
iDoRNA: An Interacting Domain-based Tool for Designing RNA-RNA Interaction Systems

 ,
,
Abstract
:
1. Introduction
2. Methodology
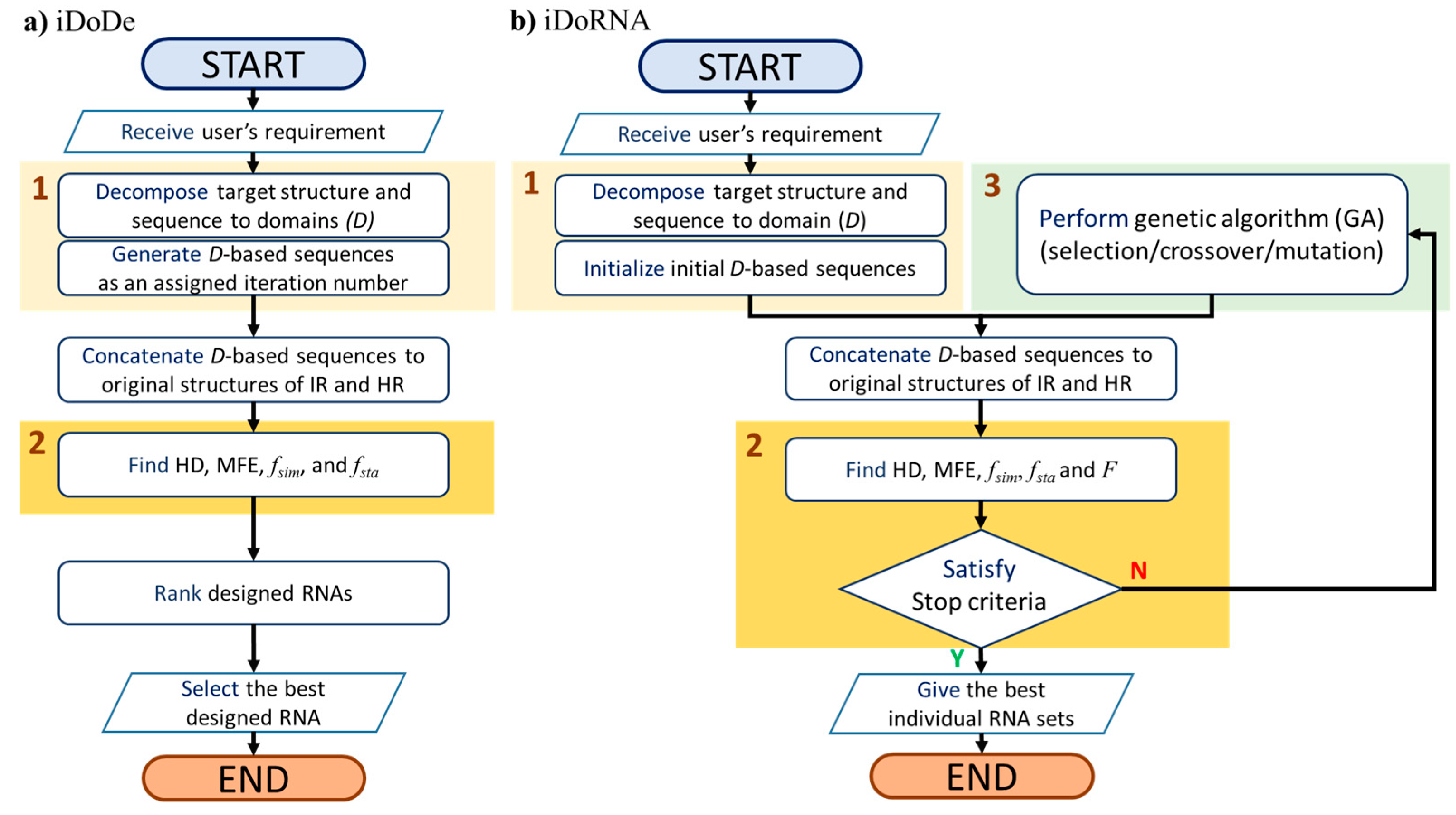
2.1. Description of the Algorithm of iDoRNA
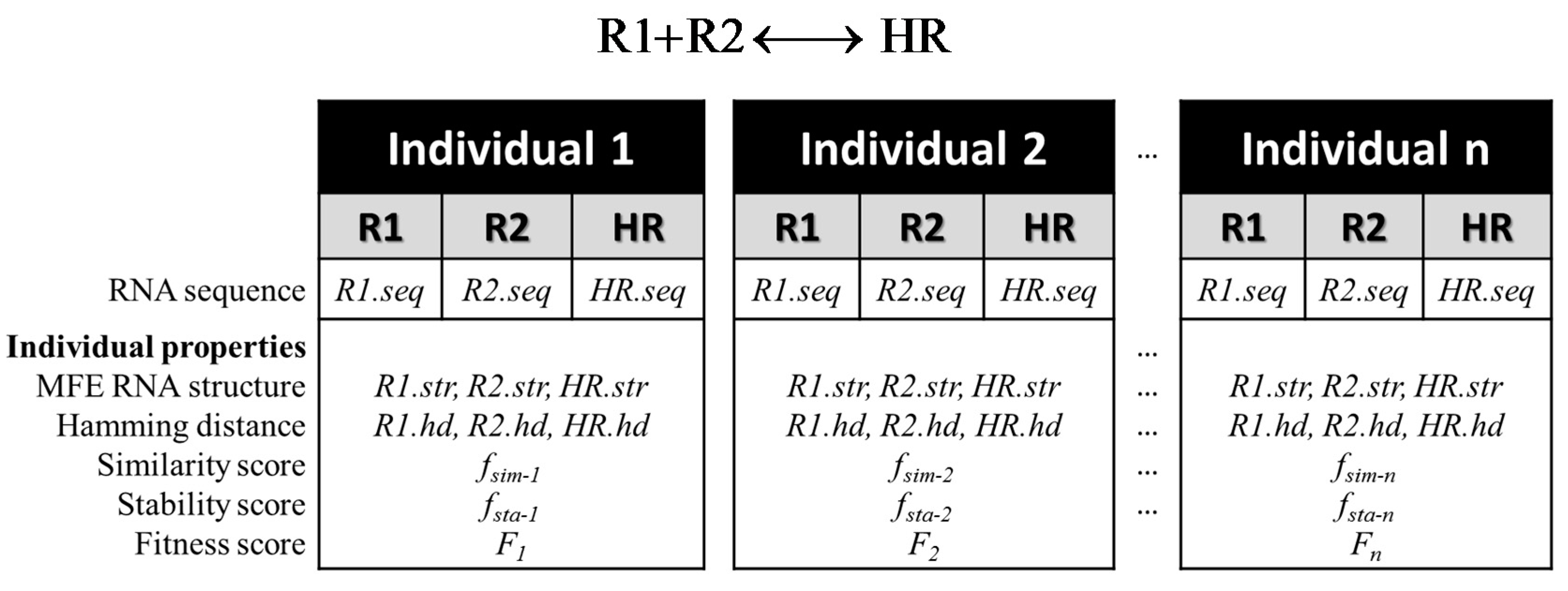
2.1.1. Representation of an RNA Individual
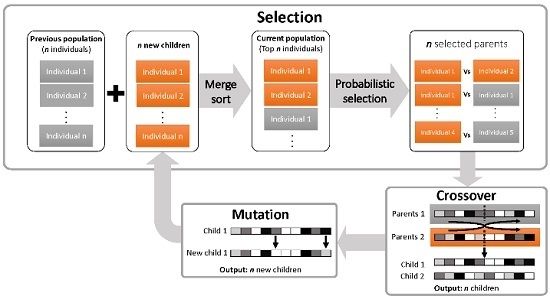
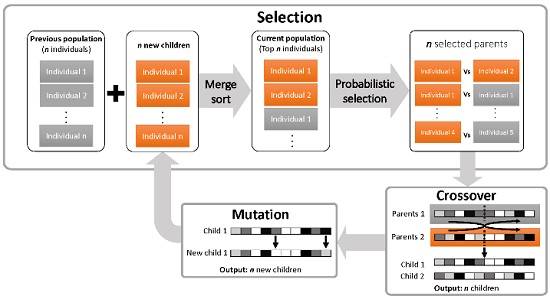
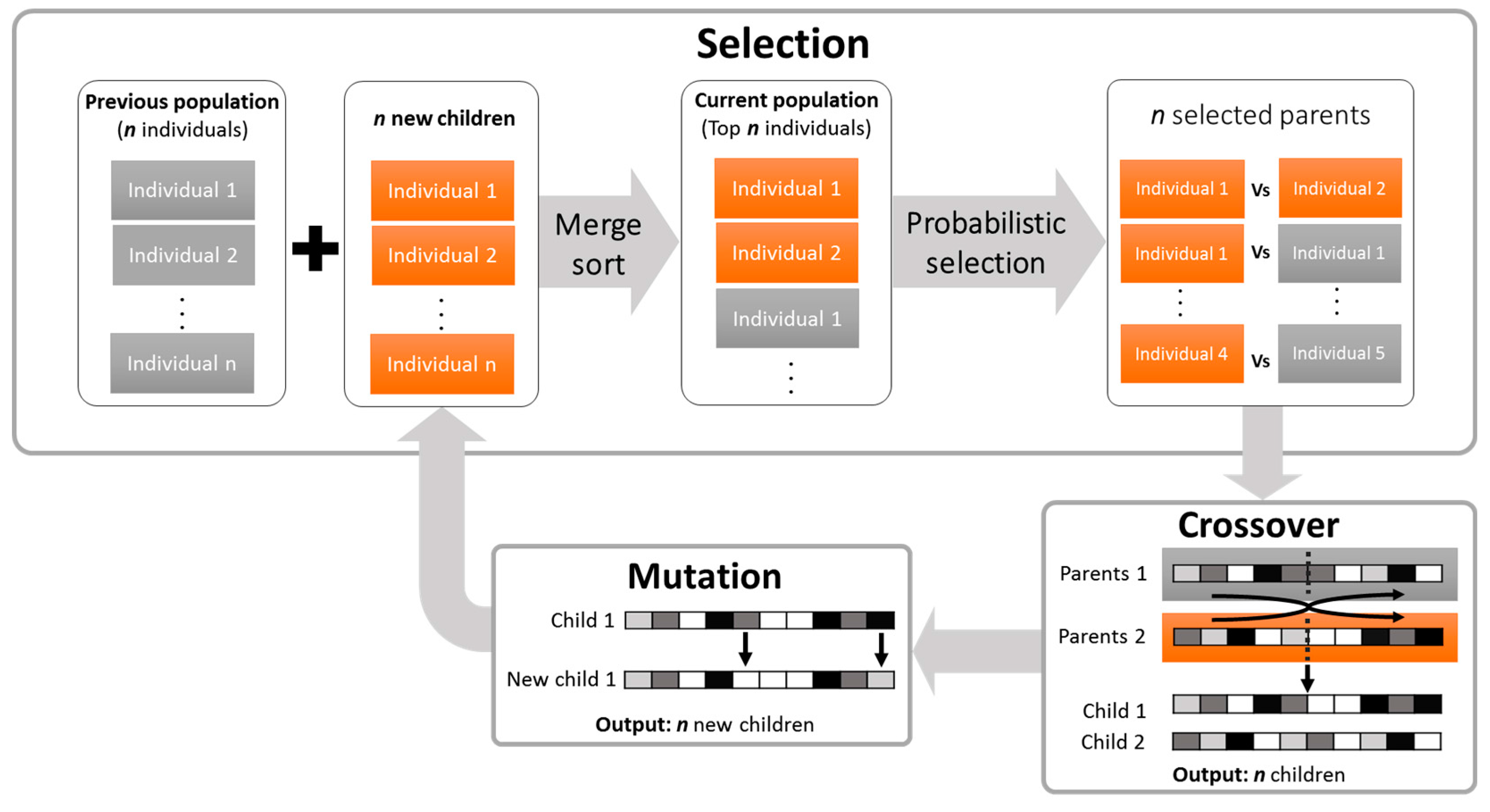
2.1.2. The iDoRNA Algorithm
Initialization
Evaluation
| Target HR structure: | .....((((((.....))))))(((((((((((&............))))))))))) |
| HR.str: | .....((((((.....))))))(((((((((((&............))))))))))) |
| HR.hd = 4 | 000000000000000000000000000000000-00000000000000000000000 |
Reproduction

2.2. Parameter Optimization
2.3. Performance Assessment
3. Results and Discussion
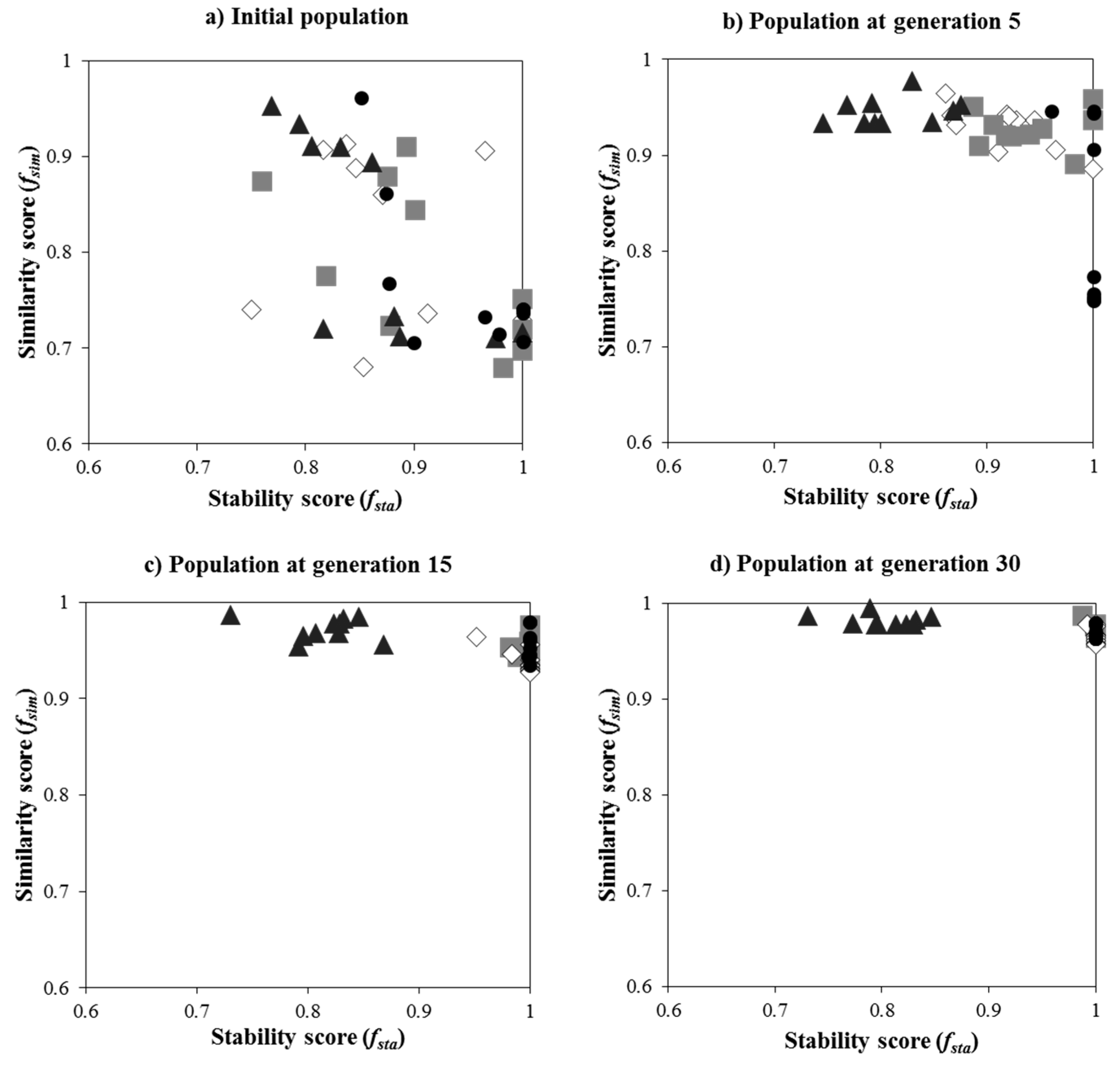
3.1. Suitable Parameters for iDoRNA
3.1.1. Effect of the Weighting Factor of Similarity
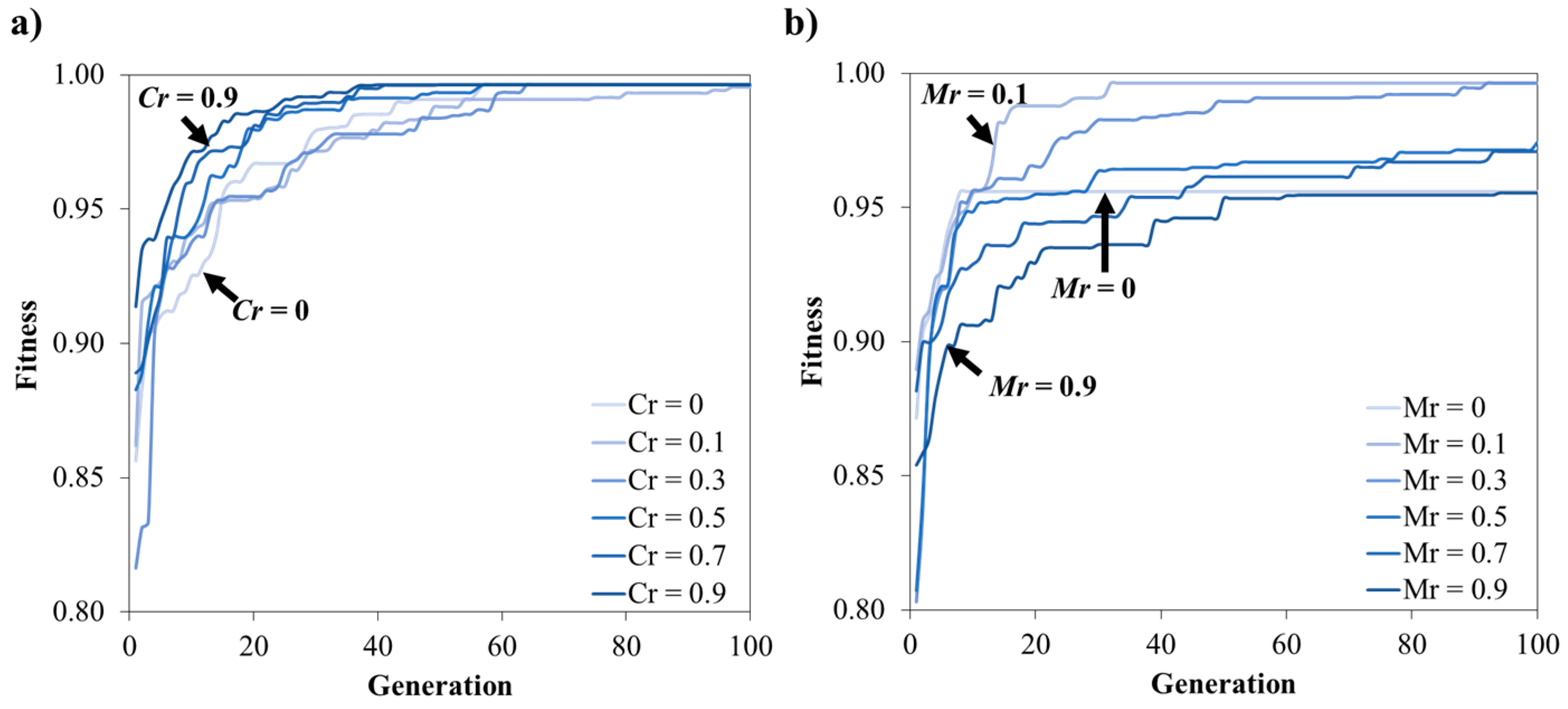
3.1.2. Effect of the Crossover and the Mutation Rates
3.1.3. Effect of the Population Size
3.2. Design Performance of iDoRNA
3.3. Comparison of the Design Performance between iDoDe and iDoRNA
3.4. Comparison of the Design Performance between iDoRNA with the other Design Tools
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Isaacs, F.J.; Dwyer, D.J.; Collins, J.J. RNA synthetic biology. Nat. Biotech. 2006, 24, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Storz, G. Regulatory RNAs in bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Inoue, T. Synthetic biology with RNA motifs. Int. J. Biochem. Cell Biol. 2009, 41, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Jung, G.Y. Synthetic regulatory RNAs as tools for engineering biological systems: Design and applications. Chem. Eng. Sci. 2013, 103, 36–41. [Google Scholar] [CrossRef]
