition of epidermal growth factor receptor signaling in malignant pleural mesothelioma. Cancer Res. 2002, 62, 5242–5247. [Google Scholar] [PubMed]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar]
- Faux, S.P.; Houghton, C.E. Cell Signaling in Mesothelial Cells by Asbestos: Evidence for the Involvement of Oxidative Stress in the Regulation of the Epidermal Growth Factor Receptor. Inhal. Toxicol. 2000, 12, 327–336. [Google Scholar] [CrossRef]
- Ohta, Y.; Shridhar, V.; Bright, R.K.; Kalemkerian, G.P.; Du, W.; Carbone, M.; Watanabe, Y.; Pass, H. VEGF and VEGF type C play an important role in angiogenesis and lymphangiogenesis in human malignant mesothelioma tumours. Br. J. Cancer 1999, 81, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Brosseau, S.; Cook, A.; Zalcman, G. Antiangiogeneic Strategies in Mesothelioma. Front. Oncol. 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Chia, P.L.; Scott, A.M.; John, T. Epidermal growth factor receptor (EGFR)-targeted therapies in mesothelioma. Expert Opin. Drug Deliv. 2019, 16, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Amatya, V.J.; Kushitani, K.; Kai, Y.; Kambara, T.; Takeshima, Y. miR-182 and miR-183 Promote Cell Proliferation and Invasion by Targeting FOXO1 in Mesothelioma. Front. Oncol. 2018, 8, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveto, S.; Alfieri, R.; Miluzio, A.; Scagliola, A.; Seclì, R.S.; Gasparini, P.; Grosso, S.; Cascione, L.; Mutti, L.; Biffo, S. A Polysome-Based microRNA Screen Identifies miR-24-3p as a Novel Promigratory miRNA in Mesothelioma. Cancer Res. 2018, 78, 5741–5753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Indovina, P.; Mattioli, E.; Forte, I.M.; Iannuzzi, C.A.; Luzzi, L.; Bellan, C.; De Summa, S.; Bucci, E.; Di Marzo, D.; et al. P53-regulated miR-320a targets PDL1 and is downregulated in malignant mesothelioma. Cell Death Dis. 2020, 11, 748. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Cedrés, S.; Ponce-Aix, S.; Zugazagoitia, J.; Sansano, I.; Enguita, A.; Navarro-Mendivil, A.; Martinez-Marti, A.; Martinez, P.; Felip, E. Analysis of Expression of Programmed Cell Death 1 Ligand 1 (PD-L1) in Malignant Pleural Mesothelioma (MPM). PLoS ONE 2015, 10, e0121071. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Roden, A.C.; Peikert, T.; Sheinin, Y.M.; Harrington, S.M.; Krco, C.J.; Dong, H.; Kwon, E.D. B7-H1 Expression in Malignant Pleural Mesothelioma is Associated with Sarcomatoid Histology and Poor Prognosis. J. Thorac. Oncol. 2014, 9, 1036–1040. [Google Scholar] [CrossRef] [Green Version]
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.; De May, R.M.; Strack, P.; Miele, L.; Bocchetta, M. Opposite Effects of Notch-1 and Notch-2 on Mesothelioma Cell Survival under Hypoxia Are Exerted through the Akt Pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.; Cheung, T.T.; Lo, C.M.; Wang, X.Q. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness due to Reduced Activation of Notch1–Stat3. Mol. Cancer Ther. 2017, 16, 1531–1543. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a selective NOTCH1 inhibitor, reduces intrahepatic cholangiocarcinoma progression by blocking VEGFA/DLL4/MMP13 axis. Cell Death Differ. 2020, 27, 2330–2343. [Google Scholar] [CrossRef] [Green Version]
- British Thoracic Society Standards of Care Committee. BTS statement on malignant mesothelioma in the UK, 2007. Thorax 2007, 62 (Suppl. S2), ii1–ii19. [Google Scholar]
- Renshaw, A.A.; Dean, B.R.; Cibas, E.S.; Antman, K.H.; Sugarbaker, D.J. The Role of Cytologic Evaluation of Pleural Fluid in the Diagnosis of Malignant Mesothelioma. Chest 1997, 111, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Creaney, J.; Yeoman, D.; Demelker, Y.; Segal, A.; Musk, A.; Skates, S.J.; Robinson, B.W. Comparison of Osteopontin, Megakaryocyte Potentiating Factor, and Mesothelin Proteins as Markers in the Serum of Patients with Malignant Mesothelioma. J. Thorac. Oncol. 2008, 3, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Creaney, J.; Robinson, B.W. Detection of Malignant Mesothelioma in Asbestos-Exposed Individuals: The Potential Role of Soluble Mesothelin-Related Protein. Hematol. Clin. N. Am. 2005, 19, 1025–1040. [Google Scholar] [CrossRef]
- Creaney, J.; Segal, A.; Olsen, N.; Dick, I.M.; Musk, A.W.; Skates, S.J.; Robinson, B.W. Pleural Fluid Mesothelin as an Adjunct to the Diagnosis of Pleural Malignant Mesothelioma. Dis. Markers 2014, 2014, 413946. [Google Scholar] [CrossRef] [Green Version]
