Estimation of Activity Interaction Parameters in Fe-S-j Systems


Abstract
:1. Introduction
2. Calculation Method
2.1. Basic Relations
2.2. Miedema Model
2.3. Hillert-Toop Geometric Model
2.4. Calculation Model
- If the asymmetric component is solute, the activity interaction parameter can be calculated by:
- If the solvent is an asymmetric component, the activity interaction parameter is:wherehere:where the αij is identical to Equation (5).
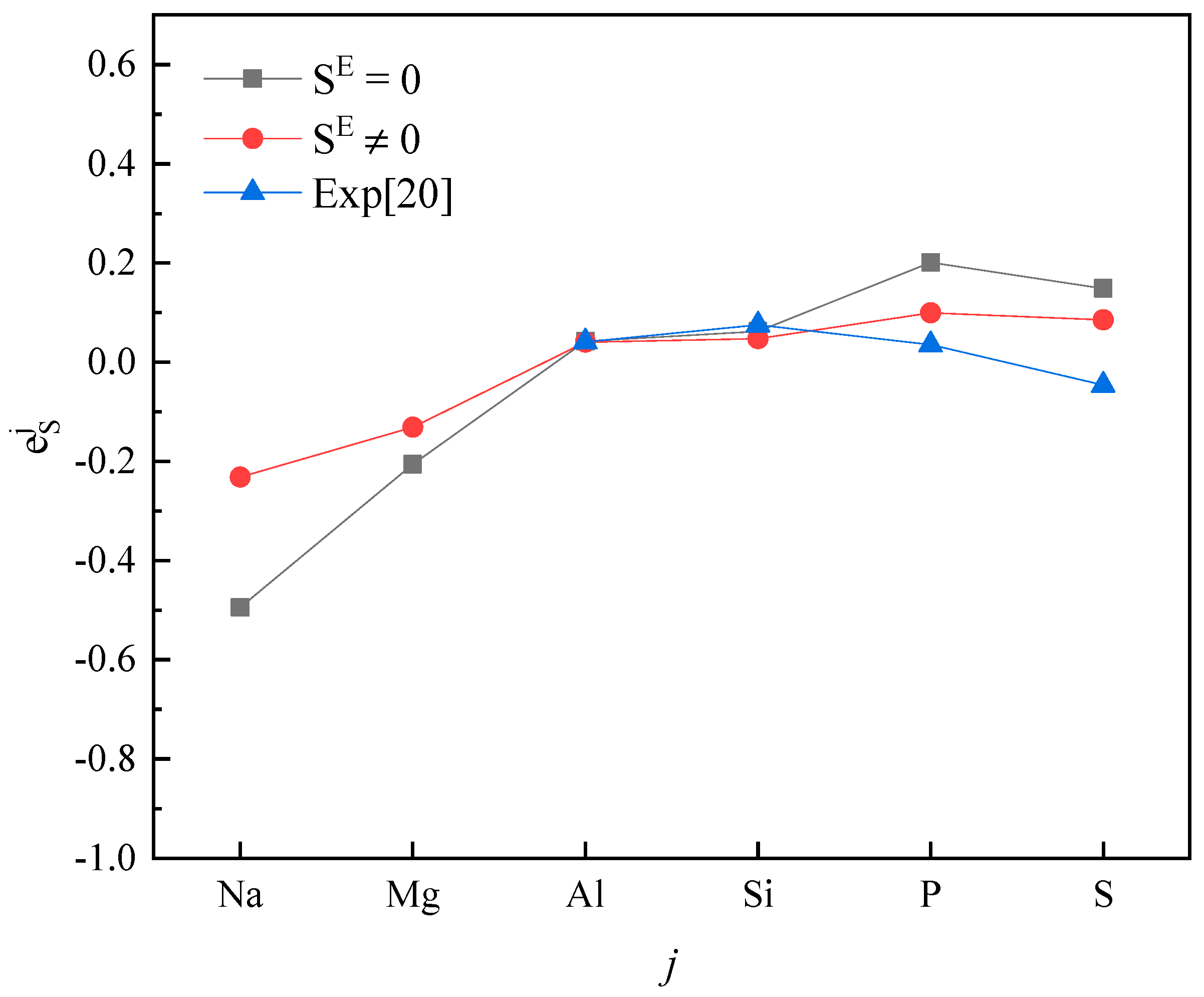
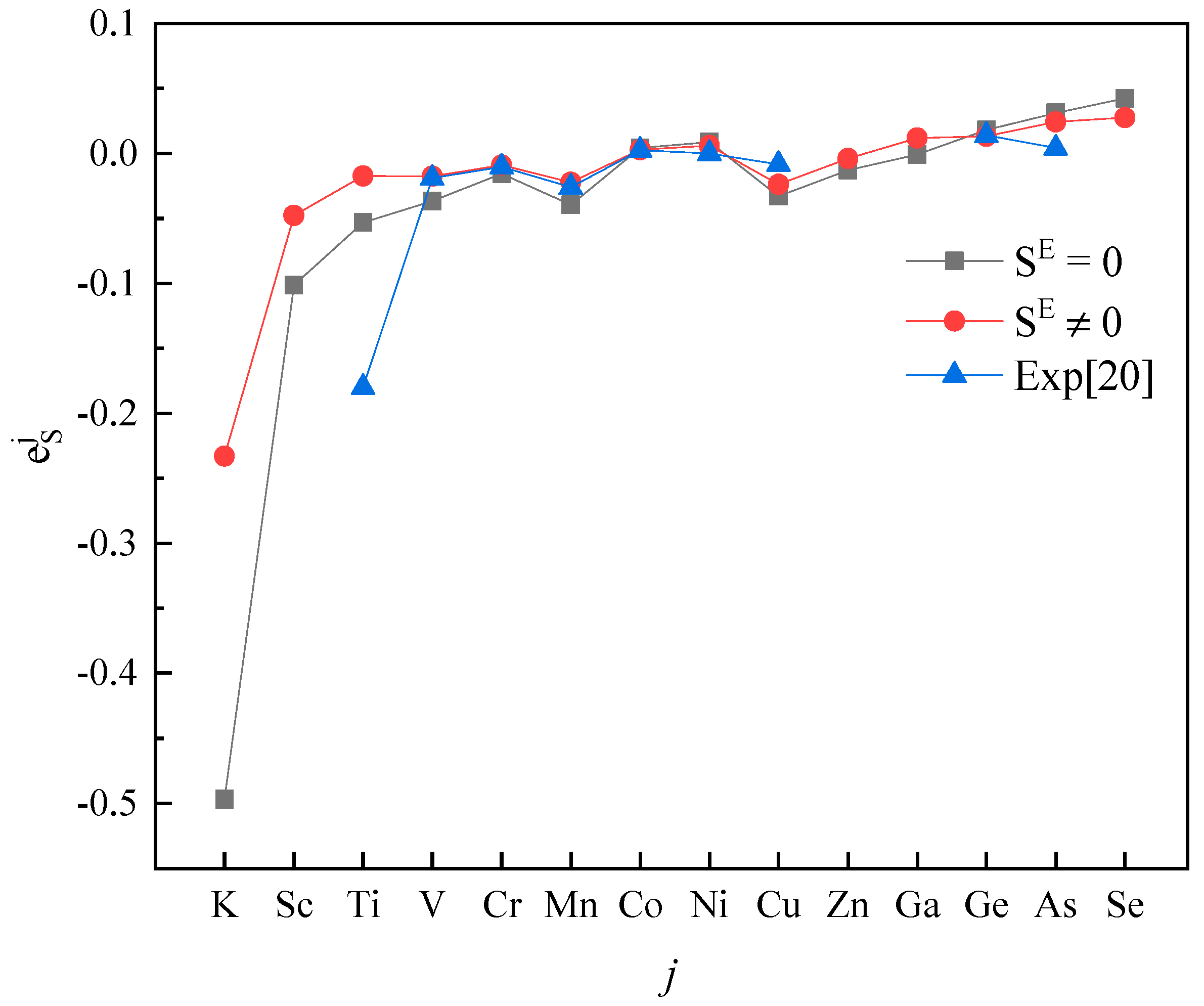
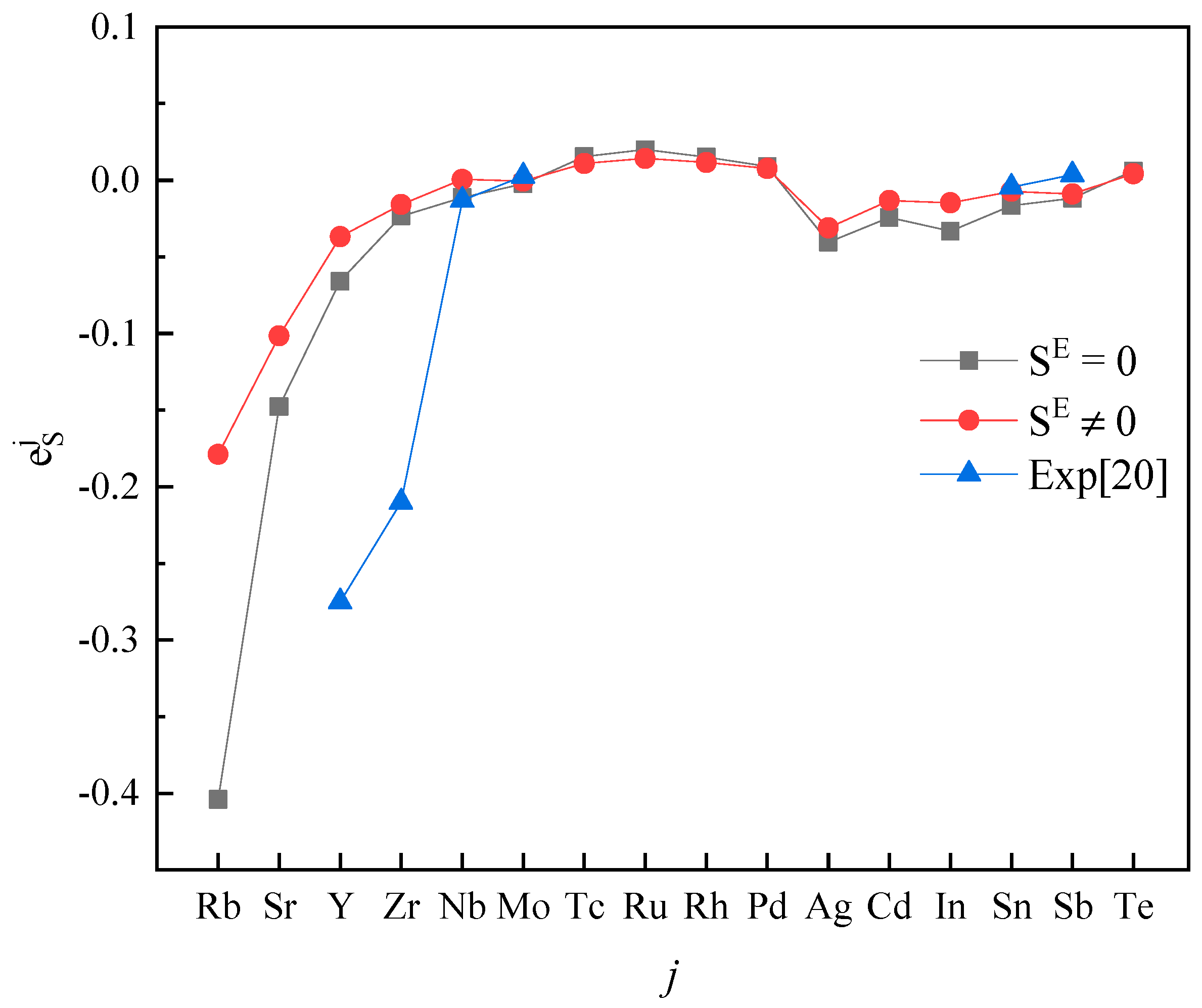
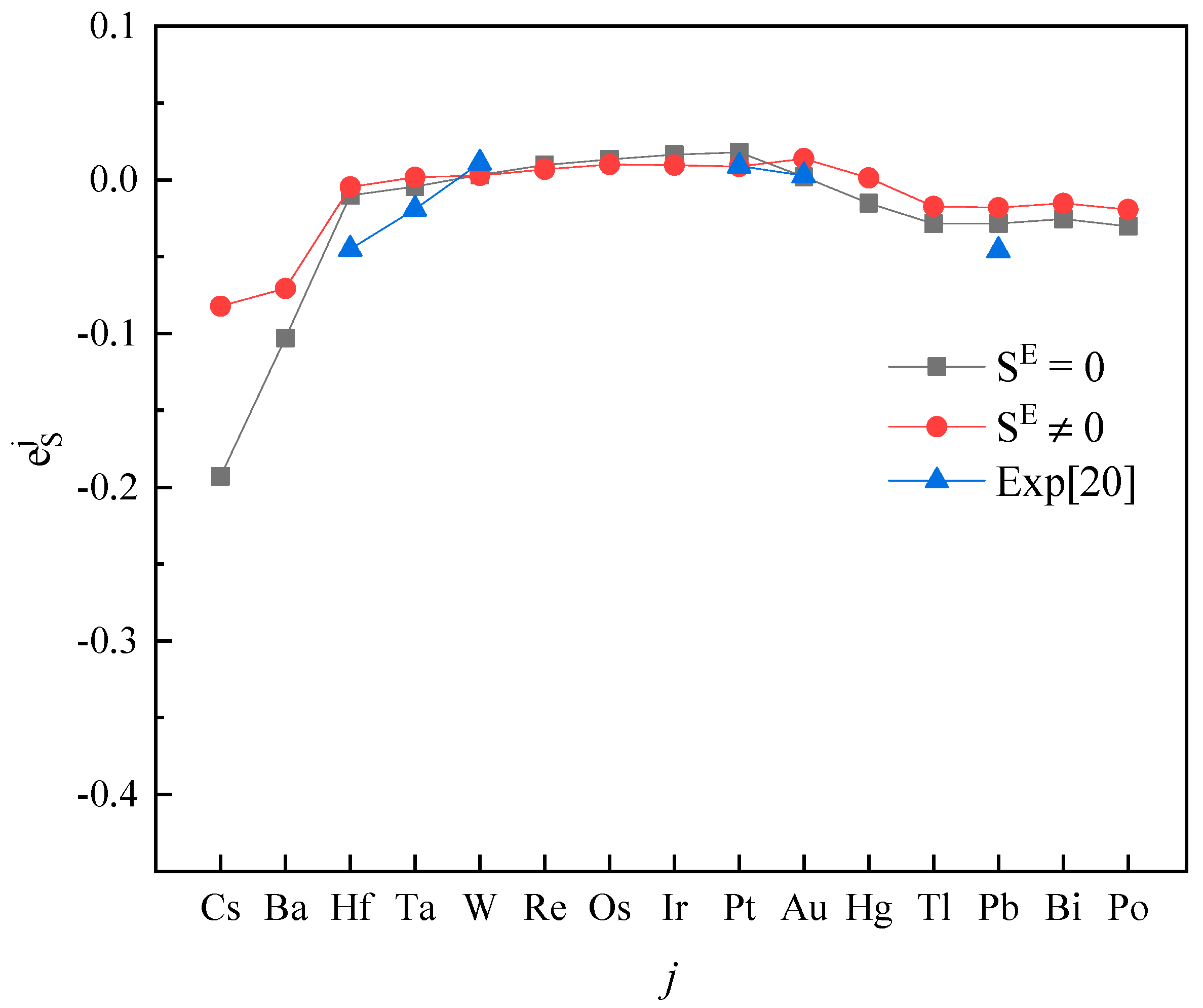
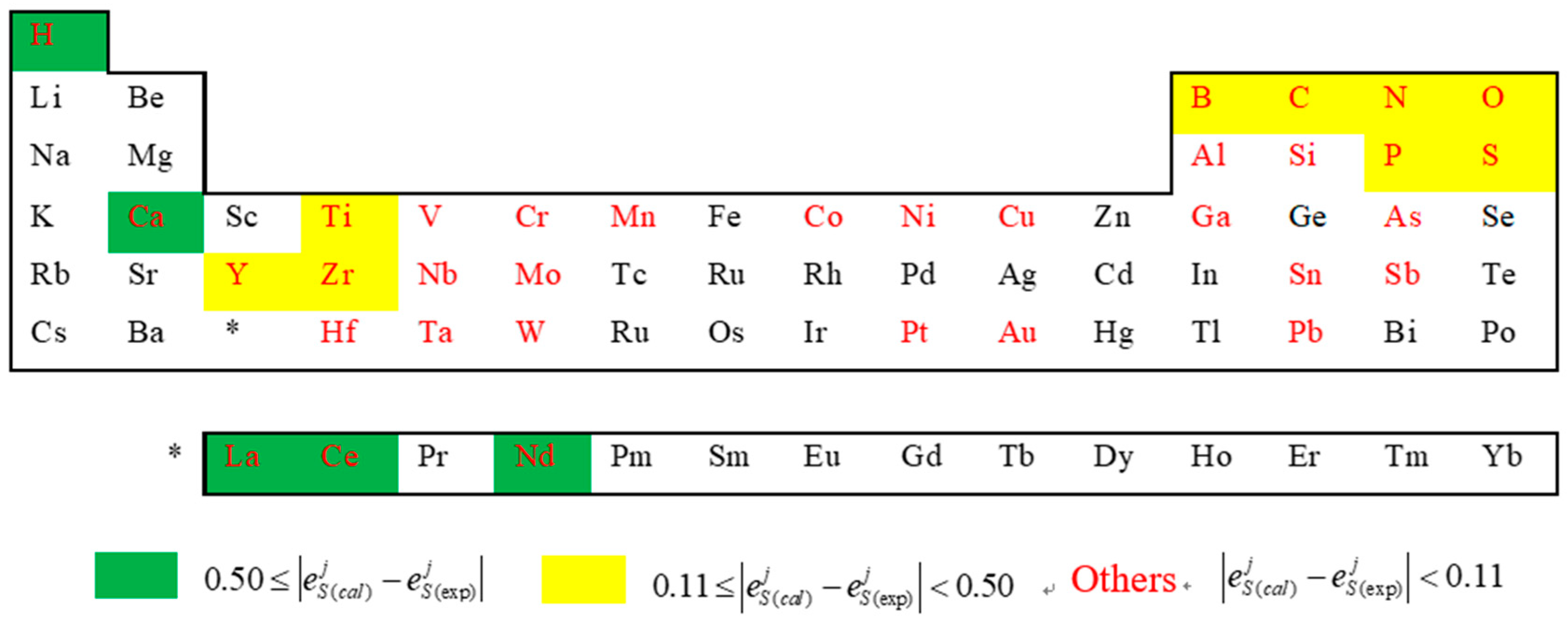
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mintz, B. The influence of composition on the hot ductility of steels and to the problem of transverse cracking. ISIJ Int. 1999, 39, 833–855. [Google Scholar] [CrossRef]
- Ryan, M.P.; Williams, D.E.; Chater, R.J.; Hutton, B.M.; McPhail, D.S. Why stainless steel corrodes. Nature 2002, 415, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Costa e Silva, A. Thermodynamic aspects of inclusion engineering in steels. Rare Met. 2006, 25, 412–419. [Google Scholar] [CrossRef]
- Gollapalli, V.; Rao, M.B.V.; Karamched, P.S.; Borra, C.R.; Roy, G.G.; Srirangam, P. Modification of oxide inclusions in calcium-treated Al-killed high sulphur steels. Ironmak. Steelmak. 2018, 1–8. [Google Scholar] [CrossRef]
