
Modeling the Dynamics of T-Cell Development in the Thymus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
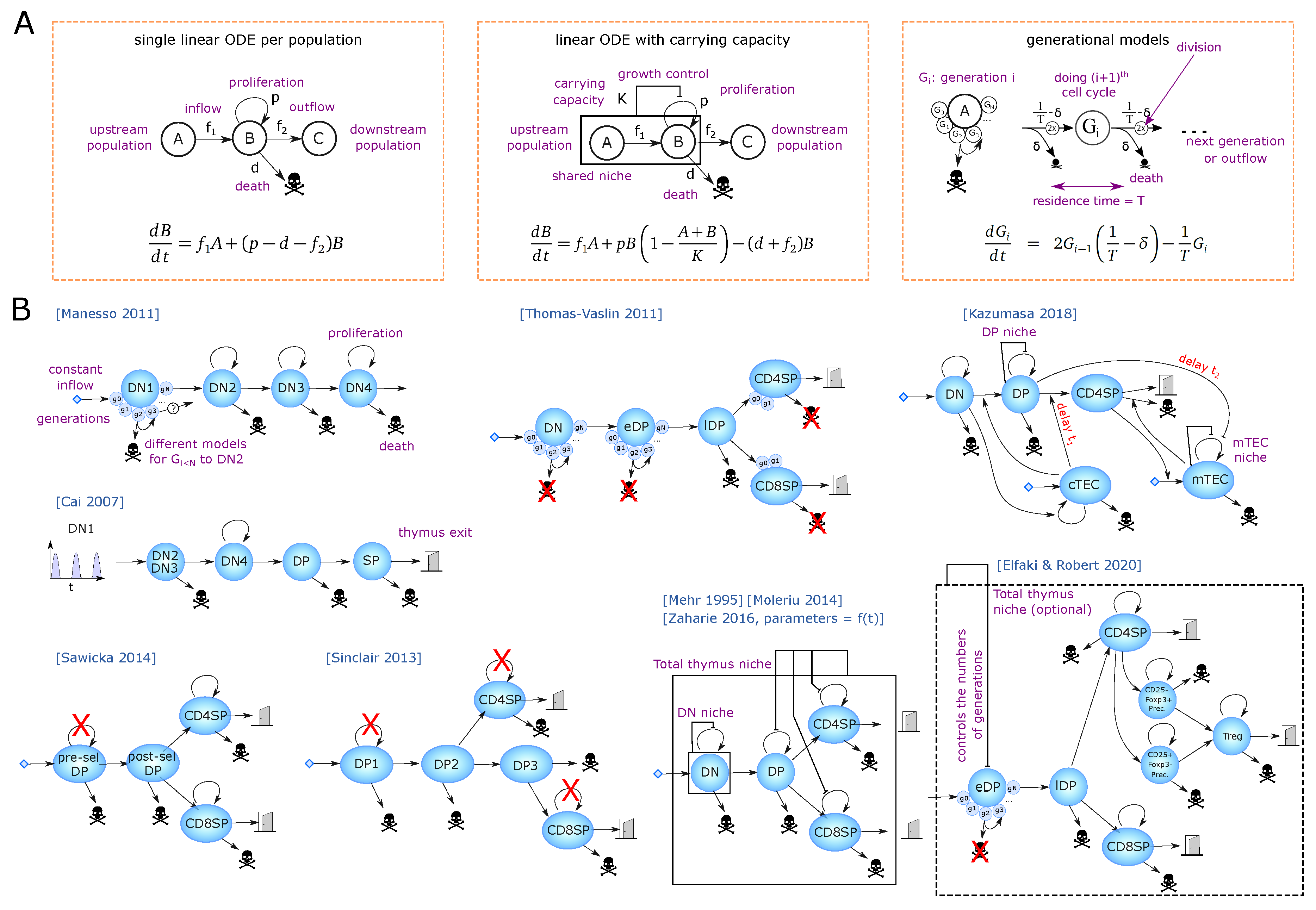
2. A Journey through Population Models of T-Cell Development
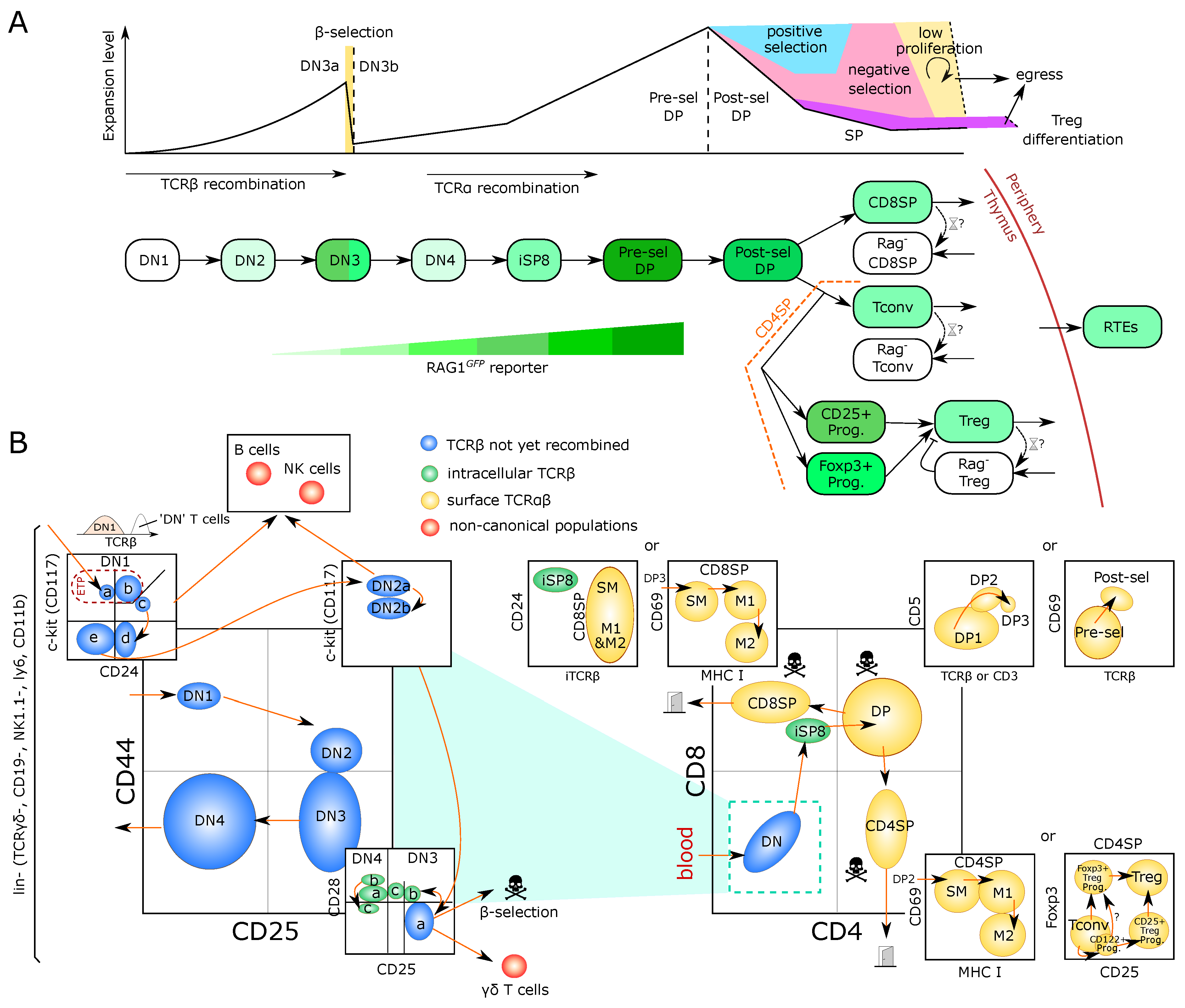
2.1. Early Steps of T-Cell Development
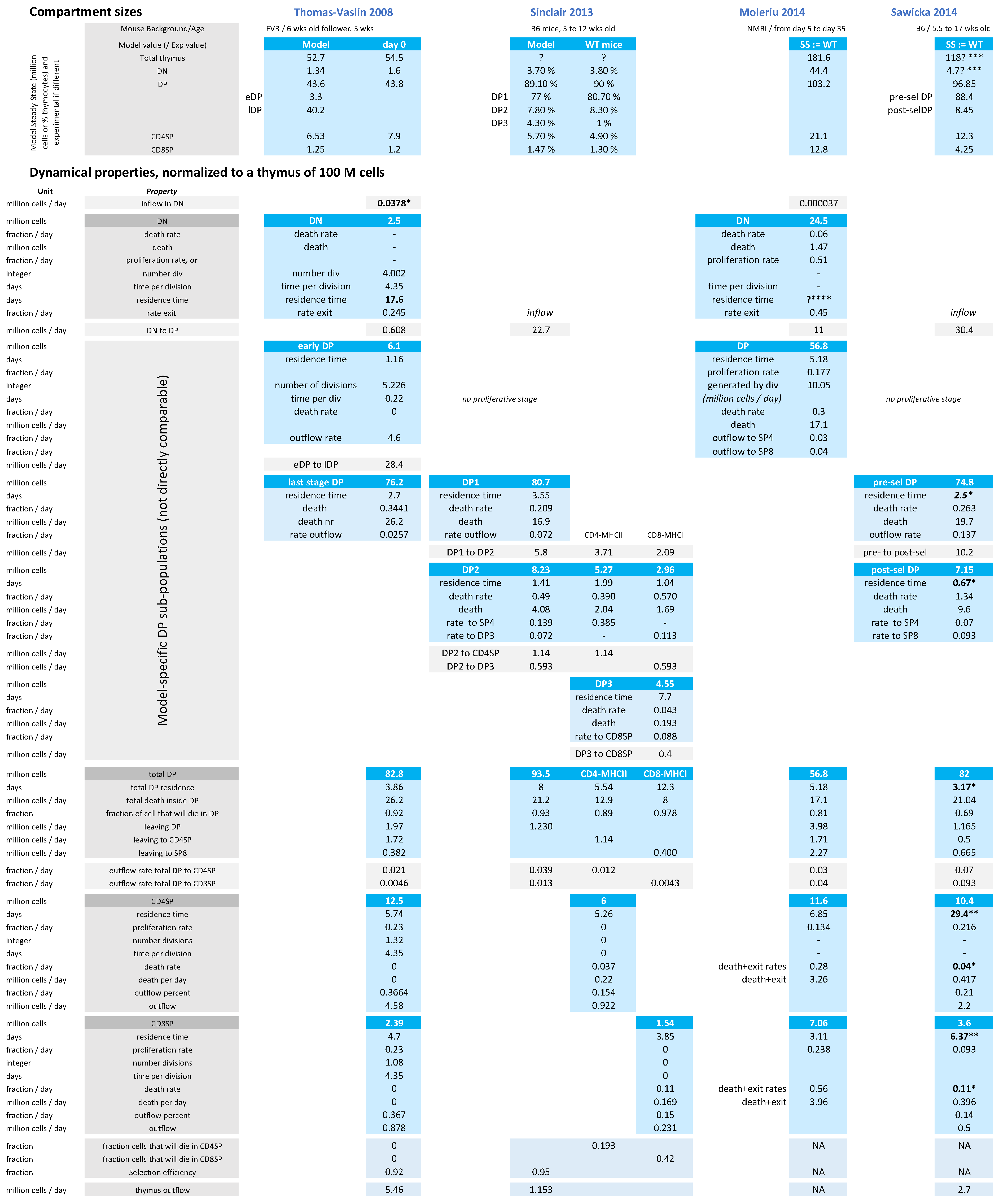
2.2. Estimation of the Flow between Compartments at Steady State Using Larger Models
2.3. Models for Thymus Involution and Shrinkage
2.4. Regulations between Thymic Populations
3. Estimation of In Vivo Cell Proliferation in the Thymus
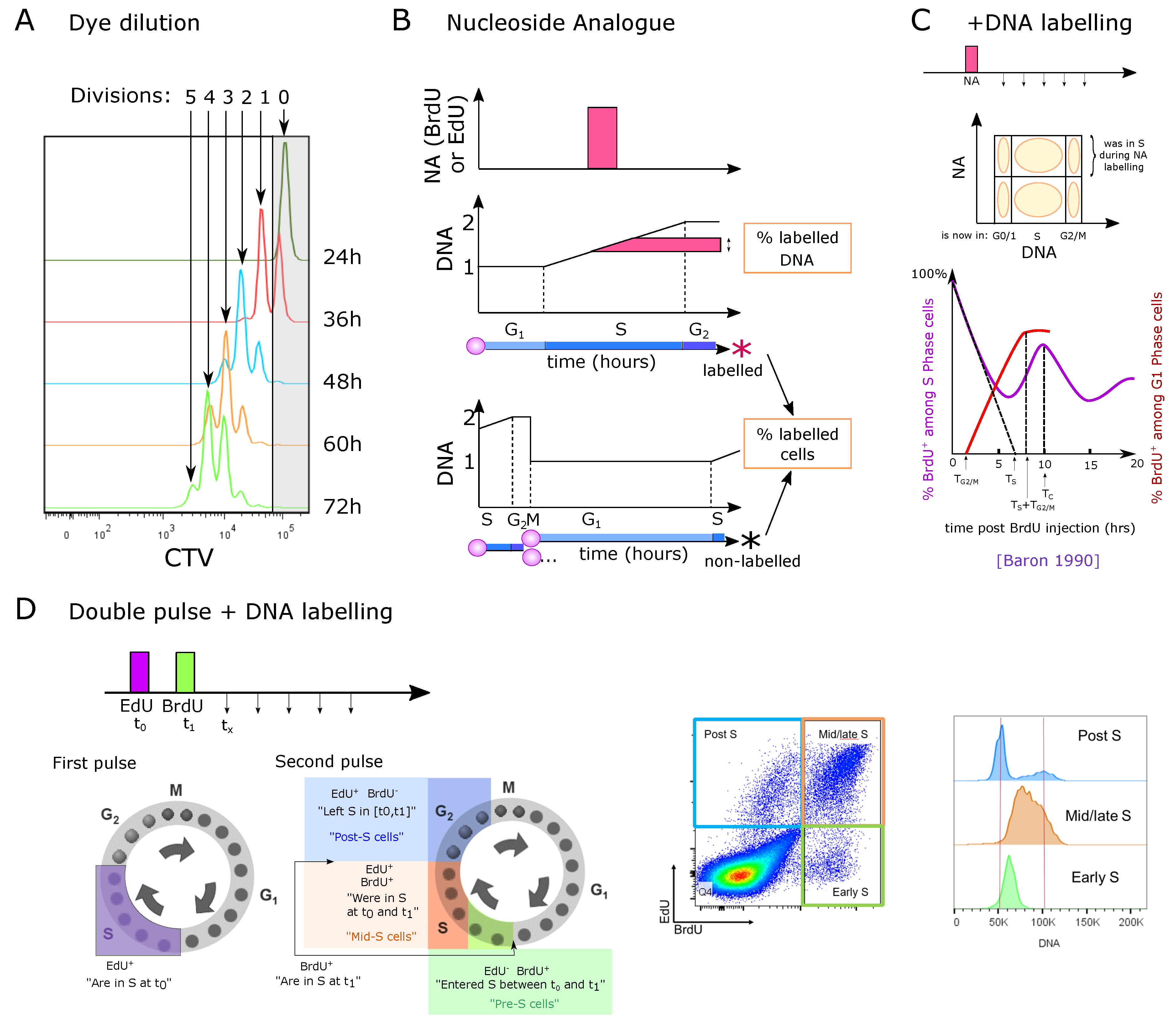
3.1. Measuring the Number of Divisions by Label Dilution
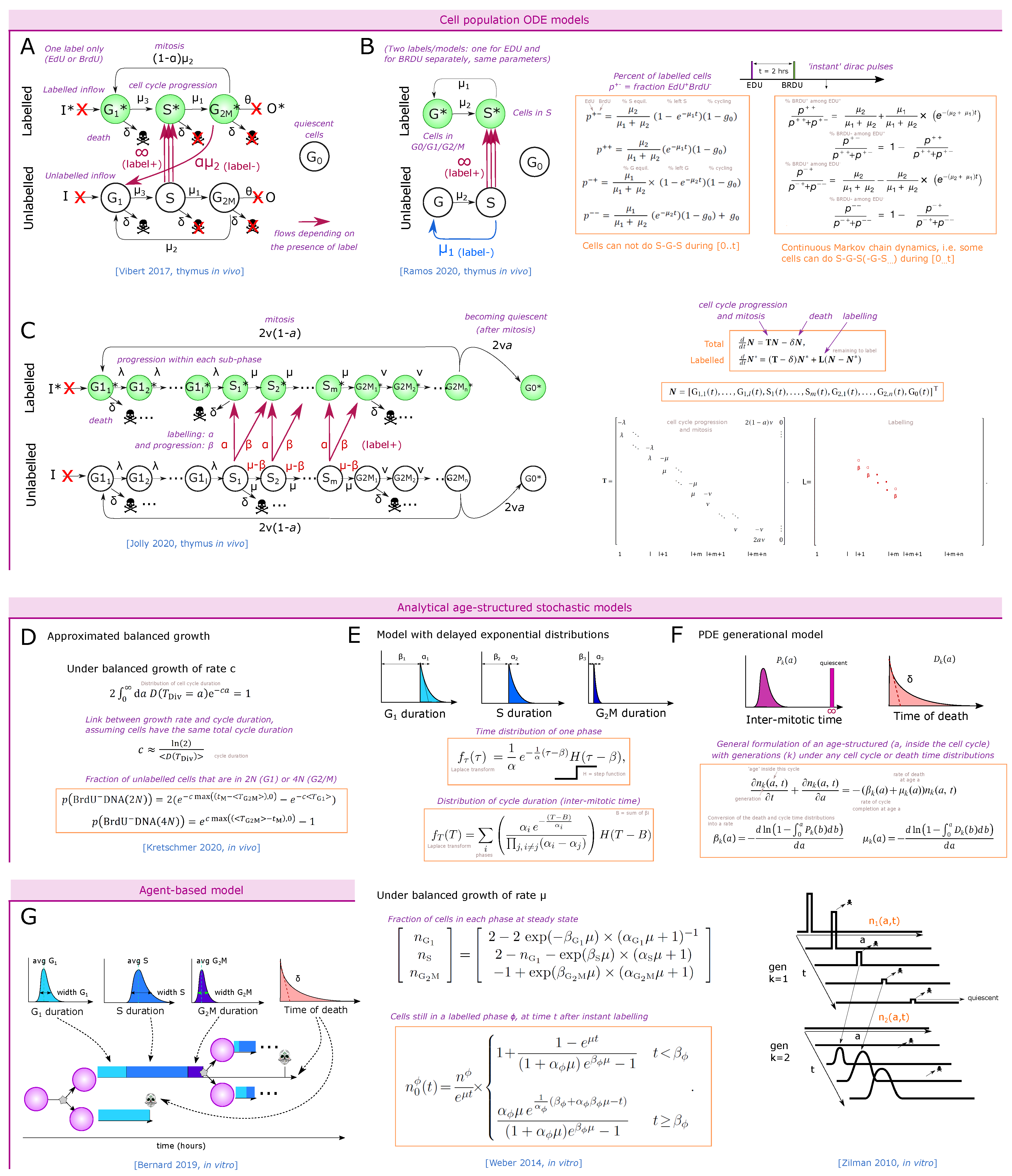
3.2. Nucleoside Analogue Incorporation during S Phase
3.2.1. Direct EdU or BrdU Staining
3.2.2. One-Point EdU or BrdU Pulse Followed by DNA Staining at Different Time-Points
3.2.3. Dual Labeling with EdU and BrdU at Different Time-Points
3.3. Future Models and Finding the Optimal Experimental Set-Up
4. Estimation of In Vivo Cell Death in the Thymus
5. Multi-Scale Considerations on Thymic Dynamics
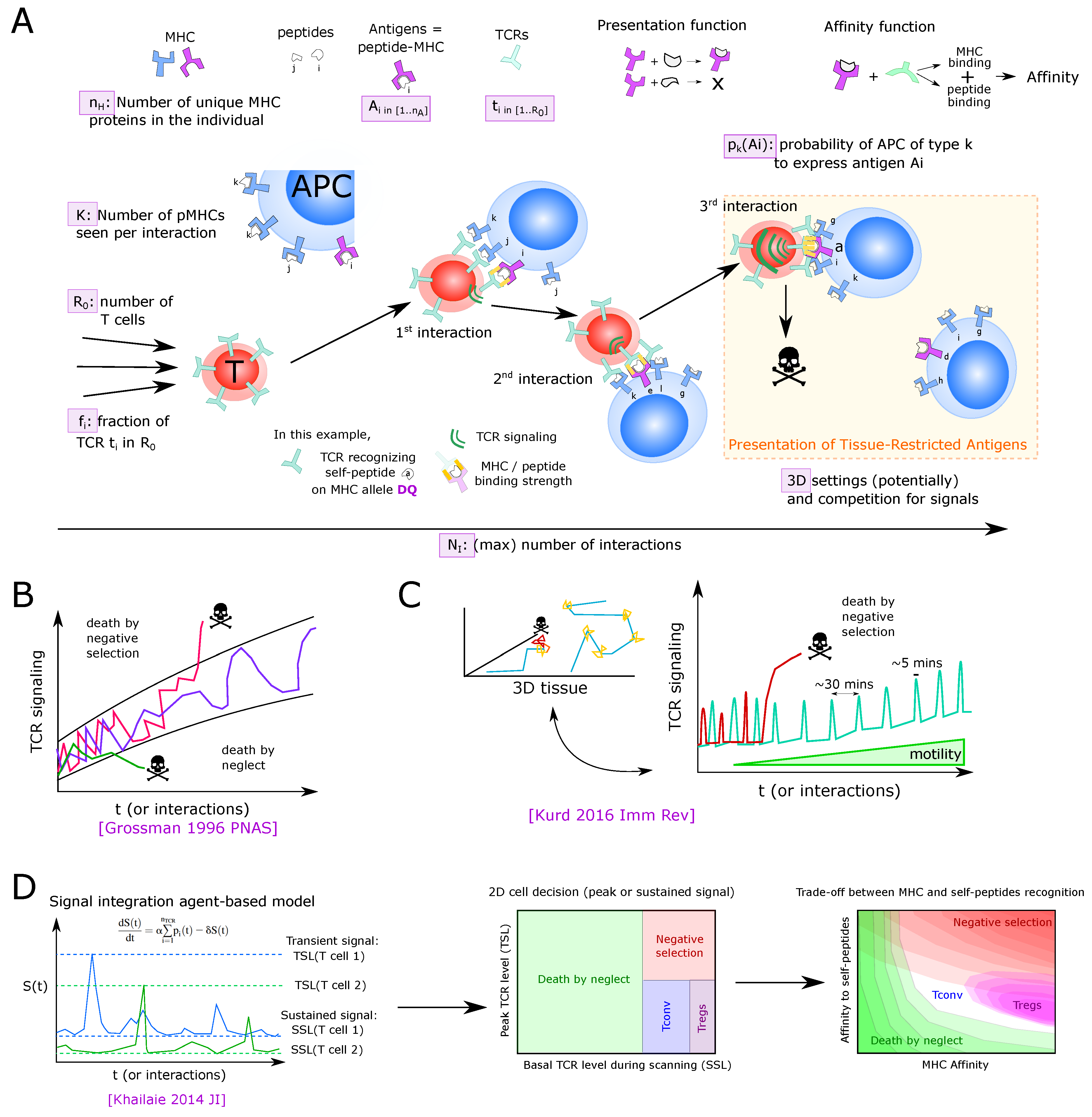
5.1. Linking the History of TCR Signaling to Cell Fate
5.2. 3D Models of Thymic Development, APC Types and Antigen Spatial Compartmentalization
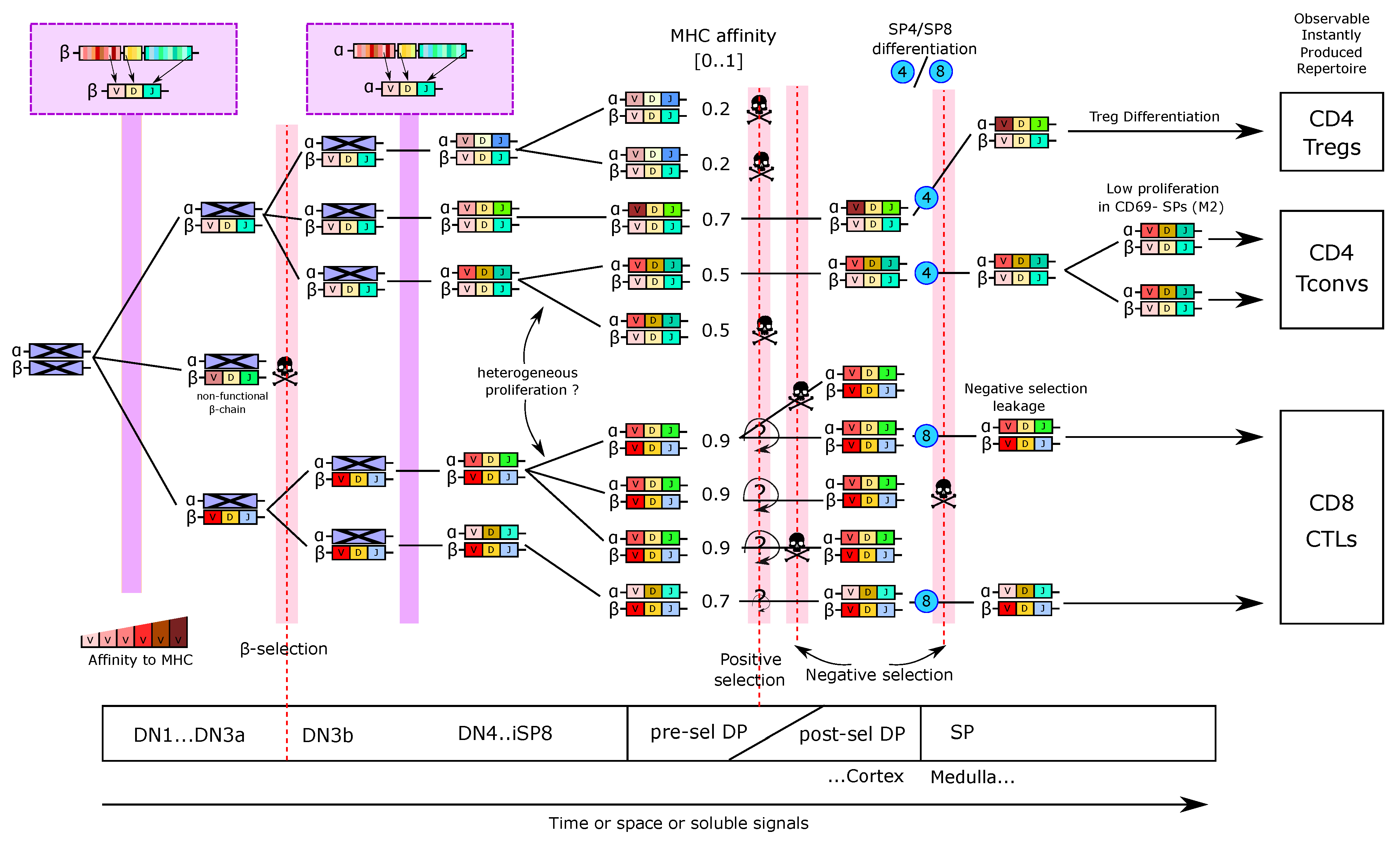
5.3. Thymus Dynamical Models Can Help the Analysis of TCR Repertoires
5.4. Future Types of Multi-Scale Models
6. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | Antigen-Presenting Cell |
| BrdU | Bromodeoxyuridine |
| CLP | Common Lymphoid Progenitor |
| CD4SP | Single-Positive CD4CD8 thymocyte |
| CD8SP | Single-Positive CD4CD8 thymocyte |
| CTV | Cell Trace Violet |
| DC | Dendritic Cell |
| DN | Double-Negative thymocyte |
| DP | Double-Positive thymocyte |
| EdU | 5-Ethynyl-2’-deoxyuridine |
| ETP | Early T-lineage Progenitor |
| iSP8 | Immature Single-Positive CD8 thymocyte |
| LMPP | Lymphoid-Primed Multipotent Progenitors |
| MHC | Major Histocompatibility Complex |
| ODE | Ordinary Differential Equation |
| SP | Single-Positive thymocyte |
| TCR | T-cell Receptor |
| TRA | Tissue Restricted Antigen |
| Treg | Regulatory T cell |
References
- Krueger, A. Thymus colonization: Who, how, how many? Arch. Immunol. Ther. Exp. 2018, 66, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Peaudecerf, L.; Lemos, S.; Galgano, A.; Krenn, G.; Vasseur, F.; Di Santo, J.P.; Ezine, S.; Rocha, B. Thymocytes may persist and differentiate without any input from bone marrow progenitors. J. Exp. Med. 2012, 209, 1401–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, V.C.; Ruggiero, E.; Schlenner, S.M.; Madan, V.; Schmidt, M.; Fink, P.J.; von Kalle, C.; Rodewald, H.R. Thymus-autonomous T cell development in the absence of progenitor import. J. Exp. Med. 2012, 209, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, A.R.; Liu, H. Acute thymic involution and mechanisms for recovery. Arch. Immunol. Ther. Exp. 2017, 65, 401–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114. [Google Scholar] [CrossRef] [PubMed]
