
Serendipitous Discovery of Desert Hairy Scorpion Mitogenomes as Bycatch in Venom Data via Nanopore Sequencing


 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruiz-Mendoza, P.X.; Jasso-Martínez, J.M.; Gutiérrez-Rodríguez, J.; Samacá-Sáenz, E.; Zaldívar-Riverón, A. Mitochondrial genome characterization and mitogenome phylogenetics in the central Mexican Stenopelmatus talpa complex (Orthoptera: Stenopelmatidae: Stenopelmatini). Rev. Mex. Biodivers. 2023, 94, e945094. [Google Scholar] [CrossRef]
- Kong, D.; Gan, Z.; Li, X. Phylogenetic relationships and adaptation in deep-sea carideans revealed by mitogenomes. Gene 2023, 896, 148054. [Google Scholar] [CrossRef] [PubMed]
- Galen, S.C.; Borner, J.; Perkins, S.L.; Weckstein, J.D. Phylogenomics from transcriptomic “bycatch” clarify the origins and diversity of avian trypanosomes in North America. PLoS ONE 2020, 15, e0240062. [Google Scholar] [CrossRef] [PubMed]
- Ghanavi, H.R.; Twort, V.G.; Duplouy, A. Exploring bycatch diversity of organisms in whole genome sequencing of Erebidae moths (Lepidoptera). Sci. Rep. 2021, 11, 24499. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Graham, M.R.; Sharma, P.P.; Ortiz, E.; Possani, L.D. Hadrurid scorpion toxins: Evolutionary conservation and selective pressures. Toxins 2019, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ojanguren-Affilastro, A.A.; Sharma, P.P. Another one bites the dust: Taxonomic sampling of a key genus in phylogenomic datasets reveals more non-monophyletic groups in traditional scorpion classification. Invertebr. Syst. 2020, 34, 133–143. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; González-Santillán, E.; Monod, L.; Sharma, P.P. Phylogenomics facilitates stable scorpion systematics: Reassessing the relationships of Vaejovidae and a new higher-level classification of Scorpiones (Arachnida). Mol. Phylogenet. Evol. 2019, 135, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ojanguren-Affilastro, A.A.; Graham, M.R.; Sharma, P.P. Congruence between ultraconserved element-based matrices and phylotranscriptomic datasets in the scorpion Tree of Life. Cladistics 2023, 39, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Shimwell, C.; Atkinson, L.; Graham, M.R.; Murdoch, B. A first molecular characterization of the scorpion telson microbiota of Hadrurus arizonensis and Smeringurus mesaensis. PLoS ONE 2023, 18, e0277303. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 474, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Ban, X.C.; Shao, Z.K.; Wu, L.J.; Sun, J.T.; Xue, X.F. Highly diversified mitochondrial genomes provide new evidence for interordinal relationships in the Arachnida. Cladistics 2022, 38, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, W.T.; Zhang, Q.L.; Liu, M.; Xing, C.W.; Cao, Y.; Luo, F.Z.; Yuan, M.L. Mitochondrial phylogenomics provides insights into the phylogeny and evolution of spiders (Arthropoda: Araneae). Zool. Res. 2022, 43, 566–584. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeeseler, A.; Lanfear, R. IQTREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Revell, L. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Fet, V.; Soleglad, M.E.; Barker, M.D. Phylogenetic analysis of the “hirsutus” group of the genus Hadrurus Thorell (Scorpiones: Iuridae) based on morphology and mitochondrial DNA. In Scorpions 2001. In Memoriam Gary A. Polis; Fet, V., Selden, P.A., Eds.; British Arachnological Society: Burnham Beeches, UK, 2001; pp. 139–160. [Google Scholar]