- Wagner, C. Thermodynamics of Alloys; Addison-Wesley Press: Boston, MA, USA, 1952. [Google Scholar]
- Lupis, C.H.P.; Elliott, J.F. Generalized interaction coefficients. Acta Metall. 1966, 14, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Darken, L. Thermodynamics of binary metallic solutions. Trans. Met. Soc. AIME 1967, 239, 80–89. [Google Scholar]
- Pelton, A.D.; Bale, C.W. A modified interaction parameter formalism for non-dilute solutions. Metall. Trans. A 1986, 17, 1211–1215. [Google Scholar] [CrossRef]
- Bale, C.W.; Pelton, A.D. The unified interaction parameter formalism: Thermodynamic consistency and applications. Metall. Trans. A 1990, 21, 1997–2002. [Google Scholar] [CrossRef]
- Costa e Silva, A. Interaction parameters of oxygen and deoxidants in liquid iron. J. Min. Metall. Sect. B Metall. 2016, 52, 41–46. [Google Scholar] [CrossRef]
- Waseda, Y. Interaction parameters in metallic solutions estimated from liquid structure and the heat of solution at infinite dilution. High Temp. Mater. Process. 2012, 31, 203–208. [Google Scholar] [CrossRef]
- Ding, X.Y.; Fan, P.; Wang, W.Z. Thermodynamic calculation for alloy systems. Metall. Mater. Trans. B 1999, 30, 271–277. [Google Scholar] [CrossRef]