- Hollevoet, K.; Reitsma, J.B.; Creaney, J.; Grigoriu, B.D.; Robinson, B.W.; Scherpereel, A.; Cristaudo, A.; Pass, H.; Nackaerts, K.; Portal, J.A.R.; et al. Serum Mesothelin for Diagnosing Malignant Pleural Mesothelioma: An Individual Patient Data Meta-Analysis. J. Clin. Oncol. 2012, 30, 1541–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creaney, J.; Olsen, N.J.; Brims, F.; Dick, I.M.; Musk, A.W.; de Klerk, N.; Skates, S.J.; Robinson, B.W. Serum Mesothelin for Early Detection of Asbestos-Induced Cancer Malignant Mesothelioma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2238–2246. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzada, T.; Reid, G.; Kao, S. Biomarkers in malignant pleural mesothelioma: Current status and future directions. J. Thorac. Dis. 2018, 10, S1003–S1007. [Google Scholar] [CrossRef] [Green Version]
- Creaney, J.; Dick, I.M.; Meniawy, T.; Leong, S.L.; Leon, J.S.; Demelker, Y.; Segal, A.; Musk, A.W.; Lee, Y.C.G.; Skates, S.J.; et al. Comparison of fibulin-3 and mesothelin as markers in malignant mesothelioma. Thorax 2014, 69, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Pass, H.I.; Levin, S.M.; Harbut, M.R.; Melamed, J.; Chiriboga, L.; Donington, J.; Huflejt, M.; Carbone, M.; Chia, D.; Goodglick, L.; et al. Fibulin-3 as a Blood and Effusion Biomarker for Pleural Mesothelioma. N. Engl. J. Med. 2012, 367, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pass, H.I.; Lott, D.; Lonardo, F.; Harbut, M.; Liu, Z.; Tang, N.; Carbone, M.; Webb, C.; Wali, A. Asbestos Exposure, Pleural Mesothelioma, and Serum Osteopontin Levels. N. Engl. J. Med. 2005, 353, 1564–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriu, B.-D.; Scherpereel, A.; Devos, P.; Chahine, B.; Letourneux, M.; LeBailly, P.; Grégoire, M.; Porte, H.; Copin, M.-C.; Lassalle, P. Utility of Osteopontin and Serum Mesothelin in Malignant Pleural Mesothelioma Diagnosis and Prognosis Assessment. Clin. Cancer Res. 2007, 13, 2928–2935. [Google Scholar] [CrossRef] [Green Version]
- Gillezeau, C.N.; Van Gerwen, M.; Ramos, J.; Liu, B.; Flores, R.; Taioli, E. Biomarkers for malignant pleural mesothelioma: A meta-analysis. Carcinogenesis 2019, 40, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Ostroff, R.M.; Mehan, M.R.; Stewart, A.; Ayers, D.; Brody, E.N.; Williams, S.A.; Levin, S.; Black, B.; Harbut, M.; Carbone, M.; et al. Early Detection of Malignant Pleural Mesothelioma in Asbestos-Exposed Individuals with a Noninvasive Proteomics-Based Surveillance Tool. PLoS ONE 2012, 7, e46091. [Google Scholar] [CrossRef] [PubMed]
- Tsou, J.A.; Galler, J.S.; Wali, A.; Ye, W.; Siegmund, K.D.; Groshen, S.; Laird, P.W.; Turla, S.; Koss, M.N.; Pass, H.I.; et al. DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer 2007, 58, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Martinez, V.D.; Minatel, B.C.; Pewarchuk, M.E.; Marshall, E.A.; Macaulay, G.M.; Hubaux, R.; Pearson, D.D.; Goodarzi, A.A.; Dellaire, G.; et al. Genomics and Epigenetics of Malignant Mesothelioma. High-Throughput 2018, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Guled, M.; Lahti, L.; Lindholm, P.; Salmenkivi, K.; Bagwan, I.; Nicholson, A.G.; Knuutila, S. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma—A miRNA microarray analysis. Genes Chromosom. Cancer 2009, 48, 615–623. [Google Scholar] [CrossRef]
- Taniguchi, T.; Karnan, S.; Fukui, T.; Yokoyama, T.; Tagawa, H.; Yokoi, K.; Ueda, Y.; Mitsudomi, T.; Horio, Y.; Hida, T.; et al. Genomic profiling of malignant pleural mesothelioma with array-based comparative genomic hybridization shows frequent non-random chromosomal alteration regions including JUN amplification on 1p32. Cancer Sci. 2007, 98, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Bononi, I.; Comar, M.; Puozzo, A.; Stendardo, M.; Boschetto, P.; Orecchia, S.; Libener, R.; Guaschino, R.; Pietrobon, S.; Ferracin, M.; et al. Circulating microRNAs found dysregulated in ex-exposed asbestos workers and pleural mesothelioma patients as potential new biomarkers. Oncotarget 2016, 7, 82700–82711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, H.; Lebanony, D.; Rosenwald, S.; Cohen, L.; Gibori, H.; Barabash, N.; Ashkenazi, K.; Goren, E.; Meiri, E.; Morgenstern, S.; et al. A Diagnostic Assay Based on MicroRNA Expression Accurately Identifies Malignant Pleural Mesothelioma. J. Mol. Diagn. 2010, 12, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Gee, G.; Koestler, D.; Christensen, B.C.; Sugarbaker, D.J.; Ugolini, D.; Ivaldi, G.P.; Resnick, M.B.; Houseman, E.A.; Kelsey, K.T.; Marsit, C.J. Downregulated microRNAs in the differential diagnosis of malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 2859–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, A.