- Gantenbein, B.; Fet, V.; Largiader, C.; Scholl, A. First DNA phylogeny of the genus Euscorpius Thorell 1876 (Scorpiones, Euscorpiidae) and its bearing on the taxonomy and biogeography of this genus. Biogeographica 1999, 75, 59–72. [Google Scholar]
- Gantenbein, B.; Largiadèr, C.R. The phylogeographic importance of the Strait of Gibraltar as a gene flow barrier in terrestrial arthropods: A case study with the scorpion Buthus occitanus as model organism. Mol. Phylogenet. Evol. 2003, 28, 119–130. [Google Scholar] [CrossRef]
- Graham, M.R.; Jaeger, J.R.; Prendini, L.; Riddle, B.R. Phylogeography of the Arizona hairy scorpion (Hadrurus arizonensis) supports a model of biotic assembly in the Mojave Desert and adds a new Pleistocene refugium. J. Biogeogr. 2013, 40, 1298–1312. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Aharon, S.; Ballesteros, J.A.; Gainett, G.; Baker, C.M.; González-Santillán, E.; Harvey, M.S.; Hassan, M.K.; Almaaty, A.H.A.; Monod, L.; et al. Phylogenomics of scorpions reveal contemporaneous diversification of scorpion mammalian predators and mammal-active sodium channel toxins. Syst. Biol. 2022, 71, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Ghanavi, H.R.; Twort, V.; Hartman, T.J.; Zahiri, R.; Wahlberg, N. The (non) accuracy of mitochondrial genomes for family-level phylogenetics in Erebidae (Lepidoptera). Zool. Scr. 2022, 51, 695–707. [Google Scholar] [CrossRef]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore adaptive sampling: A tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef]
