1. Introduction
In thermodynamics, gas dynamics, chemical kinetics, control theory, information theory, economics, ecology, biology, and other sciences, it is increasingly important to determine the non-equilibrium time-dependent behavior of the state of a system, for which the description of the equilibrium states is known and given by the solution of a constrained maximization problem. The most accurate way of doing this consists of specifying the state variables, developing a detailed kinetic model (DKM) of the non-equilibrium system, obtaining a full set of rate equations for all the state variables based on the postulated DKM, and integrating this set of equations to yield the time-dependent behavior of the non-equilibrium state. Since this approach requires, in principle, modeling all the detailed mechanisms, a potential concern is to obtain the kinetic rate data for all the detailed mechanisms. However, for example in the context of chemical kinetics, such data are quite uncertain and involve a lot of guess work. Moreover, when the DKM method must be coupled to transport equations and turbulence models or interface dynamics, the modeling and computational tasks often become formidable because of the intrinsic presence of a wide range of time and length scales, which may result in the well-known stiffness difficulties.
In the framework of chemical kinetics, such difficulties have motivated the development of numerous model order reduction techniques during the past two decades [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26]. Moreover, for micro- and nano-systems in which the small numbers of particles entail that discreteness and stochasticity play an important role on the time evolution of the species population, similar reduction methods have been proposed within the growing field of Stochastic Chemical Kinetics [
27,
28,
29] and Stochastic Simulation Algorithms [
30]. One common feature shared by the aforementioned techniques is that they all need a large DKM as the starting point. However, even though theoretically the most accurate description of the kinetics, independent of the context, comes from the corresponding DKM, a general concern in modeling complex chemical systems for which the underlying DKM involves a large number of reaction mechanisms stems from the fact that the rates of a vast majority of reactions are unknown or, at best, highly uncertain.
An alternative method which can help reduce the modeling and computational efforts is the Rate-Controlled Constrained-Equilibrium (RCCE) method, in which the state of the system at any time is that which maximizes an entropy function or minimizes an appropriate form of free energy function subject to a small number of constraints, namely, state functionals. At each instant in time, the state of the system, which is described in terms of internal variables such as the reaction coordinates (in the chemical kinetics context), is assumed to be at equilibrium with respect to all the internal dynamical mechanisms that cannot alter the values of a small number of rate-controlling constraining functionals. The mechanisms that instead do contribute to changing the values of the constraints control the time evolution through their impact on the rate of change of the values of the constraining functionals. For a closed homogeneous reacting system, the resulting time evolution (trajectory in state space) is a continuous shifting of the assumed constrained equilibrium state towards higher entropies until the highest entropy (stable equilibrium) state is reached. For an open heterogeneous system subject to stationary boundary conditions, transport phenomena will also contribute to the rate of change of the local values of the constraints, and the continuous shifting of the assumed constrained local equilibrium states will evolve until the (stable) steady state is reached. The choice in RCCE can also be viewed as the best pragmatic choice according to Bayesian Inference in the framework of Jaynes’ Maximum Entropy Principle (see, e.g., [
31,
32,
33]).
Two familiar examples of RCCE approach are the assumptions of locally complete and locally frozen chemical equilibrium. The former is characterized by atomic elements as the only constraints to the composition, subject to which pressure, temperature and Gibbs free energy are defined. The latter instead treat the entire chemical composition as a constant of the motion. Even in the absence of chemical reactions, another familiar example of the RCCE approach is the assumption of local thermodynamic equilibrium employed in the basic equations of fluid dynamics to describe non-equilibrium states by means of local temperature, pressure, and chemical potentials. Obviously, this assumption breaks down in the presence of fast diffusion processes, e.g., across a shock wave.
An application of RCCE reduces the total number of equations (algebraic + differential) to the total number of constraints, which for systems including heavy hydrocarbons can be dramatically smaller than the number of species in a corresponding DKM. Nonetheless, the dynamic evolution of all “left-out” species can be determined based on the requirement of constrained equilibrium. This often provides a good first order estimate of how kinetically important a species is when RCCE is used for model development. RCCE calculations have provided accurate results in relatively complex hydrocarbon combustion problems [
34,
35]. Two typical examples are discussed at the end of the paper. The effectiveness of the RCCE method and the accuracy of its results depend on the identification of a proper set of rate-controlling constraining functionals, which requires a fundamental understanding of the detailed dynamics of the system. In other words, the deeper the physical insight into the system’s dynamics, the more effective and structurally consistent a set of constraining functionals can be identified. In each particular field of application, a specific expertise must be developed in order to identify the controlling mechanisms and to group them into classes which must each be characterized by one or more functionals that are to become the rate-controlling constraints.
The objective in RCCE is to identify and group the rate limiting mechanisms into classes each characterized by a functional that is time-invariant within the class and which at least at some stage during the time evolution is slowly varying with respect to the full dynamics. In general, due to time-scale diversity and time-varying competition between different mechanisms, not all the constraining functionals are necessarily slowly varying and rate-controlling during the entire dynamic evolution. By identifying which constraining functionals are rate controlling during a given stage (for example, the ignition delay stage in a combustion process) it is possible to identify the corresponding underlying class of rate limiting mechanisms and gain useful physical insight.
Furthermore, despite what it may imply, a DKM is a reduced model itself for the simple reason that it neither includes all possible species in the reactor nor all the complex reactions governing them. From this point of view, we note that RCCE could be to a large extent independent of the level of approximation with which the rates of the less important reaction schemes are usually estimated in a DKM. The reason is that RCCE assumes at each instant in time the complete chemical equilibrium composition (highest entropy) compatible with the instantaneous local values of the state variables and the assumed constraining functionals. In other words,
an informed application of RCCE puts these reactions in equilibrium, and it is well known that the complete chemical equilibrium composition is independent of the underlying detailed kinetic scheme ([
36], p. 578). It is true that many of such reactions do not impose rate limiting constraints on the system’s evolution and are, therefore, eliminated in most DKM-based model order reduction techniques. But eliminating a reaction means freezing it. Instead, in RCCE the majority of such reactions are put in (constrained) equilibrium rather than being frozen.
The rates of change of the constraining functionals are to be estimated in principle from the full dynamics, but in practice only the faster mechanisms within the corresponding class are sufficient. In general, these faster mechanisms are often the most important in the fully detailed scheme and, therefore, they are the ones for which the most reliable experimental data on reaction rates are most likely available. Moreover, the physical insight, which is gained when a new successful constraining functional is identified and understood in connection with the corresponding underlying class of rate limiting mechanisms, is likely to be useful also to further improve, simplify, and complete the DKM.
In reacting gas dynamics, the RCCE method has been applied successfully for the description and simplification of many specific non-equilibrium problems. Sugden and co-workers [
37], Kaskan [
38], Schott [
39], and Franciscus and Lezberg [
40] treated the hydrogen-oxygen combustion problem. Keck and Gillespie [
41] (see also [
42,
43]) developed a general rate-controlled constrained-equilibrium method for reacting gas mixtures. Delichatsios and Keck [
44], Sarofim and co-workers [
45], and Morr and Heywood [
46] applied the method to predict carbon monoxide and nitric oxide formation in hydrocarbon combustion. Galant and Appleton [
47] further developed the method for open reacting systems. Levine [
48] treated reactions in molecular beams. Procaccia and Ross [
49] investigated the stability in highly non-equilibrium systems. It has also formed the basis for Close Parallel Inertial Manifold (CPIM) [
50] and Inertial Constrained-Equilibrium Pre-Image Curve (ICE-PIC) [
51]. The method has been later developed and applied by several others, see e.g., [
34,
35,
50,
52,
53,
54,
55,
56,
57,
58,
59].