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2016, 8, e1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.; Sharpless, N. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.-L.; Yang, L.; Chen, L.-L. The Biogenesis of Nascent Circular RNAs. Cell Rep. 2016, 15, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pillai, R.S. MicroRNA function: Multiple mechanisms for a tiny RNA? RNA 2005, 11, 1753–1761. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yan, Y.; Zeng, S.; Dai, S.; Chen, X.; Wei, J.; Gong, Z. Circular RNAs: Clinical relevance in cancer. Oncotarget 2017, 9, 1444–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis. Int. J. Mol. Sci. 2017, 18, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Wang, X.; Wei, S.; Chen, Y.; Fan, X.; Han, S.; Wu, G. hsa_circ_0013958: A circular RNA and potential novel biomarker for lung adenocarcinoma. FEBS J. 2017, 284, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Winata, P.; Cheng, Y.; Williams, M.; Lin, R.C.Y.; Reid, G. Circular RNAs as novel biomarkers for detection of malignant pleural mesothelioma. Asia-Pac. J. Clin. Oncol. 2018, 14, 82–83. [Google Scholar]
- Nicolini, F.; Bocchini, M.; Bronte, G.; Delmonte, A.; Guidoboni, M.; Crinò, L.; Mazza, M. Malignant Pleural Mesothelioma: State-of-the-Art on Current Therapies and Promises for the Future. Front. Oncol. 2020, 9, 1519. [Google Scholar] [CrossRef]
- Fear, V.S.; Cook, A.M.; Fisher, S.A. The future of mesothelioma research: Basic science research. In Caring for Patients with Mesothelioma: Principles and Guidelines; Springer: Cham, Switzerland, 2019; pp. 203–227. [Google Scholar]
- Mutti, L.; Peikert, T.; Robinson, B.W.; Scherpereel, A.; Tsao, A.S.; de Perrot, M.; Woodard, G.A.; Jablons, D.M.; Wiens, J.; Hirsch, F.R.; et al. Scientific Advances and New Frontiers in Mesothelioma Therapeutics. J. Thorac. Oncol. 2018, 13, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.K. Chemotherapy for malignant pleural mesothelioma: A review of current management and a look to the future. Ann. Cardiothorac. Surg. 2012, 1, 508–515. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Brevet, M.; Shimizu, S.; Bott, M.J.; Shukla, N.; Zhou, Q.; Olshen, A.B.; Rusch, V.; Ladanyi, M. Coactivation of Receptor Tyrosine Kinases in Malignant Mesothelioma as a Rationale for Combination Targeted Therapy. J. Thorac. Oncol. 2011, 6, 864–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strizzi, L.; Catalano, A.; Vianale, G.; Orecchia, S.; Casalini, A.; Tassi, G.; Puntoni, R.; Mutti, L.; Procopio, A. Vascular endothelial growth factor is an autocrine growth factor in human malignant mesothelioma. J. Pathol. 2001, 193, 468–475. [Google Scholar] [CrossRef]
- Filiberti, R.; Marroni, P.; Neri, M.; Ardizzoni, A.; Betta, P.G.; Cafferata, M.A.; Canessa, P.A.; Puntoni, R.; Ivaldi, G.P.; Paganuzzi, M. Serum PDGF-AB in Pleural Mesothelioma. Tumor Biol. 2005, 26, 221–226. [Google Scholar] [CrossRef]
- Betta, P.G.; Libener, R.; Orecchia, S.; Bottero, G.; Paganuzzi, M.; Marroni, P.; Andreatta, R.; Filiberti, R.; Neri, M.; Puntoni, R. Epidermal growth factor in serum from patients with malignant pleural mesothelioma. J. Clin. Oncology. 2004, 22 (Suppl. S14), 7315. [Google Scholar] [CrossRef]
- Ceresoli, G.L.; Zucali, P.A.; Mencoboni, M.; Botta, M.; Grossi, F.; Cortinovis, D.; Zilembo, N.; Ripa, C.; Tiseo, M.; Favaretto, A.G.; et al. Phase II study of pemetrexed and carboplatin plus bevacizumab as first-line therapy in malignant pleural mesothelioma. Br. J. Cancer 2013, 109, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Dowell, J.E.; Dunphy, F.R.; Taub, R.N.; Gerber, D.E.; Ngov, L.; Yan, J.; Xie, Y.; Kindler, H.L. A multicenter phase II study of cisplatin, pemetrexed, and bevacizumab in patients with advanced malignant mesothelioma. Lung Cancer 2012, 77, 567–571. [Google Scholar] [CrossRef]
- Kindler, H.L.; Karrison, T.G.; Gandara, D.R.; Lu, C.; Krug, L.M.; Stevenson, J.P.; Jänne, P.A.; Quinn, D.; Koczywas, M.N.; Brahmer, J.R.; et al. Multicenter, Double-Blind, Placebo-Controlled, Randomized Phase II Trial of Gemcitabine/Cisplatin Plus Bevacizumab or Placebo in Patients with Malignant Mesothelioma. J. Clin. Oncol. 2012, 30, 2509–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versnel, M.A.; Claesson-Welsh, L.; Hammacher, A.; Bouts, M.J.; Van Der Kwast, T.H.; Eriksson, A.; Willemsen, R.; Weima, S.M.; Hoogsteden, H.C.; Hagemeijer, A. Human malignant mesothelioma cell lines express PDGF beta-receptors whereas cultured normal mesothelial cells express predominantly PDGF alpha-receptors. Oncogene 1991, 6, 2005–2011. [Google Scholar]