- Ding, X.; Fan, P.; Han, Q. Models of activity and activity interaction parameter in ternary metallic melt. Acta Metall. Sin. 1994, 30, 49–60. [Google Scholar]
- Ueno, S.; Waseda, Y.; Jacob, K.T.; Tamaki, S. Theoretical treatment of interaction parameters in multicomponent metallic solutions. Process Metall. 1988, 59, 474–483. [Google Scholar] [CrossRef]
- Fan, P.; Chou, K.C. A self-consistent model for predicting interaction parameters in multicomponent alloys. Metall. Mater. Trans. A 1999, 30, 3099–3102. [Google Scholar] [CrossRef]
- Wang, F.M.; Li, X.P.; Han, Q.Y.; Zhang, N.X. A model for calculating interaction coefficients between elements in liquid and iron-base alloy. Metall. Mater. Trans. B 1997, 28, 109–113. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, W.; Chen, X.; Ding, X.; Zhou, G. Modeling Activity and Interaction Coefficients of Components of Multicomponent Alloy Melts: An Example of Iron Melt. High Temp. Mater. Process. 2013, 32, 215. [Google Scholar] [CrossRef]
- Ding, X.; Fan, P.; Luo, L. Alloy Melts Thermodynamic Model: Prediction and Software Development; Northeastern University Press: Shenyang, China, 1998. (In Chinese) [Google Scholar]
- Neuhausen, J.; Eichler, B. Extension of Mediema’s Macroscopic Atom Model to the Elements of Group 16 (O, S, Se, Te, Po); Paul Scherrer Inst.: Villigen, Switzerland, 2003. [Google Scholar]