Ideas similar to RCCE have been applied in specific applications also in other fields. For example, Kerner [
60] studied speciation in ecological systems. Dornbush [
61] developed a theory of exchange rate overshooting due to a changing monetary policy based on the notion that asset market prices including exchange rates adjust to the new equilibrium in practically no time whereas market prices of goods are “sticky”, adjust in finite time and thus rate-control the relaxation of the system towards the new equilibrium state.
It should be noted [
62] that the entropy maximization done by RCCE at each instant of time does not necessarily imply that the relaxation is along the path of steepest entropy ascent [
63,
64,
65,
66]. This is consistent with the theoretical proof by Ziegler [
67] as well as a recent numerical study [
17] that maximal entropy production rate is not a general feature of the standard equations of chemical kinetics.
The objective of this paper is to present a generalized formulation of the RCCE method valid also for nonlinear constraints, together with a rigorous step-by-step derivation of its implementation in the standard framework of chemical kinetics of gas mixtures with linear constraints, a discussion about its general thermodynamic consistency, and examples about the physical meaning of several basic constraints that have proved effective in simplifying combustion modeling. The chemical kinetics implementation is presented with strong emphasis on the thermodynamics aspects, careful discussion of the assumptions, explicit indication of all functional dependences, and discussion of the most important features of the RCCE method, but the generalized formulation allows its application also in diverse nonequilibrium contexts, provided the time evolution is characterized by the existence of a Lyapunov functional which cannot be increased by the system’s internal dynamics.
The paper is structured as follows. In
Section 2, we present a generalized formulation of the mathematical structure of the problem of describing the time evolution of non-equilibrium states. In
Section 3, we discuss the general ideas behind the constrained-equilibrium method, and we effectively extend the method to the case of nonlinear constraints. In
Section 4, we present a step-by-step detailed implementation of the foregoing general formulation in the framework of nonequilibrium thermodynamics of a mixture (ideal and nonideal) subject to complex chemical kinetics. There, by giving detailed proofs and explicit dependences, we address the subtleties about the domain of validity of detailed balance and the general nonnegativity of the rate of entropy generation. Finally, in
Section 5 we discuss via two examples the physical meaning of the basic constraints that have proved most effective in reducing combustion modeling.
Section 6 draws our conclusions.
2. General Non-Equilibrium Problem
In this and the next sections, we discuss the formulation of non-equilibrium dynamics for a generic state description which captures the model structure of a large class of systems of interest in various areas of science and engineering.
Let us start by asserting that when building a mathematical model of a system (physical, biological, chemical, etc.) we explicitly or implicitly select a “level of description” which postulates the existence of a set of elementary building blocks which, for the purposes of the model, we consider as indivisible and unalienable. For example, a given physical object may be described at various levels, depending on the phenomena we set out to model. In particular, as we further discuss below, the choice of the level of description is related to the range of time scales which characterize the phenomena of interest. So, if we are not interested in chemical and nuclear reactions, i.e., we may assume that on the time scale of interest they are practically “frozen”, then we may assume that the elementary constituents are the atoms and the molecules, as we do when we study non-reacting transport phenomena. If we are interested in chemical reactions but not nuclear reactions, we may take the elements (or, more precisely, the atomic nuclei and the electrons) as the elementary constituents. If we are interested in fission and fusion nuclear reactions, we may take neutrons, protons, and electrons. Or at a deeper level, we may take quarks and so on. Again, to describe resonance interactions between electromagnetic radiation and the electrons in atoms and molecules, it is in many instances sufficient to consider as elementary constituents the photons and a limited number of electronic levels.
Once the system’s constituents have been selected, the model defines what variables identify the different possible states. Let us denote by
the vector of
variables describing the state of the system under study [
68]. Often, it is convenient to separate the vector
x into a vector of internal state variables that are not directly accessible to external control and a vector of externally controllable parameters,
i.e.,
. For example, in a closed chemically reacting system, species concentrations are internal variables controlled by the internal kinetics, energy and volume may be considered external when their changes are prescribed by heat and work interactions through the system’s boundaries and compressing or expanding displacements of the boundaries, respectively.
Next, let us assume that the dynamics of the system is described by a rate equation (equation of motion) of the form
where
represents the internal or endogenous dynamical mechanisms and
the external or exogenous mechanisms. For example, if the system is a control volume in a reacting fluid flow, the endogenous mechanisms are those responsible for the spontaneous relaxation towards a local equilibrium state whereas the exogenous mechanisms are those accounting for the transport of energy, momentum and matter across the boundaries of the control volume.
The equilibrium states of the system are those that would not be changing in time if the system were isolated,
i.e., if
. Thus, a state
is equilibrium if
In general, Equation (
2) admits more than one solution, namely, the system admits more than one equilibrium state. For example, the second law of thermodynamics requires that a physical system has at least one equilibrium state for each of the possible values of the energy [
69].
The system’s definition may impose elemental or geometrical constraints implying that a set of functionals that have values that are either fixed or externally controlled,
i.e.,
for all the states
x of the system, where
are constants or externally specified functions of time. These functionals generally specify restrictions on the geometry of the state space, or internal interconnections between internal partitions, or elemental conservation intrinsic in the chosen level of description. The reason for the superscript
∞ will become apparent in the next section. For example, if the state variables
represent probabilities then a geometrical constraint is
with
. Again, if state variables
,
,
represent the volumes of three regions of space in which the overall system is partitioned by means of two moving pistons, they may be constrained by the condition that
is constant or has an externally imposed time dependence. Another example is the set of stoichiometric relations between species concentrations which, in a detailed chemical or electrochemical kinetic model of a closed system, is imposed by the assumed scheme of reaction mechanisms.
In addition, for most systems of practical interest, such as for those of interest in thermodynamics and chemical kinetics, the system admits one or more functionals that we denote by
, which are time-invariant under the internal dynamics
,
i.e., such that for every state
x,
where we defined the gradient vectors
The functionals
are said to represent the constants of the motion of the system. For example, the energy and the number of atomic nuclei of each type are constants of the motion for a chemically reacting open system. They are time invariant under the internal dynamics, but their values may change as a result of external mechanisms such as those of work, heat, bulk flow, and diffusion interactions.
If there are
r such constants of the motion, with linearly independent gradient vectors
, the
r Equation (
4) imply that the set of Equation (
2) which define the equilibrium states are not all linearly independent and, therefore, the solution depends on at least
r arbitrary parameters. In other words, the equilibrium states form at least an
r-parameter family.
For each given state x, it is useful to identify, within the combined set of elemental constraints and constants of the motion , a complete subset that we denote simply by with , such that the gradient vectors are linearly independent. The subset must be complete in the sense that any other functional in the full set has a gradient vector which is a linear combination of the vectors .
For many systems of practical interest, there is a state functional
that is non-decreasing under the internal dynamics of the system [
70],
i.e., is such that
for every
x, where we defined the gradient vector Γ with components
Our next step is to assume that the functional
is such that in the neighborhood of each one of the equilibrium states of interest (the stable equilibrium states, in thermodynamics), the functional
when restricted to the subset of states
is a Lyapunov functional [
71] and, hence, each equilibrium state is conditionally stable [
71] with respect to perturbations that do not alter the values of the
’s.
Clearly,
can be a Lyapunov functional when restricted on the set (
9) only if
where
α is some strictly-increasing positive function with
and, hence, the equilibrium state
is the unique state which maximizes the functional
on the set Equation (
9).