- Xie, Z.; Wroblewska, L.; Prochazka, L.; Weiss, R.; Benenson, Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 2011, 333, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef]
- Van der Meer, I.M.; Stam, M.E.; van Tunen, A.J.; Mol, J.N.; Stuitje, A.R. Antisense inhibition of flavonoid biosynthesis in petunia anthers results in male sterility. Plant Cell 1992, 4, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Callura, J.M.; Dwyer, D.J.; Isaacs, F.J.; Cantor, C.R.; Collins, J.J. Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. Proc. Natl. Acad. Sci. USA 2010, 107, 15898–15903. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.; Fontana, W.; Stadler, P.; Bonhoeffer, L.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte fur Chemie 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Andronescu, M.; Fejes, A.P.; Hutter, F.; Hoos, H.H.; Condon, A. A new algorithm for RNA secondary structure design. J. Mol. Biol. 2004, 336, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Backofen, R. INFO-RNA—a server for fast inverse RNA folding satisfying sequence constraints. Nucleic Acids Res. 2007, 35, W310–W313. [Google Scholar] [CrossRef] [PubMed]
- Taneda, A. Modena: A multi-objective RNA inverse folding. Adv. Appl. Bioinform. Chem. 2011, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.; Wolfe, B.; Pierce, N. Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 2011, 32, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, J.A.; Clote, P.; Dotu, I. RNAifold: A constraint programming algorithm for RNA inverse folding and molecular design. J. Bioinform. Comput. Biol. 2013, 11, 1350001. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Breaker, R.R. Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 2004, 5, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P. Toward reprogramming bacteria with small molecules and RNA. Curr. Opin. Chem. Biol. 2007, 11, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Neupert, J.; Karcher, D.; Bock, R. Design of simple synthetic RNA thermometers for temperature-controlled gene expression in escherichia coli. Nucleic Acids Res. 2008, 36, e124. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yamada, T.; Ui-Tei, K.; Morishita, S.; Saigo, K. Sidirect: Highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Res. 2004, 32, W124–W129. [Google Scholar] [CrossRef] [PubMed]
- Henschel, A.; Buchholz, F.; Habermann, B. Deqor: A web-based tool for the design and quality control of siRNAs. Nucleic Acids Res. 2004, 32, W113–W120. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Park, S.M.; Choi, Y.C.; Lee, D.; Won, M.; Kim, Y.J. Asidesigner: Exon-based siRNA design server considering alternative splicing. Nucleic Acids Res. 2008, 36, W97–W103. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Ciais, D.; Lajaunie, C.; Charbonnier, P.; Foveau, N.; Vert, J.-P.; Vandenbrouck, Y. Dsir: Assessing the design of highly potent siRNA by testing a set of cancer-relevant target genes. PLoS One 2012, 7, e48057. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-C.; Pan, W.-J.; Chen, C.-W.; Chen, Y.-T.; Chu, Y.-W. Desi: A design engine of siRNA that integrates SVMs prediction and feature filters. Biocatal. Agric. Biotech. 2012, 1, 129–134. [Google Scholar] [CrossRef]