- Hino, M.; Ito, K. Thermodynamic Data for Steelmaking; Tohoku University Press: Sendai, Japan, 2010. [Google Scholar]
- Inoue, R.; Suito, H. Calcium desulfurization equilibrium in liquid iron. Steel Res. 1994, 65, 403–409. [Google Scholar] [CrossRef]
- Hillert, M. Empirical methods of predicting and representing thermodynamic properties of ternary solution phases. Calphad 1980, 4, 1–12. [Google Scholar] [CrossRef]
- Tanaka, T.; Morita, Z.-I.; Gokcen, N.A.; Iida, T. Thermodynamic relationship between enthalpy of mixing and excess entropy in liquid binary alloys. Zeitschrift für Metallkunde 1993, 84, 192–200. [Google Scholar]
- Miedema, A.; De Chatel, P.; De Boer, F. Cohesion in alloys—Fundamentals of a semi-empirical model. Physica B+C 1980, 100, 1–28. [Google Scholar] [CrossRef]
- Niessen, A.; Miedema, A.; De Boer, F.; Boom, R. Enthalpies of formation of liquid and solid binary alloys based on 3d metals: IV. Alloys of cobalt. Physica B+C 1988, 151, 401–432. [Google Scholar] [CrossRef]
- Niessen, A.K.; de Boer, F.R.; Boom, R.; De Châtel, P.F.; Mattens, W.C.M.; Miedema, A.R. Model predictions for the enthalpy of formation of transition metal alloys II. Calphad 1983, 7, 51–70. [Google Scholar] [CrossRef]
- Wu, X.S.; Yan, X.H.; Ma, B.K.; Lin, Z.J.; Yang, X.Z. Calculation of the Heat of Formation of Ternary Compounds—P-Ga-as, N-Ga-as, N-Ga-P. Acta Phys. Sin. 1995, 4, 62–70. [Google Scholar]