- Yates, A. Theories and quantification of thymic selection. Front. Immunol. 2014, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, A.; Ziętara, N.; Łyszkiewicz, M. T cell development by the numbers. Trends Immunol. 2017, 38, 128–139. [Google Scholar] [CrossRef]
- Wu, L.; Scollay, R.; Egerton, M.; Pearse, M.; Spangrude, G.J.; Shortman, K. CD4 expressed on earliest T-lineage precursor cells in the adult murine thymus. Nature 1991, 349, 71–74. [Google Scholar] [CrossRef]
- Allman, D.; Sambandam, A.; Kim, S.; Miller, J.P.; Pagan, A.; Well, D.; Meraz, A.; Bhandoola, A. Thymopoiesis independent of common lymphoid progenitors. Nat. Immunol. 2003, 4, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Foss, D.L.; Donskoy, E.; Goldschneider, I. The importation of hematogenous precursors by the thymus is a gated phenomenon in normal adult mice. J. Exp. Med. 2001, 193, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziętara, N.; Łyszkiewicz, M.; Puchałka, J.; Witzlau, K.; Reinhardt, A.; Förster, R.; Pabst, O.; Prinz, I.; Krueger, A. Multicongenic fate mapping quantification of dynamics of thymus colonization. J. Exp. Med. 2015, 212, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Gossens, K.; Naus, S.; Corbel, S.Y.; Lin, S.; Rossi, F.M.; Kast, J.; Ziltener, H.J. Thymic progenitor homing and lymphocyte homeostasis are linked via S1P-controlled expression of thymic P-selectin/CCL25. J. Exp. Med. 2009, 206, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Donskoy, E.; Foss, D.; Goldschneider, I. Gated importation of prothymocytes by adult mouse thymus is coordinated with their periodic mobilization from bone marrow. J. Immunol. 2003, 171, 3568–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, D.I.; Kennedy, J.; Suda, T.; Zlotnik, A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8-triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J. Immunol. 1993, 150, 4244–4252. [Google Scholar] [PubMed]
- Ceredig, R.; Lowenthal, J.W.; Nabholz, M.; Macdonald, H.R. Expression of interleukin-2 receptors as a differentiation marker on intrathymic stem cells. Nature 1985, 314, 98–100. [Google Scholar] [CrossRef]