- Isaacs, F.J.; Dwyer, D.J.; Ding, C.; Pervouchine, D.D.; Cantor, C.R.; Collins, J.J. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotech. 2004, 22, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Mutalik, V.K.; Qi, L.; Guimaraes, J.C.; Lucks, J.B.; Arkin, A.P. Rationally designed families of orthogonal RNA regulators of translation. Nat. Chem. Biol. 2012, 8, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Thaiprasit, J.; Cheevadhanarak, S.; Waraho, D.; Meechai, A. Conceptual design of RNA-RNA interaction based devices. Procedia Comput. Sci. 2012, 11, 139–148. [Google Scholar] [CrossRef]
- Rodrigo, G.; Landrain, T.E.; Majer, E.; Daros, J.A.; Jaramillo, A. Full design automation of multi-state RNA devices to program gene expression using energy-based optimization. PLoS Comput. Biol. 2013, 9, e1003172. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Landrain, T.E.; Jaramillo, A. De novo automated design of small RNA circuits for engineering synthetic riboregulation in living cells. Proc. Natl. Acad. Sci. USA 2012, 109, 15271–15276. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, J.A.; Dotu, I.; Clote, P. RNAifold 2.0: A web server and software to design custom and Rfam-based RNA molecules. Nucleic Acids Res. 2015, 43, W513–W521. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Jaramillo, A. Ribomaker: Computational design of conformation-based riboregulation. Bioinformatics 2014, 30, 2508–2510. [Google Scholar] [CrossRef] [PubMed]
- Thaiprasit, J.; Kaewkamnerdpong, B.; Waraho, D.; Cheevadhanarak, S.; Meechai, A. Domain-based design platform of interacting RNAs: A promising tool in synthetic biology. In Proceeding of the 7th Biomedical Engineering International Conference, Fukuoka, Japan, 26–28 November 2014; pp. 1–5.