- Mathy, A.; Baas, P.; Dalesio, O.; van Zandwijk, N. Limited efficacy of imatinib mesylate in malignant mesothelioma: A phase II trial. Lung Cancer 2005, 50, 83–86. [Google Scholar] [CrossRef]
- Tsao, A.S.; Harun, N.; Lee, J.J.; Heymach, J.; Pisters, K.; Hong, W.K.; Fujimoto, J.; Wistuba, I. Phase I Trial of Cisplatin, Pemetrexed, and Imatinib Mesylate in Chemonaive Patients with Unresectable Malignant Pleural Mesothelioma. Clin. Lung Cancer 2013, 15, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.Z.; Pang, H.; Kratzke, R.A.; Otterson, G.A.; Hodgson, L.; Vokes, E.E.; Kindler, H.L. Phase II Study of Dasatinib in Patients with Previously Treated Malignant Mesothelioma (Cancer and Leukemia Group B 30601): A Brief Report. J. Thorac. Oncol. 2012, 7, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II Study of Erlotinib in Patients with Malignant Pleural Mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E., 2nd; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [Green Version]
- Takayama, Y.; Hattori, N.; Hamada, H.; Masuda, T.; Omori, K.; Akita, S.; Iwamoto, H.; Fujitaka, K.; Kohno, N. Inhibition of PAI-1 Limits Tumor Angiogenesis Regardless of Angiogenic Stimuli in Malignant Pleural Mesothelioma. Cancer Res. 2016, 76, 3285–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, N.; Hamada, H.; Masuda, T.; Akita, S.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Kohno, N. SK-216, An Inhibitor Of Plasminogen Activator Inhibitor-1, Inhibits Tumor Growth In A Mouse Model Of Pleural Mesothelioma. D23 Moonraker: Diagnosis and Management of the Pleural Space. Am. J. Respir. Crit. Care Med. 2014, 189, A5487. [Google Scholar]
- Stapelberg, M.; Gellert, N.; Swettenham, E.; Tomasetti, M.; Witting, P.K.; Procopio, A.; Neuzil, J. Alpha-tocopheryl succinate inhibits malignant mesothelioma by disrupting the fibroblast growth factor autocrine loop: Mechanism and the role of oxidative stress. J. Biol. Chem. 2005, 280, 25369–25376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Münzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast Growth Factor Receptor Inhibition Is Active against Mesothelioma and Synergizes with Radio- and Chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef]
- Shanthi, E.; Krishna, M.H.; Arunesh, G.M.; Reddy, K.V.; Kumar, J.S.; Viswanadhan, V.N. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: A patent review. Expert Opin. Ther. Patents 2014, 24, 1077–1100. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Cance, W.G. Focal Adhesion Kinase and p53 Signaling in Cancer Cells. Int. Rev. Cytol. 2007, 263, 103–153. [Google Scholar] [CrossRef]
- Moen, I.; Gebre, M.; Alonso-Camino, V.; Chen, D.; Epstein, D.; McDonald, D.M. Anti-metastatic action of FAK inhibitor OXA-11 in combination with VEGFR-2 signaling blockade in pancreatic neuroendocrine tumors. Clin. Exp. Metastasis 2015, 32, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Schunselaar, L.M.; Quispel-Janssen, J.M.; Neefjes, J.; Baas, P. A catalogue of treatment and technologies for malignant pleural mesothelioma. Expert Rev. Anticancer Ther. 2016, 16, 455–463. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Xiao, G.-H.; Gallagher, R.; Jablonski, S.; Jhanwar, S.C.; Testa, J.R. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene 2006, 25, 5960–5968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachter, J.A.; Kolev, V.N.; Schunselaar, L.; Shapiro, I.M.; Bueno, R.; Baas, P.; Xu, Q.; Weaver, D.T. Abstract 4236: FAK inhibitor VS-6063 (defactinib) targets mesothelioma cancer stem cells, which are enriched by standard of care chemotherapy. Cancer Res. 2015, 75, 4236. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin Deficiency Predicts FAK Inhibitor Sensitivity: A Synthetic Lethal Relationship. Sci. Transl. Med. 2014, 6, 237ra68. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sato, T.; Yokoi, K.; Sekido, Y. E-cadherin expression is correlated with focal adhesion kinase inhibitor resistance in Merlin-negative malignant mesothelioma cells. Oncogene 2017, 36, 5522–5531. [Google Scholar] [CrossRef]