- Taguchi, K.; Ono-Nakazato, H.; Nakai, D.; Usui, T.; Marukawa, K. Deoxidation and desulfurization equilibria of liquid iron by calcium. ISIJ Int. 2003, 43, 1705–1709. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Du, T. Thermodynamics of rare earth elements in liquid iron. J. Less-Common Met. 1985, 110, 187–193. [Google Scholar] [CrossRef]
- Zhang, R.F.; Liu, B.X. Proposed model for calculating the standard formation enthalpy of binary transition-metal systems. Appl. Phys. Lett. 2002, 81, 1219–1221. [Google Scholar] [CrossRef]
- Sun, S.P.; Yi, D.Q.; Jiang, Y.; Zang, B.; Xu, C.H.; Li, Y. An improved atomic size factor used in Miedema’s model for binary transition metal systems. Chem. Phys. Lett. 2011, 513, 149–153. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Podloucky, R. Miedema’s model revisited: The parameter for Ti, Zr, and Hf. Calphad 2006, 30, 266–269. [Google Scholar] [CrossRef]
- Chartrand, P.; Pelton, A.D. On the choice of “Geometric” thermodynamic models. J. Phase Equilib. 2000, 21, 141–147. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | μ | |||
|---|---|---|---|---|
| O | 6.97 | 2.66 | 1.70 | 0.04 |
| S | 5.60 | 4.38 | 1.46 | 0.04 |
| Se | 5.17 | 5.17 | 1.40 | 0.04 |
| Te | 4.72 | 6.44 | 1.31 | 0.04 |