- Zhang, D.Y. Towards domain-based sequence design for DNA strand displacement reactions. In Proceedings of the 16th International Conference on DNA Computing and Molecular Programming, Hong Kong, China, 14–17 June 2010; Sakakibara, Y., Mi, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. Lecture Notes in Computer Science. Volume 6518, pp. 162–175. [Google Scholar]
- Lawrence, C.; Reilly, A. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins 1990, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Mach. Learn. J. 1995, 21, 51–83. [Google Scholar] [CrossRef]
- Qi, Y.; Ye, P.; Bader, J. Genetic interaction motif finding by expectation maximization—a novel statistical model for inferring gene modules from synthetic lethality. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Pramanik, S.; Chung, M.J. Multiple sequence alignment using simulated annealing. Comput. Appl. Biosci. CABIOS 1994, 10, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Tomshine, J.; Kaznessis, Y. Optimization of a stochastically simulated gene network model via simulated annealing. Biophys. J. 2006, 91, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Stivala, A.; Stuckey, P.; Wirth, A. Fast and accurate protein substructure searching with simulated annealing and GPUs. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Kell, D. Metabolomics, modelling and machine learning in systems biology—towards an understanding of the languages of cells. Febs J. 2006, 273, 873–894. [Google Scholar] [CrossRef] [PubMed]
- Larranaga, P.; Calvo, B.; Santana, R.; Bielza, C.; Galdiano, J.; Inza, I.; Lozano, J.; Armananzas, R.; Santafe, G.; Perez, A.; et al. Machine learning in bioinformatics. Brief. Bioinform. 2006, 7, 86–112. [Google Scholar] [CrossRef] [PubMed]
- Dasika, M.; Gupta, A.; Maranas, C. A mixed integer linear programming (MILP) framework for inferring time delay in gene regulatory networks. In Proceedings of the Pacific Symposium on Biocomputing, Lihue, HI, USA, 6–10 January 2004; pp. 474–486.