- Yui, M.A.; Feng, N.; Rothenberg, E.V. Fine-scale staging of T cell lineage commitment in adult mouse thymus. J. Immunol. 2010, 185, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Taylor, A.A.; Coburn, M.Z.; Marino, J.H.; Van De Wiele, C.J.; Teague, T.K. Ten-color flow cytometry reveals distinct patterns of expression of CD124 and CD126 by developing thymocytes. BMC Immunol. 2011, 12, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porritt, H.E.; Gordon, K.; Petrie, H.T. Kinetics of steady-state differentiation and mapping of intrathymic-signaling environments by stem cell transplantation in nonirradiated mice. J. Exp. Med. 2003, 198, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Manesso, E.; Chickarmane, V.; Kueh, H.Y.; Rothenberg, E.V.; Peterson, C. Computational modelling of T-cell formation kinetics: Output regulated by initial proliferation-linked deferral of developmental competence. J. R. Soc. Interface 2013, 10, 20120774. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yui, M.A.; Williams, B.A.; Yun, J.; Wold, B.J.; Cai, L.; Rothenberg, E.V. Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst. 2019, 9, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Schlenner, S.M.; Madan, V.; Busch, K.; Tietz, A.; Läufle, C.; Costa, C.; Blum, C.; Fehling, H.J.; Rodewald, H.R. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity 2010, 32, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Saran, N.; Łyszkiewicz, M.; Pommerencke, J.; Witzlau, K.; Vakilzadeh, R.; Ballmaier, M.; von Boehmer, H.; Krueger, A. Multiple extrathymic precursors contribute to T-cell development with different kinetics. Blood J. Am. Soc. Hematol. 2010, 115, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.Q.; Landman, K.A.; Hughes, B.D.; Witt, C.M. T cell development in the thymus: From periodic seeding to constant output. J. Theor. Biol. 2007, 249, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, N.N.; Brown, D.E.; Diaz, A.I.G.; Rae, A.; Jarra, W.; Thompson, J.; Langhorne, J.; Potocnik, A.J. Induction of an IL7-R+ c-Kit hi myelolymphoid progenitor critically dependent on IFN-γ signaling during acute malaria. Nat. Immunol. 2010, 11, 477–485. [Google Scholar] [CrossRef]
- Chen, E.L.; Thompson, P.K.; Zúñiga-Pflücker, J.C. RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat. Immunol. 2019, 20, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Ungewiss, K.; Azogui, O.; Palacios, R.; Owen, M.J.; Hayday, A.C.; von Boehmer, H. A novel disulfide-linked heterodimer on pre—T cells consists of the T cell receptor β chain and a 33 kd glycoprotein. Cell 1993, 75, 283–294. [Google Scholar] [CrossRef]
- Pénit, C.; Lucas, B.; Vasseur, F. Cell expansion and growth arrest phases during the transition from precursor (CD4-8-) to immature (CD4+ 8+) thymocytes in normal and genetically modified mice. J. Immunol. 1995, 154, 5103–5113. [Google Scholar] [PubMed]