To fix ideas, by analogy with the thermodynamics context, we will assume that the functional
is twice differentiable and at least locally strictly concave in its variables, so that the Hessian matrix of generalized stiffness moduli
is well defined, negative semi-definite, and clearly symmetric by Schwarz’s theorem on mixed second derivatives. Moreover it has a nonzero determinant and, since it is also the Jacobian matrix of the system
of
relations
it follows that the relations
can be inverted to yield the system
of
relations
Using this, we can express the functional
also as a functional of the vector Γ,
i.e.,
, for which
x is the gradient vector, with components
and the symmetric, negative semi-definite Hessian matrix of generalized capacities,
is the inverse of the generalized stiffness matrix in Equation (
10).
As a result of the above assumptions, the equilibrium states can be found by solving the following constrained maximization problem
where
is either a fixed value of a geometrical constraint or the value of a constant of the motion. We have implicitly assumed that the functionals
form a complete set of geometrical constraints and constants of the motion with linearly independent gradients
. The solution
of the maximization problem satisfies the set of Euler-Lagrange equations
where
is the Lagrange multiplier associated with the
i-th constraint. Together with the constraints (
15), Equation (
16) yields an
-parameter family of equilibrium states
which are stable with respect to all perturbations [
71] if the function
is continuous in each
. As is well-known, the standard procedure to obtain
(
17) is as follows. First we invert Equation (
16) to express the state variables in terms of the multipliers,
; these relations are then inserted into (
15) to express the values of the constraints as functions of the multipliers,
; these are then inverted to express the multipliers in terms of the values of the constraints,
, which inserted into
yields
,
i.e., Equation (
17). Moreover, it is noteworthy that as a result of the last inversion, we have the identity
which can also be viewed as a consequence of the condition that the constraints have linearly independent gradients, as necessary for the assumed invertibilities.
Using Equation (
16) and Equation (
18) we obtain the relations
which express, geometrically, the general orthogonality condition between the vector
of Lagrange multipliers and the constant-
F contour surfaces in
space.
An immediate consequence of Equation (
19) and Schwarz’s theorem on mixed second derivatives is the following Maxwell relations expressing the reciprocity of the cross dependence of the constraint-potentials on the values of the constraints,
Geometrically, the Hessian matrix of
represents the curvature matrix of the
F contour surfaces in
space.
Similarly, the dual representation of the same surface in
space is obtained by defining the Legendre transform of
as follows,
where the family of stable equilibrium states is now re-parametrized in terms of the
Lagrange multipliers,
It is easy to show that in
space the orthogonality condition is between the vector
of constraint values and the constant-
G contour surfaces,
Moreover, we have the dual Maxwell relations expressing the reciprocity of the cross dependence of the values of the constraints on the constraint-potentials,
and we can readily show that the Hessian matrix of
G in Equation (
24), which represents the curvature matrix of the
G contour surfaces in
space, is the inverse of the Hessian matrix of
F in Equation (
20).
If the internal dynamics is much faster than the external, we may assume that for all practical purposes the system always passes through equilibrium states, namely,
where the values of the constants of the motion vary according to Equation (
1),
i.e., in view of Equation (
4),
For example, this is a reasonable assumption for the local description of non-reacting flow fields subject to moderate spatial gradients. Actually, recent molecular dynamics simulations [
72] have shown this to be an excellent approximation even for very large spatial gradients. But it would not be a reasonable assumption for reacting flows with reaction time scales of the same order as the transport time scales.
3. Constrained Equilibrium Method
In general, the evolution of the state,
, is obtained by solving the differential Equation (
1) for a given initial state
and the exogenous dynamics
. However, the detailed modeling of
may be a formidable task.
Upon gaining a deeper understanding of the dynamics of a system, it is often discovered that many of the governing kinetic mechanisms are of secondary importance, such that the time dependent behavior of the system is relatively insensitive to them. For example the three body reactions dominate the exothermic dynamics of the process of expansion of combustion products within a supersonic nozzle or in an internal combustion engine, while bimolecular reactions are almost equilibrated [
35,
73]. Also, the almost fuel independent burning velocity of the majority of the hydrocarbon fuels suggests the existence of an intermediate constrained-equilibrium path in which the fuel molecule breaks down into smaller fragments, mostly C1, C2 and C3, which could drastically simplify kinetic modeling of laminar flame propagation.
Ideally, by scrutinizing the features of the full endogenous dynamics of a system, we could classify the various governing mechanisms in a hierarchical structure based on their time scale as follows. We would single out all the internal mechanisms that act on a time scale shorter than a given scale
τ, and identify a complete set of functionals, additional to the
’s, that are also time invariant if the internal dynamics of the system is restricted to only those mechanisms that have a time scale shorter than
τ. We denote these additional time-invariant functionals by
and we impose the condition that their gradient vectors,
are linearly independent of the gradients
. Clearly, as
τ increases, the number
of such functionals decreases to reach
. Conversely, as
τ decreases and reaches the limit
, at which time scale no mechanism can be active, the number
increases to reach
where
is the number of state variables in vector
x and
is the number of (
τ-independent) time invariants
.
In principle, one would have to examine a hierarchy of dynamical equations of the form
where
accounts for all the internal mechanisms with time scale shorter than
τ. The functionals
are the additional constants of the motion for
with gradients
that are linearly independent of the gradients
of the constants of the motion of the full internal dynamics
. This means that no mechanism with time scale shorter than
τ is capable of altering the value of the functionals
.
If we are interested in describing the dynamics of the system only on a time scale longer than a certain scale
τ, then we are not interested in the details of the time evolution for time scales shorter than
τ. We may therefore assume that the fast mechanisms, described by
, contribute to the overall evolution by letting the functional
increase in practically no time to the highest value compatible with the instantaneous values of the functionals
—which are constants of the motion for the overall dynamics—and the functionals
—which are constants of the motion for the fast dynamics. Thus, we only need to develop a detailed model of the dynamical mechanisms acting on time scales between
τ and
∞, which we will denote by the symbol
. In other words, we assume that for all practical purposes the system is at all times in some constrained-equilibrium state
that can be found by solving the following constrained maximization problem
The solution
of the maximization problem satisfies the set of Euler-Lagrange equations
which, together with the constraints (
31) and (
32), yields an
-parameter family of constrained-equilibrium states
Again, Equation (
33) imply that, within this family, the function
satisfies the following orthogonality relations
where
and
are the constraint-potentials conjugated to the constraint functionals
and
. Therefore, the relations and geometrical representations discussed in the previous section extend here as well for the constant-
F surfaces of the constrained equilibrium states in
space and for the dual constant-
G surfaces in
space. As a result of this duality, the time dependence can be followed both in the space of the constraints and in that of the constraint-potentials.
3.1. Rate Equations for the Constraints
If the exogenous dynamics,
, operates on a time scale larger than
τ, then we see that the evolution of the system can be described by modeling only those internal mechanisms with time scale longer than
τ. Indeed,
To follow the evolution of the state variable in
space, we assume at each instant in time
where the function
is that found in Equation (
34) as a result of the maximization just discussed.
As a result, the solution of this scheme is based on the set of explicit differential Equations (36) and (37) coupled with the Equations (31)–(33), where the unknown functions of time are , , , , and .
In particular, Equation (37) shows that the only internal mechanisms that determine the rates of change of the values of the functionals
are those acting on time scales between
τ and
∞. Thus, the set of Equations (
36)–(
38) is independent of the details of the dynamical mechanisms whose time scale is shorter than the given
τ.
It is finally noteworthy that Relations (
4) and (
6) impose important restrictions on the expressions of
that can be adopted to model the rate-controlling slow endogenous dynamics of the system. The first condition that we have already used in Equation (
36) is that for every constrained-equilibrium state
,
namely, the functionals
must be time invariants of the endogenous dynamics also in the constrained-equilibrium scheme. The second, more demanding condition is that the state functional
must be non-decreasing under the endogenous dynamics. Setting the exogenous term
in Equation (
36) and (37) and using Relations (
35), this condition requires that at any constrained-equilibrium state
along the assumed time evolution,
where in writing the second equality we used Equation (
33) and Equation (
39), and in the third we used Equation (37).