- Wohlers, I.; Petzold, L.; Domingues, F.; Klau, G. Paul: Protein structural alignment using integer linear programming and lagrangian relaxation. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef]
- Huang, T.; He, Z. A linear programming model for protein inference problem in shotgun proteomics. Bioinformatics 2012, 28, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D. SAGA: Sequence alignment by genetic algorithm. Nucleic Acids Res. 1996, 24, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Taneda, A. Cofolga: A genetic algorithm for finding the common folding of two RNAs. Comput. Biol. Chem. 2005, 29, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Gopal, S. Genetic algorithm learning as a robust approach to RNA editing site prediction. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef]
- Taneda, A. An efficient genetic algorithm for structural RNA pairwise alignment and its application to non-coding RNA discovery in yeast. BMC Bioinformatics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Montaseri, S.; Zare-Mirakabad, F.; Moghadam-Charkari, N. RNA-RNA interaction prediction using genetic algorithm. Algorithms Mol. Biol. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; O’Brien, E.; Higgins, D. RAGA: RNA sequence alignment by genetic algorithm. Nucleic Acids Res. 1997, 25, 4570–4580. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.-Y.; Tong, K.-K.; Lee, K.-H.; Leung, K.-S. RIPGA: RNA-RNA interaction prediction using genetic algorithm. In Proceedings of the 2013 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Singapore, Singapore, 16–19 April 2013; pp. 148–153.