- Abramson, N.I.; Bodrov, S.Y.; Bondareva, O.V.; Genelt-Yanovskiy, E.A.; Petrova, T.V. A mitochondrial genome phylogeny of voles and lemmings (Rodentia: Arvicolinae): Evolutionary and taxonomic implications. PLoS ONE 2021, 16, e0248198. [Google Scholar] [CrossRef]
- Pan, D.; Shi, B.; Du, S.; Gu, T.; Wang, R.; Xing, Y.; Zhang, Z.; Chen, J.; Cumberlidge, N.; Sun, H. Mitogenome phylogeny reveals Indochina Peninsula origin and spatiotemporal diversification of freshwater crabs (Potamidae: Potamiscinae) in China. Cladistics 2022, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Dietrich, C.H.; Yu, X.; Jiao, M.; Dai, R.; Yang, M. Mitogenomic phylogeny of Typhlocybinae (Hemiptera: Cicadellidae) reveals homoplasy in tribal diagnostic morphological traits. Ecol. Evol. 2022, 12, e8982. [Google Scholar] [CrossRef]
- Ding, L.; Zhou, Q.; Sun, Y.; Feoktistova, N.Y.; Liao, J. Two novel cricetine mitogenomes: Insight into the mitogenomic characteristics and phylogeny in Cricetinae (Rodentia: Cricetidae). Genomics 2020, 112, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; Uribe, J.E.; Eernisse, D.J.; Zardoya, R. A mitogenomic phylogeny of chitons (Mollusca: Polyplacophora). BMC Evol. Biol. 2020, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Catanese, G.; Morey, G.; Verger, F.; Grau, A.M. The Nursehound Scyliorhinus stellaris mitochondrial genome—Phylogeny, relationships among Scyliorhinidae and variability in waters of the Balearic Islands. Int. J. Mol. Sci. 2022, 23, 10355. [Google Scholar] [CrossRef] [PubMed]