- Stritesky, G.L.; Xing, Y.; Erickson, J.R.; Kalekar, L.A.; Wang, X.; Mueller, D.L.; Jameson, S.C.; Hogquist, K.A. Murine thymic selection quantified using a unique method to capture deleted T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 4679–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au-Yeung, B.B.; Melichar, H.J.; Ross, J.O.; Cheng, D.A.; Zikherman, J.; Shokat, K.M.; Robey, E.A.; Weiss, A. Quantitative and temporal requirements revealed for Zap70 catalytic activity during T cell development. Nat. Immunol. 2014, 15, 687–694. [Google Scholar] [CrossRef]
- Mariathasan, S.; Zakarian, A.; Bouchard, D.; Michie, A.M.; Zúñiga-Pflücker, J.C.; Ohashi, P.S. Duration and strength of extracellular signal-regulated kinase signals are altered during positive versus negative thymocyte selection. J. Immunol. 2001, 167, 4966–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werlen, G.; Hausmann, B.; Palmer, E. A motif in the αβ T-cell receptor controls positive selection by modulating ERK activity. Nature 2000, 406, 422–426. [Google Scholar] [CrossRef]
- McNeil, L.K.; Starr, T.K.; Hogquist, K.A. A requirement for sustained ERK signaling during thymocyte positive selection in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 13574–13579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.A.; Teixeiro, E.; Gill, J.; Hausmann, B.; Roubaty, D.; Holmberg, K.; Werlen, G.; Holländer, G.A.; Gascoigne, N.R.; Palmer, E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 2006, 444, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.; Naeher, D. Affinity threshold for thymic selection through a T-cell receptor–co-receptor zipper. Nat. Rev. Immunol. 2009, 9, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Föhse, L.; Reinhardt, A.; Oberdörfer, L.; Schmitz, S.; Förster, R.; Malissen, B.; Prinz, I. Differential postselection proliferation dynamics of αβ T cells, Foxp3+ regulatory T cells, and invariant NKT cells monitored by genetic pulse labeling. J. Immunol. 2013, 191, 2384–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Campion, A.; Lucas, B.; Dautigny, N.; Léaument, S.; Vasseur, F.; Pénit, C. Quantitative and qualitative adjustment of thymic T cell production by clonal expansion of premigrant thymocytes. J. Immunol. 2002, 168, 1664–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olariu, V.; Yui, M.; Krupinski, P.; Zhou, W.; Deichmann, J.; Rothenberg, E.; Peterson, C. Multi-scale dynamical modelling of T-cell development from an early thymic progenitor state to lineage commitment. Cell Rep. 2020, 34, 108622. [Google Scholar] [CrossRef] [PubMed]