For example, like in the application of RCCE to chemical kinetics that we discuss in the next section, a fully consistent assumption is to use, in Equation (37),
,
i.e., to use the full internal dynamics (the detailed kinetic scheme) to evaluate the rates of change of the slow constraints once the constrained equilibrium state
is known. Then, the nonnegativity of Equation (
40) is guaranteed in general by the assumed nonnegativity of Equation (
6). See also note [
62].
3.2. Rate Equations for the Constraint-Potentials
Although direct integration of the rate-equations (
36) and (37) for the constraints is relatively straightforward and simple to implement, it has proved to be relatively inefficient and time consuming if, at each time step during the integration, the constrained-equilibrium values (
36) of the state variables corresponding to the values of the constraints are recalculated by solving Equation (
33) using a general purpose maximization code that does not accept as initial guess the composition computed at the previous time step [
52]. For the purpose of an implementation of the method, also an alternative method has been proposed [
53] and implemented [
34,
50,
54,
74] which, instead of the direct integration of the rate-equations for the constraint-potentials, solves the dual problem in terms of an implicit set of rate equations for the constraint-potentials
. A general advantage of this method is the reduction by
(from
to
) of the number of implicit equations that need to be solved during the integration.
For simplicity, let us rewrite the constraints without separating them into two sets, so that (
31) and (
32) are united in the single set of constraints,
and the Euler-Lagrange equations are
and implicitly define the relation
By differentiating Equation (
42) with respect to time and using (
43) in the right-hand side, we may write
Defining for convenience the matrix
multiplying both sides of (
44) by
, summing over
m and recalling that relations (
11) and (
12) are one the inverse of the other, so that
, we obtain
We now use Equation (
1) with
x given by Equation (
43) to evaluate
in Equation (
46), we multiply both sides by
, sum over
j, and after a minor rearrangement and re-inserting all dependences explicitly, we obtain the rate equations
which form a set of
differential equations for the
constraint potentials
.
In summary, the comparison between the dual solution methods presented in this and the preceding section may be clarified by writing the equations to be solved in compact vector notation. The method based on the rate equations for the constraints requires the solution of a set of
explicit differential equations coupled with
explicit equations plus
implicit (usually transcendental) equations, with the forms
for the unknown functions
,
, and
.
The method based on the rate equations for the constraint-potentials requires the solution of a set of
implicit differential equations coupled with
implicit (usually transcendental) equations, with the forms
for the unknown functions
and
.
4. Rate-Controlled Constrained-Equilibrium in Chemical Kinetics of Gas Mixtures
The development of models for describing the dynamic evolution of chemically reacting systems is a fundamental objective of chemical kinetics. As explained in the introduction, the most accurate way of doing this involves: (i) specifying the state and species variables to be included in the model; (ii) developing a detailed kinetic model (DKM) that is comprehensive, hierarchical, and includes all the important reaction pathways governing the state variables; (iii) obtaining accurate rate data for each individual reaction in the DKM either using experimental techniques or using state-of-the-art quantum chemistry methods [
75]; (iv) compiling a “full set” of rate-equations for these variables based on the postulated DKM; and (v) integrating this set of equations to obtain the time-dependent behavior of the system.
For hydrocarbon oxidation, the problem is that the DKMs involving C/H/O/N molecules can easily involve thousands of chemical species and isomers, and tens of thousands of chemical reactions for heavy molecules. Aside from the expensive computational effort required to treat such models in realistic systems involving reacting turbulent flows, one major modeling challenge is to assign reliable rate data to every individual reaction, especially the intermediate reactions involving heavy radicals and branched molecules, for which even the current most accurate quantum chemistry calculations offer factor-of-ten accuracies, and virtually many of which are irrelevant after all.
It is anticipated that the RCCE method, which was originally developed and is mainly understood and applied as a model order reduction technique, could significantly reduce the modeling effort required in developing a DKM. One hidden, not very well received promising potential of RCCE is the ability of bottom-up modeling. One starts with an exhaustive list of species and elemental constraints, which are sufficient to fix the equilibrium state. No kinetic mechanism is required at this stage in the RCCE model development, whereas the minimum number of linearly independent reactions in the corresponding DKM is equal to the number of species only to fix the equilibrium state. The appreciable gain at this very first stage of modeling is more pronounced when heavy molecules including several thousands of species are considered, formation of PAH and soot for example. More constraints and more reactions can be added to improve the accuracy to the desired level. If the only constraints are those imposed by slowly changing state variables, the RCCE method is equivalent to a “shifting” Local Thermodynamic (complete chemical) Equilibrium (LTE) calculation. If the number of constraints in an RCCE model is identical to the number of species in a DKM model, then they are, for all practical purposes, identical.
RCCE fundamentals will be summarized below by starting from the standard treatment of chemical reactions, briefly reviewed in
Section 4.1 to
Section 4.6, and then by focusing on the RCCE formulation for a homogeneous system (
Section 4.7) and on two examples (
Section 5).
4.1. Equilibrium Properties of Non-Reacting Gas Mixtures
In homogeneous, closed-reactor chemical kinetics or RCCE, where the usual frozen chemical equilibrium assumption is made to calculate the thermodynamic properties, the Lyapunov functional is the stable-equilibrium fundamental relation for the entropy
S of a non-reacting system with the instantaneous values of the internal energy
U, the volume
V, and the numbers
of particles of each different species,
where
with
Avogadro’s number and
Boltzmann’s constant. Therefore, the state vector is
. In this context, the components of the gradient vector Γ are the potentials conjugated with the state variables,
where
T is the temperature,
p the pressure, and
the chemical potential or partial molar Gibbs free energy of species
j in the mixture. Under the typical simple-system approximation ([
36], p. 263) whereby the function
is homogeneous of first degree in all its variables, the validity of the Euler relation,
, entails a relation among the potentials, which are, therefore, not all independent of one another. For this reason, it is often more convenient to select specific properties as state variables,
i.e., depending on the context, either
on a molar basis with
and
,
,
,
,
, so that in this context the state vector is
and it is easy to show that the gradient vector is
on a mass basis with
and
,
,
is the density,
and
the mass fraction and molar mass of species
j, and
the mass, so that in this context the state vector is
and it is easy to show that the gradient vector is
where
; or
on a volume basis with
and
,
,
is the concentration of species
j, so that in this context the state vector is
, and it is easy to show that the gradient vector is
This latter case is convenient in non-homogeneous, reacting and non-reacting flows, where fluid mechanics and chemical kinetics or RCCE are combined with the methods of non-equilibrium thermodynamics. The pressure is obtained from the Euler relation,
.
We start, therefore, with a system consisting of different species confined in a volume V, with numbers of particles, denoted by , sufficiently large so that the simple system approximation applies.
For a non-reacting mixture in the absence of external fields, the stable-equilibrium-state fundamental relation for a simple system with volume as the only parameter has the form
where the subscript “off” is a reminder [
76] that the functional relation is that of a non-reacting mixture (all reactions “turned off”).