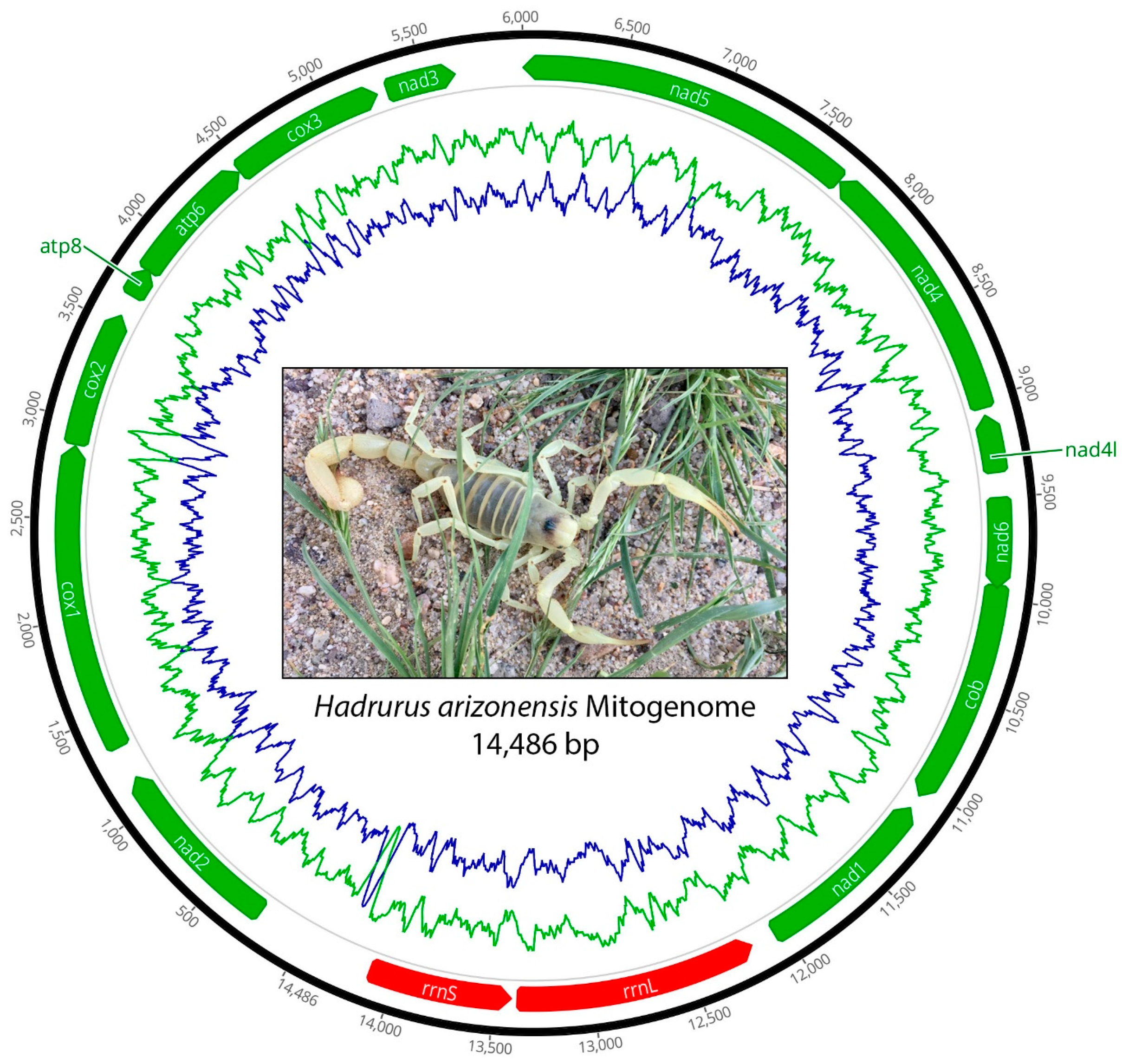{kind=link}
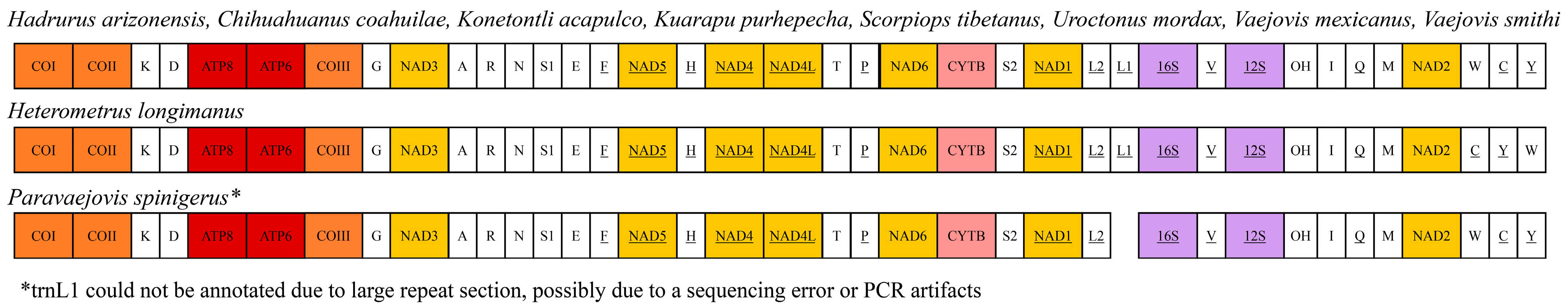{kind=link}
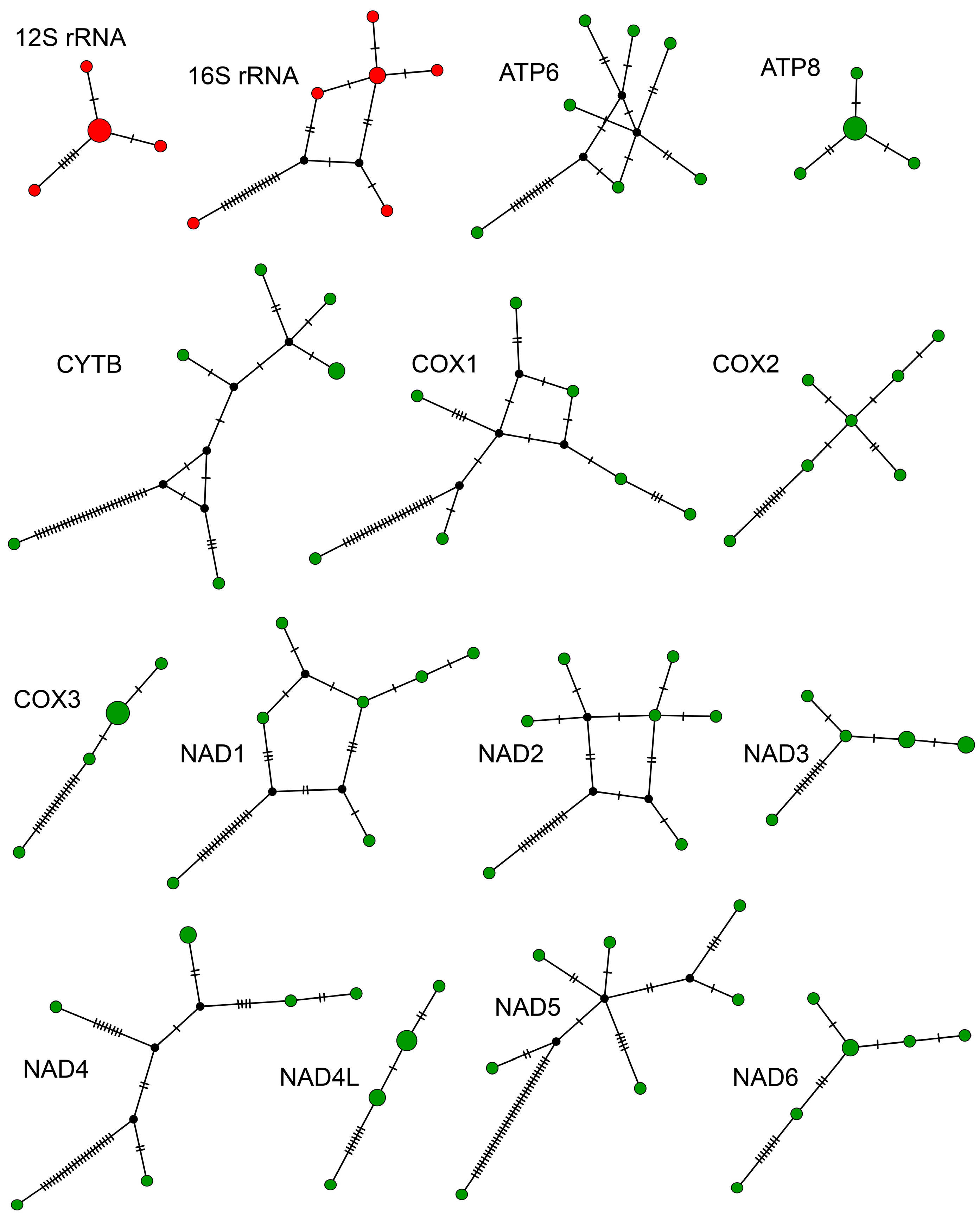{kind=link}
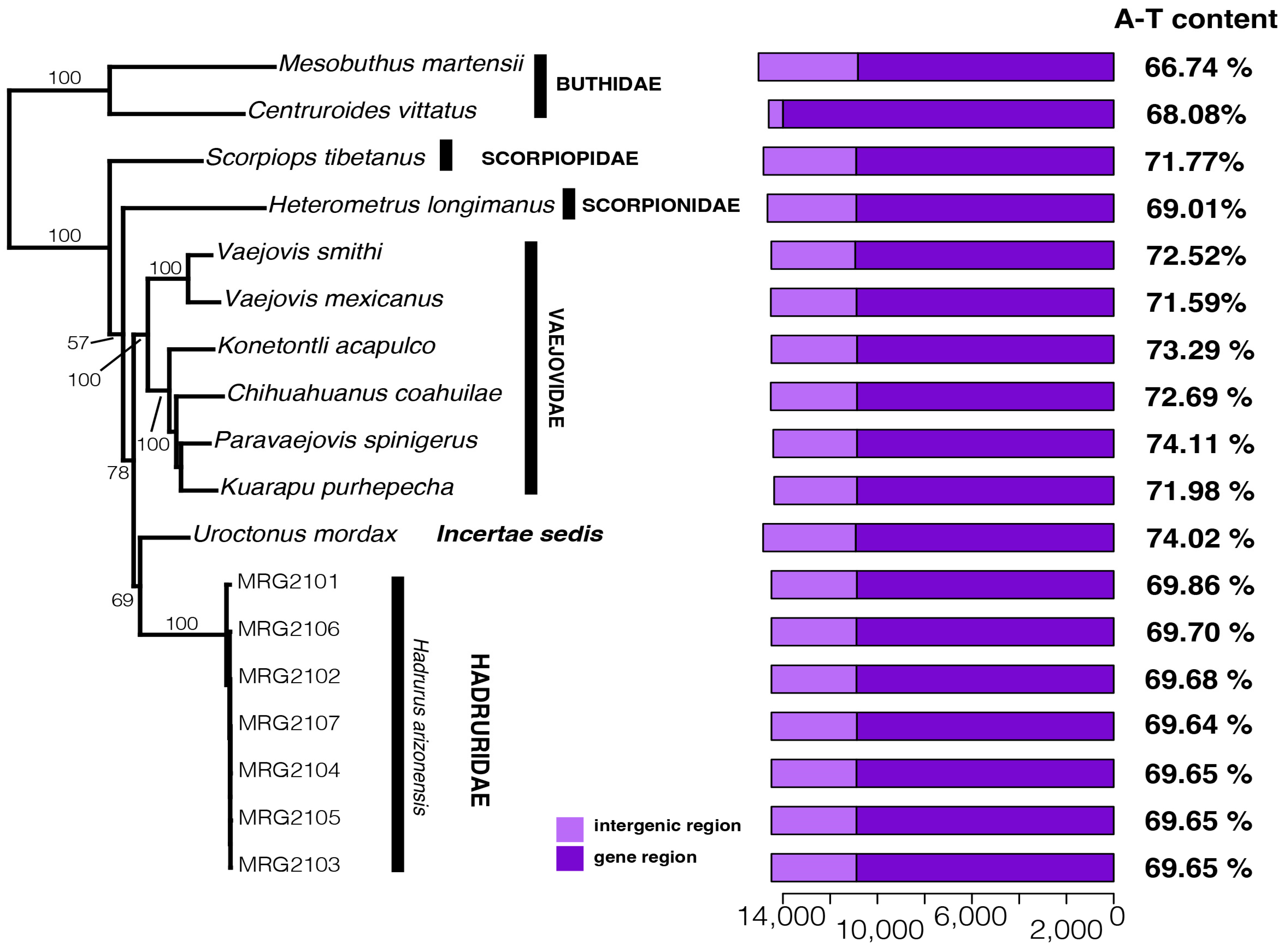{kind=link}
| Identifier | Passed Reads | Trimmed Reads | Trimmed Read Length | Mapped Reads | Coverage |
|---|---|---|---|---|---|
| MRG2101 | 444,000 | 435,340 | 200–36,686 | 1411 | 23–89 |
| MRG2102 | 1,272,000 | 515,045 | 200–30,400 | 1220 | 29–93 |
| MRG2103 | 372,000 | 362,438 | 200–40,124 | 1327 | 38–83 |
| MRG2104 | 236,000 | 235,969 | 50–25,996 | 709 | 14–64 |
| MRG2105 | 972,000 | 892,446 | 200–48,837 | 7838 | 39–145 |
| MRG2106 | 844,000 | 841,533 | 105–34,665 | 2693 | 81–170 |
| MRG2107 | 1,760,000 | 892,394 | 500–82,653 | 2563 | 84–227 |
| Gene | Full Gene Name | Length (bp) | Variable Sites (S) | Percent Variable | Haplotypes (h) | Haplotype Diversity (Hd) | Nucleotide Diversity (π) |
|---|---|---|---|---|---|---|---|
| 12S | 12S ribosomal RNA | 721 | 8 | 1.11% | 4 | 0.714 | 0.00317 |
| 16S | 16S ribosomal RNA | 1197 | 23 | 1.92% | 6 | 0.952 | 0.00597 |
| ATP6 | ATP synthase membrane subunit 6 | 666 | 22 | 3.30% | 7 | 1.000 | 0.01044 |
| ATP8 | ATP synthase membrane subunit 8 | 156 | 4 | 2.56% | 4 | 0.714 | 0.00733 |
| CYTB | Cytochrome b | 1119 | 42 | 3.75% | 6 | 0.952 | 0.01170 |
| COI | Cytochrome c oxidase I | 1539 | 39 | 2.53% | 7 | 1.000 | 0.00792 |
| COII | Cytochrome c oxidase II | 670 | 15 | 2.24% | 7 | 1.000 | 0.00739 |
| COIII | Cytochrome c oxidase III | 781 | 20 | 2.56% | 4 | 0.714 | 0.00756 |
| NAD1 | NADH dehydrogenase subunit 1 | 924 | 26 | 2.81% | 7 | 1.000 | 0.00948 |