| Po | 4.44 | 7.04 | 1.15 | 0.04 |
| j | Current Model | Ding Model [12] | JSPS | j | Current Model | Ding Model [12] | JSPS | ||
|---|---|---|---|---|---|---|---|---|---|
| H | 4.0396 | 4.5582 | 1.3551 | 0.41 | Pd | 0.0089 | 0.0077 | −0.0192 | |
| Li | −0.8793 | −0.3694 | −0.7415 | Ag | −0.0403 | −0.0310 | −0.0107 | ||
| Be | 0.0297 | 0.0334 | −0.0046 | Cd | −0.0242 | −0.0131 | −0.0138 | ||
| B | 0.4360 | 0.2808 | 0.3909 | 0.134 | In | −0.0332 | −0.0147 | −0.0044 | |
| C | 0.5800 | 0.3849 | 0.3770 | 0.111 | Sn | −0.0166 | −0.0072 | 0.0009 | −0.0044 |
| N | 0.5449 | −0.1025 | 0.3709 | 0.01 | Sb | −0.0117 | −0.0090 | 0.0101 | 0.0037 |
| O | 0.3075 | −0.3524 | 0.3574 | −0.27 | Te | 0.0062 | 0.0042 | −0.0738 | |
| Na | −0.4944 | −0.2318 | −0.2903 | Cs | −0.1929 | −0.0822 | −0.0567 | ||
| Mg | −0.2058 | −0.1314 | −0.1475 | Ba | −0.1029 | −0.0707 | −0.0222 | ||
| Al | 0.0426 | 0.0405 | 0.0112 | 0.041 | Hf | −0.0103 | −0.0047 | −0.0116 | −0.045 |
| Si | 0.0621 | 0.0471 | 0.0941 | 0.075 | Ta | −0.0044 | 0.0016 | 0.0032 | −0.019 |
| P | 0.2013 | 0.0995 | 0.1907 | 0.035 | W | 0.0032 | 0.0028 | 0.0094 | 0.011 |
| S | 0.1488 | 0.0852 | 0.1496 | −0.0461 | Re | 0.0096 | 0.0068 | 0.0117 | |
| K | −0.4969 | −0.2329 | −0.2276 | Os | 0.0133 | 0.0099 | 0.0127 | ||
| Ca | −0.2508 | −0.1668 | −0.1753 | −110.0 | Ir | 0.0164 | 0.0094 | 0.0121 | |