The Gibbs relation, which is the differential of (
61),
allows us to write the rate equation which relates the rates of change of entropy, energy, volume, and composition when the system is modeled as evolving along a continuous time sequence of states, each of which would be a stable equilibrium state if the all reactions were inhibited. Introducing, for convenience, the “dimensionless entropy”
, the “entropic temperature”
β, and the dimensionless “entropic chemical potential”
of species
j,
and assuming that
U,
V, and the
’s are functions of time, and at any instant in time the entropy is given by Equation (
61), it is easy to see that Equation (
62) implies the following relation among the time rates of change,
When applied to an infinitesimal control volume in a flow field, the set of assumptions which lead to Equation (
64) constitutes the so-called “local equilibrium” assumption of fluid mechanics. In this and the following sections, instead, we restrict the treatment to the simpler case of a homogeneous system. But it is important to note that the “shifting equilibrium” assumption is adopted also when we deal with reacting mixtures, for which the thermodynamic properties are assumed to be those of a non-reacting mixture at stable (frozen) equilibrium with the same energy, volume, and composition.
4.2. Balance Equations for Species, Energy, and Entropy
Let us now write the rate equations expressing the species, energy, and entropy balances, using the following general notation
where
,
, and
represent the net rates of outflow (inflow if negative) of species, energy, and entropy due to interactions across the system’s boundaries such as heat, bulk flow, diffusion, and work other than the expansion (or compression) work (which is accounted for explicitly in view of the “shifting equilibrium” assumption). Moreover,
, Φ, and
σ represent the net rates per unit volume of “generation within the system” of particles of type
j, internal energy, and entropy due, respectively, to chemical reactions, viscous dissipation, and irreversibilities.
Substitution of Equations (65)–(67) in Equation (
64) yields the following expression for the rate of entropy generation by irreversibility per unit volume,
where
and, of course,
.
The principle of entropy non-decrease, which follows from the Second Law, imposes that. The reason why we write the stronger condition that , , and must be independently non-negative, is to be found in the Curie principle of nonequilibrium thermodynamics, whereby chemical reactions, which are scalar processes, cannot couple to heat and mass fluxes, which are vectorial, nor to momentum transfer, which is tensorial.
4.3. Complete Chemical Equilibrium
If the composition is allowed to vary subject only to conservation of the atomic nuclei present in the initial composition
, thereby including the formation mechanisms of every possible chemical species that can be conceivably assembled with the given atomic nuclei, then the corresponding stable-equilibrium composition is the so-called “complete chemical equilibrium state”. For a given value of the energy
U, the volume
V, and the initial numbers of particles
of the different species, the complete chemical equilibrium state is found by maximizing the entropy subject to the following constraints, which express conservation of the numbers of atomic nuclei of each kind that of course no chemical reaction mechanism can change,
where
is the number of nuclei of type
i in one molecule of species
j,
is the number of elemental species which can be formed with the given initial set of particles, and the superscript
∞ is used here because we will see below that in RCCE these parameters appear in elemental constraints of the model.
For a closed system, the value of each is determined once and for all by the “initial” composition , and it cannot be altered by the chemistry.
In terms of the terminology of
Section 2 and the examples discussed at the beginning of
Section 4.1, to proceed without making further modeling assumptions, it is convenient to adopt the state variables
. Therefore, the complete chemical equilibrium state is obtained by maximizing the function
as given by Equation (
53) subject to given values of
U and
V and the constraint Equation (
72). Introducing multipliers
,
, and
for each constraint, and maximizing the Lagrangian
yields the necessary conditions
Because in general
, the latter conditions imply the relations
which can be inverted at fixed
U and
V to give
When these are inserted in the constraints (
72), the resulting relations can be inverted at fixed
U and
V to yield
which, in turn, inserted into Equation (
78) yields the complete chemical composition corresponding to the given values of
U,
V, and initial number of nuclei
,
So far, no specific assumption has been made regarding the relations between the stable-equilibrium properties of the mixture and those of the component species when pure. This is where further assumptions, for example to model a particular non-ideal mixture behavior valid in dense gas, liquid phase, or phase change, should be considered as additional “elemental” constraints of the model. In the next section, we exemplify this by assuming an ideal mixture of ideal gases, so as to recover the standard treatment of chemical kinetics and the original formulation of RCCE.
It is noteworthy that because of conditions (
74) and (
75), the term
in the right-hand side of the Lagrangian (
73) coincides with
where
G is the Gibbs free energy. Therefore, maximizing
subject to given values of
U and
V is equivalent to minimizing the dimensionless Planck function
. Thus, again, for non-ideal mixture behavior, we must proceed by adopting one of the many model expressions available in the literature (see, e.g., [
77,
78]) for the Gibbs free energy
G or the Helmholtz free energy
as functions of the other state variables.
4.4. Complete Chemical Equilibrium for Ideal Gas Mixtures
Let us now make and discuss the following standard assumptions in the treatment of ideal gas mixtures: (i) simple system model; (ii) Gibbs-Dalton ideal mixture behavior; (iii) ideal gas behavior for each of the component species when pure and at the same temperature of the mixture, in the entire range between the standard reference pressure and the pressure of the mixture.
The simple system model ([
36], p. 266) implies the validity of the Euler relation which can be written either as
or as
The Gibbs–Dalton ideal mixture model ([
36], p. 481) implies the following relations for the mixture properties
S,
U,
V, and the pure substance properties of the component species,
where
denotes the partial pressure of species
j in the mixture and
,
, and
the mole specific properties of pure species
j, where the double-
j subscript is a reminder that these properties refer to the species
j when pure. Under this model, the Dalton law applies,
.
Finally, assuming ideal gas behavior,
, for every species
j and all values of
β and
p, then the partial pressures are given by
, and Equations (82)–(84) may be written in the form
where
is the standard reference pressure. We further note that, in our notation, the relations
,
translate into
so that we may rewrite Equation (
85) as
As a result, a natural set of state variables in this context is
and so the complete chemical equilibrium state is obtained by maximizing the function
as given by Equation (
85) subject to the constraints given by Equations (
72), (86), and (87). Introducing multipliers
,
, and
for these constraints, and maximizing the Lagrangian
yields the necessary conditions
where we made use of the relations
,
,
, where of course
and
. Using Equation (87) to eliminate
N in Equation (
94), finally yields the relation
Equations (
72), (86), (87), and (
95) form a system of
implicit equations which, for given values of
U,
V, and the
’s, can be solved to yield the complete chemical equilibrium values of the
state variables
β,
p,
, and the
constraint-potentials
.
If the values of the energy U, the volume V, and the number of atomic nuclei of each type, , are allowed to vary in time, the system will evolve through a sequence of complete-equilibrium states at a rate controlled by the imposed rates. This is the well-known case of “shifting complete chemical equilibrium”, already denoted by LTE.
For completeness, it is convenient to recall and write in more standard notation, that each (dimensionless)
in Equation (
95) is computed from the molar Gibbs free energies of formation of the
j-th species at standard conditions and the temperature dependence of its molar specific heat capacity at constant pressure
, via the relation
Similarly each molar specific internal energy
may be computed via the relation
4.5. Chemical Equilibrium of Ideal Gas Mixtures with Respect to a Set of Reactions
If the composition is allowed to vary according to one or more chemical reaction mechanisms, the maximization is to be done for specified values of
U and
V and the allowed stoichiometric variations in composition. So, let us assume that the DKM is based on the following
reaction mechanisms,
where
denotes the chemical symbol of the
j-th species, and the net stoichiometric coefficients
satisfy the atomic nuclei conservation identities
where again
is the number of nuclei of type
i in one molecule of species
j.
As a result of this assumption, the term
in the species balance equation (
65), which expresses the rate of formation (consumption, if negative) of particles of species
j due to the allowed chemical reaction mechanisms, must satisfy the so-called proportionality relations
where
is the so-called coordinate or degree of advancement of the
ℓ-th reaction.