- Sarun, K.C.Y.; Schelch, K.; Reid, R. Combination of MicroRNAs provide a synergistic effect on growth inhibition on MPM cells. In Proceedings of the COSA’s 45th Annual Scientific Meeting, Mesothelioma and Gastro-Intestinal Cancers: Technology and Genomics, Perth, Australia, 13–15 November 2018. [Google Scholar]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lee, H.-W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R. Role of the INK4a Locus in Tumor Suppression and Cell Mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Guazzelli, A.; Meysami, P.; Bakker, E.; Demonacos, C.; Giordano, A.; Krstic-Demonacos, M.; Mutti, L. BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment. Int. J. Mol. Sci. 2019, 20, 429. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, G.; Sidhu, G.S.; Williamson, E.A.; Jaiswal, A.S.; Najmunnisa, N.; Wilcoxen, K.; Jones, D.; George, T.J.; Hromas, R. Synthetic lethality in malignant pleural mesothelioma with PARP1 inhibition. Cancer Chemother. Pharmacol. 2017, 80, 861–867. [Google Scholar] [CrossRef] [Green Version]
- Rathkey, D.; Khanal, M.; Murai, J.; Zhang, J.; Sengupta, M.; Jiang, Q.; Morrow, B.; Evans, C.N.; Chari, R.; Fetsch, P.; et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 but Is Enhanced by Temozolomide in Cells with High-Schlafen 11 and Low-O6-methylguanine-DNA Methyltransferase Expression. J. Thorac. Oncol. 2020, 15, 843–859. [Google Scholar] [CrossRef]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Szlosarek, P.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Jahan, T.; Koczywas, M.; Forster, M.; et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J. Clin. Oncol. 2018, 36, 8515. [Google Scholar] [CrossRef]
- Yang, C.-T.; You, L.; Yeh, C.-C.; Chang, J.W.-C.; Zhang, F.; McCormick, F.; Jablons, D.M. Adenovirus-mediated p14(ARF) gene transfer in human mesothelioma cells. J. Natl. Cancer Inst. 2000, 92, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Fukuda, A.; Nakamichi, T.; Nakamura, A.; Kuroda, A.; Hashimoto, M.; Takuwa, T.; Kondo, N.; Hasegawa, S. CDK4/6 inhibitor and radiation therapy in malignant pleural mesothelioma. J. Clin. Oncol. 2018, 36 (Suppl. S15), e24326. [Google Scholar] [CrossRef]
- Frizelle, S.P.; Grim, J.; Zhou, J.; Gupta, P.; Curiel, D.T.; Geradts, J.; Kratzke, R.A. Re-expression of p16INK4a in mesothelioma cells results in cell cycle arrest, cell death, tumor suppression and tumor regression. Oncogene 1998, 16, 3087–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.C.; Fulham, M.; Wong, K.; Cooper, W.; Brahmbhatt, H.; MacDiarmid, J.; Pattinson, S.; Sagong, J.O.; Huynh, Y.; Leslie, F.; et al. A Significant Metabolic and Radiological Response after a Novel Targeted MicroRNA-based Treatment Approach in Malignant Pleural Mesothelioma. Am. J. Respir. Crit. Care Med. 2015, 191, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Reid, G.; Johnson, T.G.; Van Zandwijk, N. Manipulating microRNAs for the Treatment of Malignant Pleural Mesothelioma: Past, Present and Future. Front. Oncol. 2020, 10, 105. [Google Scholar] [CrossRef]
- Rump, A.; Morikawa, Y.; Tanaka, M.; Minami, S.; Umesaki, N.; Takeuchi, M.; Miyajima, A. Binding of Ovarian Cancer Antigen CA125/MUC16 to Mesothelin Mediates Cell Adhesion. J. Biol. Chem. 2004, 279, 9190–9198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbels, J.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol. Cancer 2006, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Ebel, W.; Routhier, E.L.; Patel, R.; Kline, J.B.; Zhang, J.; Chao, Q.; Jacob, S.; Turchin, H.; Gibbs, L.; et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007, 7, 1–10. [Google Scholar]
- Hassan, R.; Schweizer, C.; Lu, K.F.; Schuler, B.; Remaley, A.T.; Weil, S.C.; Pastan, I. Inhibition of mesothelin–CA-125 interaction in patients with mesothelioma by the anti-mesothelin monoclonal antibody MORAb-009: Implications for cancer therapy. Lung Cancer 2010, 68, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Lerner, M.R.; Benbrook, R.; Lightfoot, S.A.; Brackett, D.J.; Wang, Q.-C.; Pastan, I. Antitumor activity of SS(dsFv)PE38 and SS1(dsFv)PE38, recombinant antimesothelin immunotoxins against human gynecologic cancers grown in organotypic culture in vitro. Clin. Cancer Res. 2002, 8, 3520–3526. [Google Scholar]
- Li, Q.; Verschraegen, C.F.; Mendoza, J.; Hassan, R. Cytotoxic activity of the recombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards tumor cell lines established from ascites of patients with peritoneal mesotheliomas. Anticancer. Res. 2004, 24, 1327–1336. [Google Scholar]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.-F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In Vitro and In Vivo Activity of the Low-Immunogenic Antimesothelin Immunotoxin RG7787 in Pancreatic Cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef] [Green Version]