| Sc | −0.1013 | −0.0477 | −0.1258 | Pt | 0.0179 | 0.0086 | 0.0056 | 0.0089 | |
| Ti | −0.0532 | −0.0174 | −0.0820 | −0.18 | Au | 0.0018 | 0.0137 | −0.0039 | 0.0028 |
| V | −0.0367 | −0.0174 | −0.0463 | −0.019 | Hg | −0.0153 | 0.0011 | −0.0091 | |
| Cr | −0.0156 | −0.0089 | −0.0173 | −0.0103 | Tl | −0.0286 | −0.0174 | −0.0068 | |
| Mn | −0.0395 | −0.0221 | −0.0393 | −0.026 | Pb | −0.0284 | −0.0182 | −0.0051 | −0.046 |
| Co | 0.0042 | 0.0029 | 0.0036 | 0.0026 | Bi | −0.0254 | −0.0154 | −0.0042 | |
| Ni | 0.0087 | 0.0061 | 0.0071 | 0 | Po | −0.0302 | −0.0194 | −0.0443 | |
| Cu | −0.0329 | −0.0238 | −0.0214 | −0.0084 | La | −0.0491 | −0.0270 | −0.0320 | −18.3 |
| Zn | −0.0130 | −0.0040 | −0.0095 | Ce | −0.0346 | −0.0176 | −0.0309 | −9.10 | |
| Ga | −0.0009 | 0.0119 | −0.0031 | Pr | −0.0316 | −0.0158 | −0.0301 | ||
| Ge | 0.0181 | 0.0133 | 0.0142 | 0.014 | Nd | −0.0307 | −0.0156 | −0.0290 | −0.76 |
| As | 0.0313 | 0.0241 | 0.0431 | 0.0041 | Pm | −0.0271 | −0.0122 | −0.0279 | |
| Se | 0.0423 | 0.0274 | 0.0456 | Sm | −0.0269 | −0.0130 | −0.0497 | ||
| Rb | −0.4041 | −0.1789 | −0.1711 | Eu | −0.0800 | −0.0500 | −0.0265 | ||
| Sr | −0.1478 | −0.1015 | −0.0872 | Gd | −0.0255 | −0.0128 | −0.0254 | ||
| Y | −0.0657 | −0.0368 | −0.0674 | −0.275 | Tb | −0.0231 | −0.0109 | −0.0248 | |