- Lyngso, R.B.; Anderson, J.W.; Sizikova, E.; Badugu, A.; Hyland, T.; Hein, J. Frnakenstein: Multiple target inverse RNA folding. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Reeves, C.R.; Rowe, J.E. Genetic Algorithms—Principles and Perspectives: A Guide to GA Theory; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Lorenz, R.; Bernhart, S.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.; Hofacker, I. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Guo, P. Overview of methods in RNA nanotechnology: Synthesis, purification, and characterization of RNA nanoparticles. In RNA Nanotechnology and Therapeutics; Guo, P., Haque, F., Eds.; Springer: New York, NY, USA, 2015; Volume 1297, pp. 1–19. [Google Scholar]
- Rivas, E.; Klein, R.; Jones, T.; Eddy, S. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001, 11, 1369–1373. [Google Scholar] [CrossRef]
- Knuth, D. Sorting and searching. In The Art of Computer Programming; Addison-Wesley: Reading, MA, USA, 1998; pp. 158–168. [Google Scholar]
- Cayrol, B.; Fortas, E.; Martret, C.; Cech, G.; Kloska, A.; Caulet, S.; Barbet, M.; Trepout, S.; Marco, S.; Taghbalout, A.; et al. Riboregulation of the bacterial actin-homolog mreB by DsrA small noncoding RNA. Integr. Biol. 2015, 7, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, T.A.; Touati, D. Hfq, a new chaperoning role: Binding to messenger RNA determines access for small RNA regulator. EMBO J. 2004, 23, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Will, W.R.; Frost, L.S. Hfq is a regulator of F-plasmid traJ and tram synthesis in Escherichia coli. J. Bacteriol. 2006, 188, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Udekwu, K.I.; Darfeuille, F.; Vogel, J.; Reimegard, J.; Holmqvist, E.; Wagner, E.G. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 2005, 19, 2355–2366. [Google Scholar] [CrossRef] [PubMed]
- Majdalani, N.; Cunning, C.; Sledjeski, D.; Elliott, T.; Gottesman, S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 12462–12467. [Google Scholar] [CrossRef] [PubMed]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Size | Computational Time (s) * | Optimal Fitness * |
|---|---|---|
| 4 | 11.50 ± 1.90 | 0.993 ± 0.006 |
| 6 | 16.10 ± 2.42 | 0.995 ± 0.003 |
| 8 | 22.30 ± 3.77 | 0.996 ± 0.000 |
| 10 | 26.00 ± 5.19 | 0.996 ± 0.000 |
| 20 | 48.30 ± 6.68 | 0.996 ± 0.000 |
| 50 | 106.80 ± 7.87 | 0.996 ± 0.000 |
| 100 | 202.00 ± 14.52 | 0.996 ± 0.000 |
| Model No. | RNA Complexity | Computational Time (s) * | Optimal Fitness * | |
|---|---|---|---|---|
| Length of R1/R2 (Nucleotides) | Number of ID | |||
| A01 | 10/10 | 3 | 4.8 ± 0.53 | 1.000 ± 0.000 |
| A02 | 15/15 | 3 | 4.5 ± 0.50 | 1.000 ± 0.000 |
| A03 | 10/20 | 4 | 6.3 ± 0.87 | 1.000 ± 0.000 |
| A04 | 15/30 | 5 | 6.4 ± 0.56 | 1.000 ± 0.000 |
| N01 | 35/67 | 17 | 17.1 ± 4.61 | 0.994 ± 0.001 |
| N02 | 55/71 | 24 | 21.3 ± 3.76 | 0.996 ± 0.002 |
| Model No. | Time(s) | iDoRNA | iDoDe * | iDoDe ** | ||||
|---|---|---|---|---|---|---|---|---|
| Optimal Fitness | Success Rate | Optimal Fitness | Success Rate | Time (s) | Optimal Fitness | Success Rate | ||
| A01 | 5 | 1.000 | 10/10 | 1.000 | 1/28 | 177 | 1.000 | 7/1000 |
| A02 | 5 | 1.000 | 10/10 | 1.000 | 1/31 | 173 | 1.000 | 42/1000 |
| A03 | 6 | 1.000 | 10/10 | 0.875 | 0/23 | 237 | 0.922 | 0/1000 |
| A04 | 6 | 1.000 | 10/10 | 0.938 | 0/22 | 301 | 0.968 | 0/1000 |
| N01 | 17 | 1.000 | 10/10 | 0.949 | 0/86 | 495 | 0.967 | 0/1000 |