- Alewine, C.; Xiang, L.; Yamori, T.; Niederfellner, G.; Bosslet, K.; Pastan, I. Efficacy of RG7787, a Next-Generation Mesothelin-Targeted Immunotoxin, against Triple-Negative Breast and Gastric Cancers. Mol. Cancer Ther. 2014, 13, 2653–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab Ravtansine: A Novel Mesothelin-Targeting Antibody–Drug Conjugate Cures Tumors with Heterogeneous Target Expression Favored by Bystander Effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scales, S.J.; Gupta, N.; Pacheco, G.; Firestein, R.; French, D.M.; Koeppen, H.; Rangell, L.; Barry-Hamilton, V.; Luis, E.; Chuh, J.; et al. An Antimesothelin-Monomethyl Auristatin E Conjugate with Potent Antitumor Activity in Ovarian, Pancreatic, and Mesothelioma Models. Mol. Cancer Ther. 2014, 13, 2630–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, R.; Alley, E.; Kindler, H.; Antonia, S.; Jahan, T.; Honarmand, S.; Nair, N.; Whiting, C.C.; Enstrom, A.; Lemmens, E.; et al. Clinical Response of Live-Attenuated, Listeria monocytogenes Expressing Mesothelin (CRS-207) with Chemotherapy in Patients with Malignant Pleural Mesothelioma. Clin. Cancer Res. 2019, 25, 5787–5798. [Google Scholar] [CrossRef]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. JBIC J. Biol. Inorg. Chem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Gotoh, T. Regulation of Nitric Oxide Production by Arginine Metabolic Enzymes. Biochem. Biophys. Res. Commun. 2000, 275, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Szlosarek, P.W.; Klabatsa, A.; Pallaska, A.; Sheaff, M.; Smith, P.; Crook, T.; Grimshaw, M.J.; Steele, J.P.; Rudd, R.M.; Balkwill, F.R.; et al. In vivo Loss of Expression of Argininosuccinate Synthetase in Malignant Pleural Mesothelioma Is a Biomarker for Susceptibility to Arginine Depletion. Clin. Cancer Res. 2006, 12, 7126–7131. [Google Scholar] [CrossRef] [Green Version]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation with Pegylated Arginine Deiminase in Patients with Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef]
- Beddowes, E.; Spicer, J.; Chan, P.Y.; Khadeir, R.; Corbacho, J.G.; Repana, D.; Steele, J.P.; Schmid, P.; Szyszko, T.; Cook, G.; et al. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients with Argininosuccinate Synthetase 1-Deficient Thoracic Cancers. J. Clin. Oncol. 2017, 35, 1778–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630. [Google Scholar] [CrossRef]
- Danson, S.J.; Conner, J.; Edwards, J.G.; Blyth, K.G.; Fisher, P.M.; Muthana, M.; Salawu, A.; Taylor, F.; Hodgkinson, E.; Joyce, P.; et al. Oncolytic herpesvirus therapy for mesothelioma—A phase I/IIa trial of intrapleural administration of HSV1716. Lung Cancer 2020, 150, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.; Robinson, C.; Lake, R. Localised spontaneous regression in mesothelioma—Possible immunological mechanism. Lung Cancer 2001, 32, 197–201. [Google Scholar] [CrossRef]
- Anraku, M.; Cunningham, K.S.; Yun, Z.; Tsao, M.; Zhang, L.; Keshavjee, S.; Johnston, M.R.; de Perrot, M. Impact of tumor-infiltrating T cells on survival in patients with malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2008, 135, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Leigh, R.A.; Webster, I. Lymphocytic infiltration of pleural mesothelioma and its significance for survival. S. Afr. Med. J. 1982, 61, 1007–1009. [Google Scholar]
- Balar, A.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Rigo, V.; Emionite, L.; Daga, A.; Astigiano, S.; Corrias, M.V.; Quintarelli, C.; Locatelli, F.; Ferrini, S.; Croce, M. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci. Rep. 2017, 7, 14049. [Google Scholar] [CrossRef] [Green Version]
- Currie, A.J.; Prosser, A.; McDonnell, A.; Cleaver, A.L.; Robinson, B.W.S.; Freeman, G.J.; van der Most, R.G. Dual Control of Antitumor CD8 T Cells through the Programmed Death-1/Programmed Death-Ligand 1 Pathway and Immunosuppressive CD4 T Cells: Regulation and Counterregulation. J. Immunol. 2009, 183, 7898–7908. [Google Scholar] [CrossRef] [Green Version]
- Maio, M.; Scherpereel, A.; Calabro’, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Cantini, L.; Hassan, R.; Sterman, D.H.; Aerts, J.G.J.V. Emerging Treatments for Malignant Pleural Mesothelioma: Where Are We Heading? Front Oncol. 2020, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Tallón de Lara, P.; Cecconi, V.; Hiltbrunner, S.; Yagita, H.; Friess, M.; Bode, B.; Opitz, I.; Vrugt, B.; Weder, W.; Stolzmann, P.; et al. Gemcitabine Synergizes with Immune Checkpoint Inhibitors and Overcomes Resistance in a Preclinical Model and Mesothelioma Patients. Clin. Cancer Res. 2018, 24, 6345–6354. [Google Scholar] [CrossRef] [Green Version]