The Second Law requires that at stable equilibrium the entropy be maximal within each set of states that have the same values of the energy
U and the volume
V, and compositions that are compatible (through the allowed reaction mechanisms) with the same initial composition
. The set of compositions compatible with
is the
r-parameter family of compositions found by integrating in time Equation (
65) with
and with
given by Equation (
100). This integration, if we set for convenience
, yields
Equation (
101) together with the additional trivial restriction
defines through the
r parameters
the family of compatible compositions over which the entropy maximization is to be restricted. Inserting this restriction into Equations (
85)–(87), they become,
Therefore, a natural set of state variables in this more constrained context is
and so the chemical equilibrium state is obtained by maximizing the function
as given by Equation (
102) subject only to the constraints (103) and (104). In fact, we note that the element-conservation constraint (
72) is automatically satisfied because the stoichiometric coefficients satisfy Equation (
99).
Introducing multipliers
and
for the two constraints, and maximizing the Lagrangian
yields the necessary conditions
,
as in Equation (
92) and Equation (
93), and
where we used
which follows from Equation (
101).
By defining the dimensionless equilibrium “constant” of reaction
ℓ at entropic temperature
β,
and
, Equation (
107) takes the familiar form in terms of the species concentrations
, namely, at equilibrium,
which is the well-known law of mass action. The right-hand side of Equation (
109) defines the so-called equilibrium constant based on concentrations,
whose dimensions depend on the value of
.
We finally note that the potential
conjugated to the reaction coordinate
,
is the dimensionless form of the entropic affinity of reaction
ℓ,
i.e., with the notation of [
76],
and with the usual notation
where
is the de Donder affinity of the reaction.
For the discussion in the next section, it is important to note that
may be expressed, using Equation (
102), as
Therefore, the chemical equilibrium condition (
109) is equivalently expressed by the condition
,
i.e., that the affinity of every reaction vanishes at equilibrium.
However, the entropic affinity
is well defined also off chemical equilibrium. In fact, inserting Equations (
100) in Equation (
69) and using Equation (
111), the rate of entropy generation due to the
r allowed chemical reactions, may be written as
In the next section, we show that under the standard kinetic modeling assumptions, the Second Law requirement that
be positive semi-definite implies that
can be interpreted as a (dimensionless) measure of the degree of disequilibrium with respect to the
ℓ-th reaction.
4.6. Entropy Production and Detailed Balance
The rate of change of reaction coordinate
per unit volume is usually called the net rate of the
ℓ-th reaction, denoted by
and expressed as the difference between the forward and reverse reaction rates,
So, the entropy generation rate density is also given by
For ideal gas mixture behavior, the standard model in chemical kinetics is based on the following expressions for the forward and reverse (positive) reaction rates
where
and
are the so-called (positive) rate constants of the reaction.
Recalling that
and taking the ratio of Equations (117) and (116) yields the relation
which is valid both at and off equilibrium. Inserting this relation in Equation (
112), we obtain, in general,
which inserted in Equation (
115) yields, in general,
Let us rewrite the expressions just obtained, in terms of two useful parameters. The first, suggested by one of us [
53] as a measure of the departure of reaction
ℓ from equilibrium, is the “degree of disequilibrium”
The second parameter, which we will see can be interpreted as the “degree of departure from detailed balance”, is
Clearly, Equation (
119) and Equation (
120) rewrite as
So far, we made no assumption about the functional dependence of the rate constants and , and hence of , on the state variables.
Close to chemical equilibrium, in the limit of vanishing net reaction rates
,
i.e., if
for every
ℓ, the affinities must approach their vanishing equilibrium values,
i.e.,
for every
ℓ, therefore Equation (
123) implies
, which is the so-called rate quotient law or detailed balance condition, relating the ratio of the forward and reverse rate constants for every reaction to the corresponding equilibrium constant,
In general, away from chemical equilibrium, by Equation (
122) and Equation (
123) we have
and the question of the validity of the detailed balance condition
can be addressed in various ways [
79], mainly depending on the modeling assumptions about the functional dependence of the rate constants
and
. However, the most general and restrictive condition for the thermodynamic consistency of any set of modeling assumptions is that the expression (
124) for
be positive semi-definite over the entire range of compatible compositions, no matter how far from chemical equilibrium.
For example, let us assume that the rate constants exhibit neither cross effects nor any dependence on the affinities, so that
for every
ℓ and
m. Then it follows that
is a necessary and sufficient condition for
to be positive semi-definite. That the condition is sufficient is a straightforward consequence of the nonnegativity of the function
. One way to prove that the condition is also necessary is to require that the minimum value of
given by (125) with respect to variations of Γ be nonnegative (it actually must be zero, since at equilibrium the entropy production must be zero). Denoting by
the location of the minimum, where
for every
ℓ, it is easy to show that
For this to be nonnegative, we must have
for every
ℓ, which inserted back into the condition for the minimum,
, yields
for every
ℓ, that is,
. An equivalent more direct proof obtains by differentiating Equation (
123) with respect to
under the assumed independence of
on the affinities. This yields
which, together with the condition that at chemical equilibrium both
and
, implies
and, hence,
.
As a result we find that the detailed balance condition,
holds also off equilibrium. This means that if the forward rate constant
is assumed, as typical, to be a function of temperature and pressure only, then Equation (
129) implies that also the reverse rate constant
must be a function of temperature and pressure only, with
independent of pressure.
Conversely, if we assume that the forward and reverse rate constants
and
are functions of temperature and pressure only, with
independent of pressure,
i.e., we assume that no matter how far from the chemical equilibrium composition,
is a function of
β only, then because of Condition (
126) such function must be equal to
implying that
at all compositions.
In summary, any set of assumptions about the dependences of the rate constants that implies the general validity of detailed balance off-equilibrium implies also that the expressions for the rate of entropy generation per unit volume reduce to the following equivalent positive semi-definite forms
We finally note that sometimes the term “detailed balance” is used with a different meaning, to express the condition of equality of the forward and reverse reaction rates, i.e., in our language, to the absence of disequilibrium, . However, it is clear that close enough to equilibrium .
4.7. Rate-Controlled Constrained-Equilibrium Formulation for a Closed System
The Rate-Controlled Constrained Equilibrium (RCCE) model assumes a time evolution through a sequence of states each maximizing the entropy subject not only to the constraints that define the complete chemical equilibrium state (
Section 4.3 and
Section 4.4), but also to a set of additional kinetically-controlled constraints. The number of additional constraints may be large or small.
At one extreme is the case that we call Detailed Model (DM) with the largest possible number of additional constraints. These are the number of particles of a selection of species among those considered in the underlying DKM scheme. The additional time-dependent constraints are ,..., , so that the evolution of the system is controlled by the rate-equations for the species, plus of course the prescribed time changes of U and V. DM and DKM differ in that once we have the DM calculation, then we may at once calculate the amounts of an augmented set of species, including any other species which can be made from the given initial nuclei in the system, even if not included in the underlying DKM scheme.
However, the RCCE method is most interesting when it effectively reproduces DKM results with a number of time-dependent constraints that is of course higher than in LTE but much smaller than in DM. So far, RCCE calculations have been based on identifying slow varying functionals
that are linear in the species number of moles
, and therefore can be written in the form
where
represents the marginal impact of a change in
onto changing the value of the slowly varying functional. The rate of change of the value
is obtained, for the closed system, by using the rates of change of the amounts
which result from using, in the rate-equations (
100), the reaction rates
known from the DKM scheme.
Because they have identical linear structure, we may formally combine Equation (
72) and Equation (
133), into a single set of
constraint equations,
So, the maximization procedure that determines the concentrations at time
t is formally identical to that outlined in
Section 4.3 except that the number of constraints here is higher,
instead of
. The resulting concentrations are given therefore by the expressions
which differ from Equation (
95) only in the absence of the superscript
∞, in accordance with the new, larger set of constraints (
134).