| Zr | −0.0234 | −0.0157 | −0.0521 | −0.210 | Dy | −0.0226 | −0.0107 | −0.0244 | |
| Nb | −0.0112 | 0.0005 | −0.0259 | −0.013 | Ho | −0.0229 | −0.0113 | −0.0233 | |
| Mo | −0.0024 | −0.0005 | −0.0041 | 0.0027 | Er | −0.0200 | −0.0089 | −0.0230 | |
| Tc | 0.0155 | 0.0109 | 0.0131 | Tm | −0.0198 | −0.0089 | −0.0400 | ||
| Ru | 0.0201 | 0.0142 | 0.0166 | Yb | −0.0600 | −0.04 | −0.0215 | ||
| Rh | 0.0152 | 0.0116 | 0.0111 | Lu | −0.0168 | −0.0066 | −0.0192 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, T.; Ding, X.; Zhang, Y.; Chen, W.; Cheng, X.; Wang, B.; Dai, J.; Yan, X. Estimation of Activity Interaction Parameters in Fe-S-j Systems. Entropy 2018, 20, 808. https://doi.org/10.3390/e20100808
Ju T, Ding X, Zhang Y, Chen W, Cheng X, Wang B, Dai J, Yan X. Estimation of Activity Interaction Parameters in Fe-S-j Systems. Entropy. 2018; 20(10):808. https://doi.org/10.3390/e20100808
Chicago/Turabian StyleJu, Tianhua, Xueyong Ding, Yingyi Zhang, Weiliang Chen, Xiangkui Cheng, Bo Wang, Jingxin Dai, and Xinlin Yan. 2018. "Estimation of Activity Interaction Parameters in Fe-S-j Systems" Entropy 20, no. 10: 808. https://doi.org/10.3390/e20100808
APA StyleJu, T., Ding, X., Zhang, Y., Chen, W., Cheng, X., Wang, B., Dai, J., & Yan, X. (2018). Estimation of Activity Interaction Parameters in Fe-S-j Systems. Entropy, 20(10), 808. https://doi.org/10.3390/e20100808