- Aston, W.J.; Fisher, S.A.; Khong, A.; Mok, C.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Combining chemotherapy and checkpoint blockade in thoracic cancer: How to proceed? Lung Cancer Manag. 2014, 3, 443–457. [Google Scholar] [CrossRef]
- Wu, L.; Wu, M.O.; De La Maza, L.; Yun, Z.; Yu, J.; Zhao, Y.; Cho, J.; De Perrot, M. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget 2015, 6, 12468–12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.C.; Cheng, Y.Y.; Williams, M.; Kirschner, M.B.; Madore, J.; Lum, T.; Sarun, K.H.; Linton, A.; McCaughan, B.; Klebe, S.; et al. Tumor Suppressor microRNAs Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2017, 12, 1421–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampatsa, A.; Achkova, D.Y.; Davies, D.M.; Parente-Pereira, A.C.; Woodman, N.; Rosekilly, J.; Osborne, G.; Thayaparan, T.; Bille, A.; Sheaf, M.; et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017, 393, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.; June, C.H.; Albelda, S.M. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl. Res. 2017, 187, 1–10. [Google Scholar] [CrossRef]
- Tay, B.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.; Panizza, B.; Frazer, I.; Cruz, J. Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions. Vaccines 2021, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Creaney, J.; Ma, S.; Sneddon, S.A.; Tourigny, M.R.; Dick, I.M.; Leon, J.S.; Khong, A.; Fisher, S.A.; Lake, R.A.; Lesterhuis, W.J.; et al. Strong spontaneous tumor neoantigen responses induced by a natural human carcinogen. Oncoimmunology 2015, 4, e1011492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneddon, S.; Rive, C.; Ma, S.; Dick, I.M.; Allcock, R.J.N.; Brown, S.D.; Holt, R.; Watson, M.; Leary, S.; Lee, Y.C.G.; et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. OncoImmunology 2019, 9, 1684713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pease, D.F.; Kratzke, R.A. Oncolytic Viral Therapy for Mesothelioma. Front. Oncol. 2017, 7, 179. [Google Scholar] [CrossRef]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Peng, K.-W.; Dingli, D.; Kratzke, R.A.; Russell, S.J. Oncolytic measles viruses encoding interferon β and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010, 17, 550–558. [Google Scholar] [CrossRef]
- Kucharczuk, J.C.; Randazzo, B.; Chang, M.Y.; Amin, K.M.; Elshami, A.A.; Sterman, D.; Rizk, N.P.; Molnar-Kimber, K.L.; Brown, S.M.; MacLean, A.R.; et al. Use of a “replication-restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Res. 1997, 57, 466–471. [Google Scholar]
- Danson, S.; Woll, P.; Edwards, J.; Blyth, K.; Fisher, P.; Roman, J.; Simpson, K.; Spavin, R.; Learmonth, K.; Conner, J. 366PD—Oncolytic herpesvirus therapy for mesothelioma: A phase I/IIa trial of intrapleural administration of HSV1716 (NCT01721018). Ann. Oncol. 2017, 28, v122. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, P.L.; Delaune, C.L.; Abadie, J.M.; Toups, M.; Lacour, J.; Marrero, L.; Zhong, Q.; Kolls, J.K. Efficacy of CD40 Ligand Gene Therapy in Malignant Mesothelioma. Am. J. Respir. Cell Mol. Biol. 2003, 29, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Belin, L.J.; Ady, J.W.; Lewis, C.; Marano, D.; Gholami, S.; Mojica, K.; Eveno, C.; Longo, V.; Zanzonico, P.B.; Chen, N.G.; et al. An oncolytic vaccinia virus expressing the human sodium iodine symporter prolongs survival and facilitates SPECT/CT imaging in an orthotopic model of malignant pleural mesothelioma. Surgery 2013, 154, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Biomarker | Biomarker Type | Specificity | Sensitivity | Reference |
|---|---|---|---|---|
| Mesothelin | Protein | High a (95%) | Low b (67%) | [89] |
| Fibulin-3 | Protein | High a (95%) in plasma, but low b (52%) in pleural effusion | Low b (22% and 59% in plasma and pleural effusion, respectively). | [93] |
| Osteopontin | Protein | High a (78%) for differentiating MPM from asbestos-exposed individuals without cancer | High a (86%) for differentiating asbestos-exposed individuals without cancer | |
| SOMAscan 13-protein biomarker panel | Protein | High a (91%) | High a (93%) | [98] |
| ESR1, SLC6A20 and SYK three-gene signature | Methylated genes | Moderate c (73%) | High a (92%) | [99] |
| miR-135b, miR-181a-2 *, miR-499-5p, miR-517b, miR-519d, miR-615-5p and miR-624 | MicroRNA | High a (>75%) for epithelioid subtype MPM. | Not reported | [101] |
| miR-218-2 *, miR-346, miR-377 *, miR-485-5p and miR-525-3p | MicroRNA | High a (>75%) for biphasic subtype MPM | Not reported | [101] |
| miR-301b, miR-433 and miR-543 | MicroRNA | High a (>75%) for sarcomatoid subtype MPM | Not reported | [101] |