Equations (89), (87), (
134), and (
135) form a system of
implicit equations which can be solved for given values of
U,
V, and the
’s, to yield the constrained equilibrium values of the
state variables
β,
p,
, and the
constraint-potentials
.
As the values of the energy U, the volume V, the number of atomic nuclei of each type, , and the slowly varying constraints are allowed to vary in time, the system will evolve through a sequence of constrained-equilibrium states at a rate controlled by the imposed rates.
In view of their particular functional dependences and because the constraint gradients
are constants, the RCCE equations can be integrated more efficiently by rewriting them as rate equations for the constraint-potentials and making use of the species and energy balance Equations (
65) and (66), and the kinetic Equations (
100), (
114), (
116), and (117) that determine the rate of change of the values
of the constraints. By differentiating Equations (86), (87), (
134), and (
135) with respect to time, after few rearrangements corresponding to the general procedure outlined in
Section 3.2 for the general case, we obtain the following set of rate equations
where
These
implicit differential equations together with the
Equation (
135) can be solved for given values of
,
, and the
, to yield the
state variables
,
, and
, and the
constraint-potentials
.
It is finally important to note that the thermodynamic-consistency condition (
40), that the entropy generation rate be non-negative definite under the endogenous dynamics, is always satisfied, because in evaluating the rates
we use the full internal dynamics (the detailed kinetic scheme), with forward and reverse rate constants
and
that depend on temperature and pressure only, a condition that we have seen in
Section 4.6 warrants the nonnegativity of the entropy generation rate. As previously noted (see also [
53,
76]), whether or not there are kinetically-controlled constraints in addition to element conservation, all conceivable reactions contribute to the rate of entropy production. However, in the sum in Equation (
120), the contribution of the non-constraint-changing reactions is through the adjustments in the chemical affinities of the constraint-changing reactions.
5. Selection of Constraints in Combustion Examples
The careful selection of constraints is the key to the success of the RCCE method. In general, the constraints must (i) include the elemental constraints; (ii) be linearly independent combinations of the species molar concentrations; (iii) constrain the system in the specified initial state; (iv) constrain global reactions in which reactants or intermediates go directly to products; and (v) determine the energy and entropy of the system within experimental accuracy. In addition, they should reflect whatever information is available about rate-limiting reactions which control the evolution of the system on the time scale of interest.
Under broad conditions in the gas phase, two important structural constraints are the total number of moles and the amount of free valence in a reacting system. The former is controlled by slow three-body dissociation/recombination reactions and the latter by reactions which make and break valence bonds. In connection with the ideas presented in
Section 2 and
Section 3, we observe that the total number of moles is a constant of the endogenous dynamics within the class of bimolecular reactions. Three-body reactions are generally slow in the endothermic direction because of the high activation energies required, and in the exothermic direction because of small three-body collision rates and small radical concentrations. They impose slowly varying constraints on the total molar concentration
N and the free valence of the system. We denote these two constraints by the symbol M and FV, respectively. A finite value of FV is a necessary condition for chemical reactions to proceed.
A third important structural constraint, accounting for slow O−O bond-breaking reactions, is the free oxygen, FO, defined as any oxygen atom not directly bound to another oxygen atom. An increase in FO is a necessary condition for the formation of the major reaction products of hydrocarbon oxidation, HO, CO and CO. Two additional constraints which slightly improve the agreement between RCCE and DKM calculations under some conditions are: OHO ≡ OH + O and DCO ≡ HCO + CO. The OHO constraint is a consequence of the relatively slow constraint-changing reaction RH + OH ↔ HO + R coupled with the fast reaction RH + O ↔ OH + R which equilibrates OH and O. The DCO constraint is a consequence of the slow spin-forbidden reaction CO + HO↔ CO + OH coupled with the fast reaction HCO + O↔ CO + HO which equilibrates HCO and CO.
For systems involving the elements C, H and O, the five nontrivial constraints M, FV, FO, OHO, and DCO are independent of the initial reactants and may, therefore, be considered structurally “universal” constraints. Along with the three equilibrium reactions, H + O ↔ OH + H, H + HOO ↔ HO + H, and HCO + O↔ CO + HO, they are sufficient to determine the constrained-equilibrium mole fractions of the 11 major hydrocarbon combustion products (H, O, OH, HO, H, O, HO, HO, HCO, CO and CO) under both high and low temperature conditions.
For hydrocarbon oxidation, four additional fuel-dependent constraints have proved to be relevant [
34]. The first is a constraint on the fuel, FU, imposed by slow hydrogen-abstraction reactions of the type FU + O
↔ FR + HO
and even slower dissociation/recombination of the type AB + M ↔ A + B + M. This constraint is necessary to hold the system in its initial state. The second is a constraint on fuel radicals, FR, which is necessary to prevent the equilibration of forbidden global reactions such as CH
+ 2O
+ 2H
O ↔ CO
+ 2H
O
+ H
+ H which would otherwise convert fuel radicals directly to major products. The third is a constraint on alkylperoxides, APO ≡ CH
OOH + CH
OO + CH
OOH, imposed by slow reactions which convert APO to hydroperoxides coupled with fast reactions which equilibrate the species comprising APO. The fourth is a constraint on alcohol plus formaldehyde, ALCD ≡ CH
OH + CH
O + CH
OH + CH
O imposed by relatively slow reactions which generate/remove ALCD coupled with fast reactions which equilibrate the species comprising ALCD.
The following two combustion examples show typical RCCE results under the constraints discussed above, in comparison with the corresponding DKM calculations.
Given the fact that the internal dynamics of the system and the controlling constraints depend, in general, upon the initial distance of the system from its chemical equilibrium state, we consider two extreme cases below: close-to and far-from equilibrium systems.