- Egerton, M.; Scollay, R.; Shortman, K. Kinetics of mature T-cell development in the thymus. Proc. Natl. Acad. Sci. USA 1990, 87, 2579–2582. [Google Scholar] [CrossRef] [Green Version]
- Egerton, M.; Shortman, K.; Scollay, R. The kinetics of immature murine thymocyte development in vivo. Int. Immunol. 1990, 2, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.Y. Quantitative Dissection of T Cell Negative Selection Mechanisms in the Thymus. Ph.D. Thesis, The Australian National University, Camberra, Australia, 2017. [Google Scholar]
- McCaughtry, T.M.; Wilken, M.S.; Hogquist, K.A. Thymic emigration revisited. J. Exp. Med. 2007, 204, 2513–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, C.; Seddon, B. Overlapping and asymmetric functions of TCR signaling during thymic selection of CD4 and CD8 lineages. J. Immunol. 2014, 192, 5151–5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, C.; Bains, I.; Yates, A.J.; Seddon, B. Asymmetric thymocyte death underlies the CD4: CD8 T-cell ratio in the adaptive immune system. Proc. Natl. Acad. Sci. USA 2013, 110, E2905–E2914. [Google Scholar] [CrossRef] [Green Version]
- Mehr, R.; Globerson, A.; Perelson, A.S. Modeling positive and negative selection and differentiation processes in the thymus. J. Theor. Biol. 1995, 175, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, M.; Stritesky, G.; Reynolds, J.; Abourashchi, N.; Lythe, G.; Molina-París, C.; Hogquist, K. From pre-DP, post-DP, SP4, and SP8 thymocyte cell counts to a dynamical model of cortical and medullary selection. Front. Immunol. 2014, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Ther. 2005, 108, 308–319. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, N.; Guo, Z.; Chi, F.; Song, Y.; Zhu, X. Wnt4 signaling is associated with the decrease of proliferation and increase of apoptosis during age-related thymic involution. Mol. Med. Rep. 2015, 12, 7568–7576. [Google Scholar] [CrossRef]
- Carbajosa, S.; Gea, S.; Chillón-arinas, C.; Poveda, C.; del Carmen Maza, M.; Fresno, M.; Gironès, N. Altered bone marrow lymphopoiesis and interleukin-6-dependent inhibition of thymocyte differentiation contribute to thymic atrophy during Trypanosoma cruzi infection. Oncotarget 2017, 8, 17551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoller, A.L.; Kersh, G.J. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of β-selected thymocytes. J. Immunol. 2006, 176, 7371–7378. [Google Scholar] [CrossRef] [PubMed]