| NAD2 | NADH dehydrogenase subunit 2 | 962 * | 25 | 2.60% | 7 | 1.000 | 0.00832 |
| NAD3 | NADH dehydrogenase subunit 3 | 348 | 14 | 4.02% | 5 | 0.905 | 0.01286 |
| NAD4 | NADH dehydrogenase subunit 4 | 1275 | 39 | 3.06% | 6 | 0.952 | 0.01083 |
| NAD4L | NADH dehydrogenase subunit 4L | 288 | 10 | 3.47% | 4 | 0.810 | 0.01157 |
| NAD5 | NADH dehydrogenase subunit 5 | 1698 | 53 | 3.12% | 7 | 1.000 | 0.00937 |
| NAD6 | NADH dehydrogenase subunit 6 | 453 | 14 | 3.09% | 6 | 0.952 | 0.01092 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, M.R.; Santibáñez-López, C.E.; Zehnpfennig, J.R.; Tillman, D.S.; Murdoch, B. Serendipitous Discovery of Desert Hairy Scorpion Mitogenomes as Bycatch in Venom Data via Nanopore Sequencing. Arthropoda 2024, 2, 119-129. https://doi.org/10.3390/arthropoda2020009
Graham MR, Santibáñez-López CE, Zehnpfennig JR, Tillman DS, Murdoch B. Serendipitous Discovery of Desert Hairy Scorpion Mitogenomes as Bycatch in Venom Data via Nanopore Sequencing. Arthropoda. 2024; 2(2):119-129. https://doi.org/10.3390/arthropoda2020009
Chicago/Turabian StyleGraham, Matthew R., Carlos E. Santibáñez-López, Jessica R. Zehnpfennig, Dylan S. Tillman, and Barbara Murdoch. 2024. "Serendipitous Discovery of Desert Hairy Scorpion Mitogenomes as Bycatch in Venom Data via Nanopore Sequencing" Arthropoda 2, no. 2: 119-129. https://doi.org/10.3390/arthropoda2020009
APA StyleGraham, M. R., Santibáñez-López, C. E., Zehnpfennig, J. R., Tillman, D. S., & Murdoch, B. (2024). Serendipitous Discovery of Desert Hairy Scorpion Mitogenomes as Bycatch in Venom Data via Nanopore Sequencing. Arthropoda, 2(2), 119-129. https://doi.org/10.3390/arthropoda2020009