5.1. Example 1. Close-to-Equilibrium Oxygen-Methane Combustion
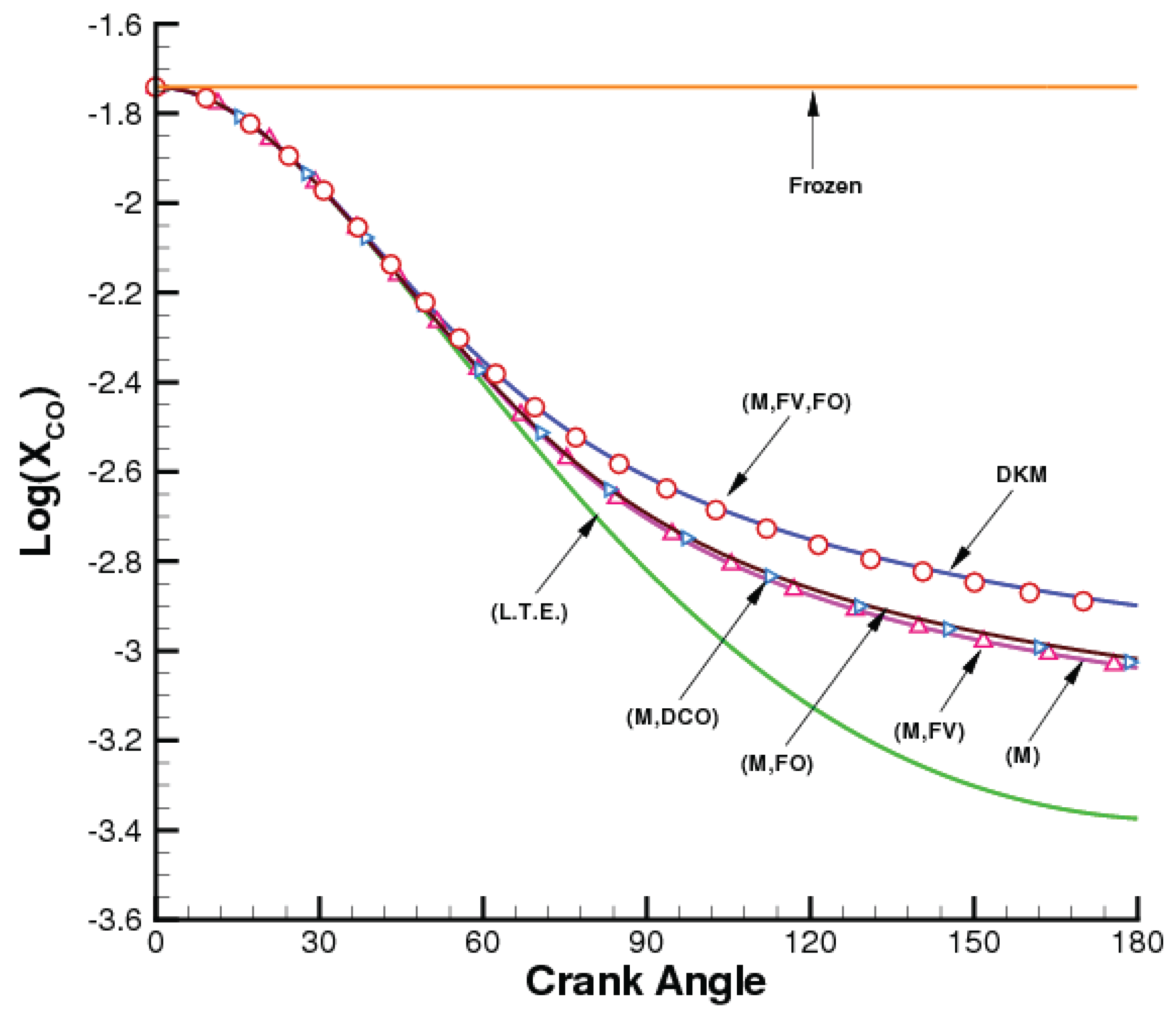
The constraints defined in the previous section are particularly useful for modeling systems driven away from an initial equilibrium by fast expansions and compressions. They are useful both to increase the speed and efficiency of calculations and to provide insight about the most important reaction mechanisms. An example of this is shown in
Figure 1 where RCCE calculations of the CO mole fraction during the expansion of combustion products in an internal combustion engine are compared with a detailed (DKM) calculation [
35]. RCCE calculations are carried out using rate equations for the constraint-potentials.
Figure 1.
(Color online) CO mole fractions as a function crank angle during the expansion stroke of an internal combustion engine.
Figure 1.
(Color online) CO mole fractions as a function crank angle during the expansion stroke of an internal combustion engine.
RCCE calculations involving only energy and volume constraints are shown by the curve labeled LTE,
i.e., the shifting equilibrium case. The DKM calculation includes 29 species and 132 reactions abstracted from the GRI-3 mechanism [
80] and the compilations in [
81,
82]. It can be seen that the slow rate of expansion during the early stages of the power stroke results in local adjustment of composition to LTE predictions. However, as the rate of expansion increases, the LTE prediction departs significantly from the DKM prediction, due to the finite rates of some of the kinetic mechanisms that determine the relaxation process. The LTE results at the end of the expansion stroke are lower than the DKM results by about an order of magnitude. Also shown in
Figure 1 is the choice-of-constraints dependence study of the RCCE predictions. Consistently with the Le Ch
telier-Braun principle, the shift of the internal dynamics towards the exothermic direction due to the cooling effect of expansion makes the recombination reactions an essential part of the energy-restoration dynamics. Just constraining the total number of moles of gas, M, shows a significant improvement with respect to the LTE predictions. Adding constraints one at a time shows that only a subset of structural constraints, namely M, FV, and FO, are sufficient to capture the nonequilibrium dynamics during expansion. Again, we note that the constraint-potentials obtained from the RCCE calculations can be used to obtain mole fractions not only for the 29 species in the detailed model but also for any species involving the same elements and for which the thermodynamic properties are known.
5.2. Example 2. Far-from-Equilibrium Oxygen-Methane Combustion
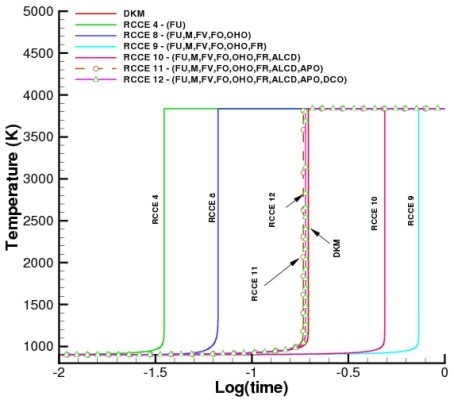
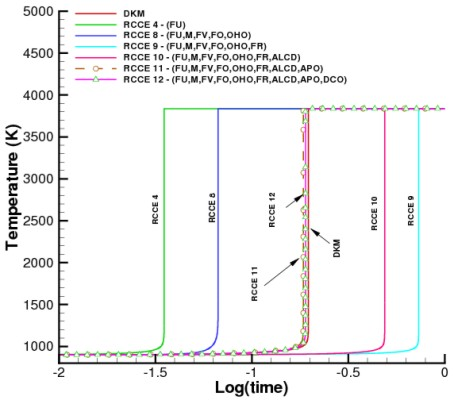
For far-from-equilibrium systems, it is necessary to use the fuel as a constraint to fix the initial state of the system. The fuel constraint together with the elemental constraints are sufficient to guarantee that the system will go to the correct final equilibrium state. However, to obtain the correct time evolution of the system, additional constraints may be needed and the accuracy of the results depends on the careful selection of these added constraints.
This case is illustrated in
Figure 2 which shows the predicted temperature profiles when constraints are added one at a time for a homogeneous stoichiometric mixtures of CH
/O
in a constant volume adiabatic chamber. The initial temperature is 900 K and the initial pressure 10 atm. The DKM calculations include 132 reactions and 29 species, and are again based on the GRI-Mech 3.0 and the other compilations previously cited. The minimum number of reactions needed in the RCCE model, however, depends on the number and the type of constraints. Nevertheless, since all such calculations involve elemental constraints, the constrained equilibrium concentration of all 29 species can be obtained. The improvement in the prediction of the temperature profile can be seen to steadily increase over the entire range of conditions studied as additional constraints are included in the model.
In general, the accuracy of RCCE calculations improves as the number of constraints increases. In the limit where the number of constraints approaches the number of species in the DKM, the RCCE and DKM calculations become almost equivalent, but RCCE allows to evaluate concentrations also of species not included in DKM.
Although strongly supported by its general thermodynamic consistency, the experience and intuition dependent “art” of choosing the most appropriate constraints for an RCCE calculation may not be as effective when experimental data and reliability of the DKM are scarce. In such cases, we suggest that systematic studies of the DKM based on the tools of dynamical systems theory, such as the eigenvalue analysis of the local Jacobians [
83] and the construction of the slow one-dimensional invariant manifold (as done in the SIM, ILDM, CSP, MIM, G and other schemes cited in the Introduction), could provide useful hints for the identification of physically meaningful structural constraining functions that maintain their rate-controlling capabilities for a broad class of problems, independently (or for a broad range) of initial thermodynamic conditions.
Figure 2.
(Color online) Constraint-dependence study of RCCE predictions of the temperature profile during the oxidation of stoichiometric methane-oxygen mixtures at K and atm in a constant volume adiabatic chamber.
Figure 2.
(Color online) Constraint-dependence study of RCCE predictions of the temperature profile during the oxidation of stoichiometric methane-oxygen mixtures at K and atm in a constant volume adiabatic chamber.

{kind=link}
{kind=link}
{kind=link}