- Zoller, A.L.; Schnell, F.J.; Kersh, G.J. Murine pregnancy leads to reduced proliferation of maternal thymocytes and decreased thymic emigration. Immunology 2007, 121, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Alves, C.; Nobrega, C.; Behar, S.M.; Correia-Neves, M. Tolerance has its limits: How the thymus copes with infection. Trends Immunol. 2013, 34, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, A.B.; Haasbach, E.; Reiling, S.J.; Droebner, K.; Klingel, K.; Planz, O. Highly pathogenic influenza virus infection of the thymus interferes with T lymphocyte development. J. Immunol. 2010, 185, 4824–4834. [Google Scholar] [CrossRef]
- Thomas-Vaslin, V.; Altes, H.K.; de Boer, R.J.; Klatzmann, D. Comprehensive assessment and mathematical modeling of T cell population dynamics and homeostasis. J. Immunol. 2008, 180, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, Y.; Robert, P.A.; Binz, C.; Falk, C.S.; Bruder, D.; Prinz, I.; Floess, S.; Meyer-Hermann, M.; Huehn, J. Influenza A virus-induced thymus atrophy differentially affects dynamics of conventional and regulatory T cell development. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Moleriu, R.D.; Zaharie, D.; Moatar-Moleriu, L.C.; Gruia, A.T.; Mic, A.A.; Mic, F.A. Insights into the mechanisms of thymus involution and regeneration by modeling the glucocorticoid-induced perturbation of thymocyte populations dynamics. J. Theor. Biol. 2014, 348, 80–99. [Google Scholar] [CrossRef]
- Hu, D.Y.; Yap, J.Y.; Wirasinha, R.C.; Howard, D.R.; Goodnow, C.C.; Daley, S.R. A timeline demarcating two waves of clonal deletion and Foxp3 upregulation during thymocyte development. Immunol. Cell Biol. 2016, 94, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.; Sinclair, C.; Tung, S.; Seddon, B. Differential requirement for IL-2 and IL-15 during bifurcated development of thymic regulatory T cells. J. Immunol. 2014, 193, 5525–5533. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.E.; Parnell, S.M.; Nakamura, K.; Caamano, J.H.; Lane, P.J.; Jenkinson, E.J.; Jenkinson, W.E.; Anderson, G. The thymic medulla is required for Foxp3+ regulatory but not conventional CD4+ thymocyte development. J. Exp. Med. 2013, 210, 675–681. [Google Scholar] [CrossRef]
- Liston, A.; Nutsch, K.M.; Farr, A.G.; Lund, J.M.; Rasmussen, J.P.; Koni, P.A.; Rudensky, A.Y. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. USA 2008, 105, 11903–11908. [Google Scholar] [CrossRef] [Green Version]
- yszkiewicz, M.; Winter, S.J.; Witzlau, K.; Föhse, L.; Brownlie, R.; Puchałka, J.; Verheyden, N.A.; Kunze-Schumacher, H.; Imelmann, E.; Blume, J.; et al. miR-181a/b-1 controls thymic selection of Treg cells and tunes their suppressive capacity. PLoS Biol. 2019, 17, e2006716. [Google Scholar]