| miR-192, miR-193a-3p and miR-200c | MicroRNA | High a (94%) for differentiating MPM from lung adenocarcinoma | High a (100%) for differentiating MPM from lung adenocarcinoma | [104] |
| Clinical Trial Code | Phase | Title | Treatment | Target | Completed (C) or In Progress (IP) | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Targeted Therapies | |||||||
| NCT02860286 | II | Study of the EZH2 Inhibitor Tazemetostat in Malignant Mesothelioma | Tazemetostat | BAP1-deficient MPM | C | Disease control was achieved in 51% of patients at 12 weeks and 25% of patients at 24 weeks | [160] |
| NCT00651456 | III | Mesothelioma Avastin Plus Pemetrexed-cisplatin Study (MAPS) | CT + bevacizumab | VEGF | C | 2.6 month increase in patient survival compared to standard CT | [7] |
| NCT03762018 | III | BEAT-meso: Bevacizumab and Atezolizumab in Malignant Pleural Mesothelioma (BEAT-meso) | CT + bevacizumab + atezolizumab | VEGF | IP | - | - |
| NCT03654833 | II | Mesothelioma Stratified Therapy (MiST): A Multi-drug Phase II Trial in Malignant Mesothelioma (MiST) | Abemaciclib | p16INK4A-deficient MPM | IP | - | - |
| NCT02369198 | I | MesomiR 1: A Phase I Study of TargomiRs as 2nd or 3rd Line Treatment for Patients with Recurrent MPM and NSCLC | TargomiRs | EGFR | C | Objective response achieved for 1 patient and stable disease achieved for 15 patients in a cohort of 27 patients | [164,165] |
| NCT01675765 | I | Safety and Efficacy of Listeria in Combination with Chemotherapy as Front-line Treatment for Malignant Pleural Mesothelioma | CT + CRS-207 vaccine | Mesothelin | C | Disease control achieved in 89% of patients, with 31% showing a reduction in tumour size | [178] |
| NCT01279967 | II | A Clinical Trial of ADI-PEG 20TM in Patients with Malignant Pleural Mesothelioma (ADAM) | Adi-PEG 20 | ASS1-deficient MPM | C | 3.2 months PFS achieved for Adi-PEG 20-treated patients compared to 2 months for patients without Adi-PEG 20 treatment | [183] |
| NCT02709512 | II/III | Ph 2/3 Study in Subjects with MPM to Assess ADI-PEG 20 with Pemetrexed and Cisplatin (ATOMIC) | CT + Adi-PEG 20 | ASS1-deficient MPM | IP | - | - |
| Immunotherapies | |||||||
| NCT02054806 | I | Study of Pembrolizumab (MK-3475) in Participants with Advanced Solid Tumors (MK-3475-028/KEYNOTE-28) | Pembrolizumab | PD-1 | C | Objective response was achieved in 28% of patients and stable disease achieved in 48% | [185] |
| NCT02991482 | III | Pembrolizumab Immunotherapy Versus Standard Chemotherapy for Advanced Pre-treated Malignant Pleural Mesothelioma (PROMISE-meso) | CT + pembrolizumab | PD-1 | IP | - | - |
| NCT02899299 | III | Study of Nivolumab Combined with Ipilimumab Versus Pemetrexed and Cisplatin or Carboplatin as First Line Therapy in Unresectable Pleural Mesothelioma Patients (CheckMate743) | CT + nivolumab + ipilimumab | PD-1 and CTLA-4 | IP | A median overall patient survival of 18 months was achieved for patients treated with CT in combination with nivolumab and ipilimumab, compared to 14 months for patients receiving CT alone | [123] |
| NCT01722149 | I | Re-directed T Cells for the Treatment (FAP)-Positive Malignant Pleural Mesothelioma | FAP-specific CAR T cells | FAP | C | - | - |
| Virotherapies | |||||||
| NCT01721018 | I/IIa | Intrapleural Administration of HSV1716 to Treat Patients with Malignant Pleural Mesothelioma. (1716-12) | HSV-1716 | MPM tumour cells | C | A median survival of 15 months and 18 months was achieved in HSV-1716-treated patients and HSV-1716-treated patients that exhibited evidence of anti-tumour immunogenicity. | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, B.; Lee, K.; Cheng, Y.Y. Pre-Clinical Research Advancements Relating to Improving the Diagnosis and Treatment of Malignant Pleural Mesothelioma: A Review. Onco 2021, 1, 49-82. https://doi.org/10.3390/onco1020006
Johnson B, Lee K, Cheng YY. Pre-Clinical Research Advancements Relating to Improving the Diagnosis and Treatment of Malignant Pleural Mesothelioma: A Review. Onco. 2021; 1(2):49-82. https://doi.org/10.3390/onco1020006
Chicago/Turabian StyleJohnson, Ben, Kenneth Lee, and Yuen Yee Cheng. 2021. "Pre-Clinical Research Advancements Relating to Improving the Diagnosis and Treatment of Malignant Pleural Mesothelioma: A Review" Onco 1, no. 2: 49-82. https://doi.org/10.3390/onco1020006
APA StyleJohnson, B., Lee, K., & Cheng, Y. Y. (2021). Pre-Clinical Research Advancements Relating to Improving the Diagnosis and Treatment of Malignant Pleural Mesothelioma: A Review. Onco, 1(2), 49-82. https://doi.org/10.3390/onco1020006