| N02 | 21 | 0.996 | 0/10 | 0.934 | 0/54 | 252 | 0.881 | 0/1000 |
| Features | NUPACK | RNAiFold 2.0 | RiboMaker | iDoRNA | |
|---|---|---|---|---|---|
| Service: | |||||
| Source code | C | C++ | C++ | C | |
| Web server | ✓ | ✓ | ✓ | ✗ | |
| Input: | |||||
| Target structures: | Single stranded RNA | ✓ (Web server); ✗ (Source code) | ✗ | ✓ | ✓ |
| Hybridized RNA | ✓ | ✓ | ✓ | ✓ | |
| Constrained sequences | ✓ | ✓ | ✓ | ✓ | |
| IUPAC codes | IUPAC codes | IUPAC codes | A C G U N | IUPAC codes | |
| Pseudo-knot | ✗ | ✗ | ✗ | ✗ | |
| Specification: | |||||
| Folding temperature | ✓ | ✓ | ✗ | ✓ | |
| GC-content | ✓ | ✓ | ✗ | ✓ | |
| Consecutive nucleotides prevention | ✓ | ✓ | ✗ | ✗ | |
| Solution per run | ✓ | ✓ | ✗ | ✗ | |
| Alternative energy parameters | ✓ | ✗ | ✗ | ✗ | |
| Output: | |||||
| Designed sequences | ≥ 2 strands (Web server) 2 strands (Source code) | 2 strands | 2 strands | 2 strands | |
| Predicted structures | Dot-bracket or Dot-plus-parentheses notation | ✓ | ✓ | ✓ | ✓ |
| Graphical structure | ✗ | ✓ | ✓ | ✓ | |
| Design method: | |||||
| Design objective | min ensemble defect | Constraint-based | min Fobj | max Fitness | |
| Decomposition | Hierarchical Structure decomposition | Tree-like decomposition | ✗ | Interacting domain-based decomposition | |
| Optimization* | EO | CP, LNS | MCSA | GA | |
| Folding prediction | NUPACK | RNAcofold | RNAfold, RNAup | RNAfold, RNAcofold | |
| HR-length/# of ID | Model No. | Average Ensemble Defect | Average Fitness Score | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NUPACK | RNAiFold | RiboMaker | iDoRNA | NUPACK | RNAiFold | RiboMaker | iDoRNA | ||
| Actual models | |||||||||
| 102-212/ 17-35 | N01-07 | 0.00-0.02 | 0.02-0.07 | 0.60-0.84 | 0.05-0.18 | N/A | N/A | 0.18-0.44 | 0.90-1.00 |
| Artificial models | |||||||||
| 10-49/ 3-11 | A01-06 | 0.01-0.03 | 0.01-0.19 | 0.64-0.79 | 0.02-0.21 | N/A | N/A | 0.14-0.87 | 0.94-1.00 |
| 50-99/ 12-20 | A07-16 | 0.00-0.01 | 0.01-0.19 | 0.67-0.86 | 0.05-0.21 | N/A | N/A | 0.34-0.66 | 0.92-1.00 |
| 100-149/ 6-30 | A17-28 | 0.00-0.02 | 0.00-0.09 | 0.64-0.81 | 0.00-0.28 | N/A | N/A | 0.34-0.87 | 0.87-0.99 |
| 150-199/ 22-51 | A29-35 | 0.00-0.02 | 0.01-0.08 | 0.54-0.76 | 0.00-0.31 | N/A | N/A | 0.24-0.57 | 0.87-0.99 |
| 200-250/ 22-30 | A36-37 | 0.01-0.04 | 0.01-0.02 | 0.64-0.59 | 0.10-0.18 | N/A | N/A | 0.43-0.44 | 0.95-0.98 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thaiprasit, J.; Kaewkamnerdpong, B.; Waraho-Zhmayev, D.; Cheevadhanarak, S.; Meechai, A. iDoRNA: An Interacting Domain-based Tool for Designing RNA-RNA Interaction Systems. Entropy 2016, 18, 83. https://doi.org/10.3390/e18030083
Thaiprasit J, Kaewkamnerdpong B, Waraho-Zhmayev D, Cheevadhanarak S, Meechai A. iDoRNA: An Interacting Domain-based Tool for Designing RNA-RNA Interaction Systems. Entropy. 2016; 18(3):83. https://doi.org/10.3390/e18030083
Chicago/Turabian StyleThaiprasit, Jittrawan, Boonserm Kaewkamnerdpong, Dujduan Waraho-Zhmayev, Supapon Cheevadhanarak, and Asawin Meechai. 2016. "iDoRNA: An Interacting Domain-based Tool for Designing RNA-RNA Interaction Systems" Entropy 18, no. 3: 83. https://doi.org/10.3390/e18030083
APA StyleThaiprasit, J., Kaewkamnerdpong, B., Waraho-Zhmayev, D., Cheevadhanarak, S., & Meechai, A. (2016). iDoRNA: An Interacting Domain-based Tool for Designing RNA-RNA Interaction Systems. Entropy, 18(3), 83. https://doi.org/10.3390/e18030083