- Owen, D.L.; Mahmud, S.A.; Sjaastad, L.E.; Williams, J.B.; Spanier, J.A.; Simeonov, D.R.; Ruscher, R.; Huang, W.; Proekt, I.; Miller, C.N.; et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat. Immunol. 2019, 20, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Zaharie, D.; Moleriu, R.D.; Mic, F.A. Modeling the development of the post-natal mouse thymus in the absence of bone marrow progenitors. Sci. Rep. 2016, 6, 36159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Freeden-Jeffry, U.; Vieira, P.; Lucian, L.A.; McNeil, T.; Burdach, S.; Murray, R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 1995, 181, 1519–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.R.; Borghans, J.A.; Freitas, A.A. T Cell HomeostasisThymus Regeneration and Peripheral T Cell Restoration in Mice with a Reduced Fraction of Competent Precursors. J. Exp. Med. 2001, 194, 591–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotoff, D.A.; Sambandam, A.; Logan, T.D.; Bell, J.J.; Schwarz, B.A.; Bhandoola, A. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood J. Am. Soc. Hematol. 2010, 115, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, A.; Willenzon, S.; Łyszkiewicz, M.; Kremmer, E.; Förster, R. CC chemokine receptor 7 and 9 double-deficient hematopoietic progenitors are severely impaired in seeding the adult thymus. Blood J. Am. Soc. Hematol. 2010, 115, 1906–1912. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.V.; Ballesteros-Arias, L.; Silva, J.G.; Paiva, R.A.; Nogueira, M.F.; Carneiro, J.; Gjini, E.; Martins, V.C. Cell Competition, the Kinetics of Thymopoiesis, and Thymus Cellularity Are Regulated by Double-Negative 2 to 3 Early Thymocytes. Cell Rep. 2020, 32, 107910. [Google Scholar] [CrossRef]
- Apert, C.; Romagnoli, P.; van Meerwijk, J.P. IL-2 and IL-15 dependent thymic development of Foxp3-expressing regulatory T lymphocytes. Protein Cell 2018, 9, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiault, N.; Darrigues, J.; Adoue, V.; Gros, M.; Binet, B.; Perals, C.; Leobon, B.; Fazilleau, N.; Joffre, O.P.; Robey, E.A.; et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat. Immunol. 2015, 16, 628–634. [Google Scholar] [CrossRef]
- Klein, L.; Robey, E.A.; Hsieh, C.S. Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 2019, 19, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.B.; Tateishi, R.; Miyao, T.; Takakura, Y.; Akiyama, N.; Yokota, R.; Akiyama, T.; Kobayashi, T.J. Quantitative analysis reveals reciprocal regulations underlying recovery dynamics of thymocytes and thymic environment in mice. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.C.; Hernandez-Vargas, E.A.; Meyer-Hermann, M. Reducing complexity: An iterative strategy for parameter determination in biological networks. Comput. Phys. Commun. 2015, 190, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Bandara, S.; Schlöder, J.P.; Eils, R.; Bock, H.G.; Meyer, T. Optimal experimental design for parameter estimation of a cell signaling model. PLoS Comput. Biol. 2009, 5, e1000558. [Google Scholar] [CrossRef] [PubMed]
- Graziano, M.; St-Pierre, Y.; Beauchemin, C.; Desrosiers, M.; Potworowski, E.F. The Fate of Thymocytes Labeled in vivo with CFSE. Exp. Cell Res. 1998, 240, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kreslavsky, T.; Gleimer, M.; Miyazaki, M.; Choi, Y.; Gagnon, E.; Murre, C.; Sicinski, P.; von Boehmer, H. β-Selection-induced proliferation is required for αβ T cell differentiation. Immunity 2012, 37, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Hare, K.J.; Wilkinson, R.W.; Jenkinson, E.J.; Anderson, G. Identification of a developmentally regulated phase of postselection expansion driven by thymic epithelium. J. Immunol. 1998, 160, 3666–3672. [Google Scholar] [PubMed]
- Quackenbush, R.; Shields, A. Local re-utilization of thymidine in normal mouse tissues as measured with iododeoxyuridine. Cell Prolif. 1988, 21, 381–387. [Google Scholar] [CrossRef]
- Hagan, M.C. Cell Proliferation Kinetics Analyzed with BrdU and Near-UV Light Treatment1. In Current Methodology in Experimental Hematology; Karger Publishers: Basel, Switzerland, 1984; Volume 48, pp. 384–401. [Google Scholar]
- Matiašová, A.; Ševc, J.; Mikeš, J.; Jendželovskỳ, R.; Daxnerová, Z.; Fedoročko, P. Flow cytometric determination of 5-bromo-2-deoxyuridine pharmacokinetics in blood serum after intraperitoneal administration to rats and mice. Histochem. Cell Biol. 2014, 142, 703–712. [Google Scholar] [CrossRef]
- Vogel, K.U.; Bell, L.S.; Galloway, A.; Ahlfors, H.; Turner, M. The RNA-binding proteins Zfp36l1 and Zfp36l2 enforce the thymic β-selection checkpoint by limiting DNA damage response signaling and cell cycle progression. J. Immunol. 2016, 197, 2673–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonhoeffer, S.; Mohri, H.; Ho, D.; Perelson, A.S. Quantification of cell turnover kinetics using 5-bromo-2-deoxyuridine1. J. Immunol. 2000, 164, 5049–5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, C.; Pénit, C. Study of the thymocyte cell cycle by bivariate analysis of incorporated bromodeoxyuridine and DNA content. Eur. J. Immunol. 1990, 20, 1231–1236. [Google Scholar] [CrossRef]
- Vibert, J.; Thomas-Vaslin, V. Modelling T cell proliferation: Dynamics heterogeneity depending on cell differentiation, age, and genetic background. PLoS Comput. Biol. 2017, 13, e1005417. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.V.; Ballesteros-Arias, L.; Silva, J.G.; Nogueira, M.; Gjini, E.; Martins, V.C. Cell competition regulates the kinetics of thymopoiesis and thymus cellularity. bioRxiv 2019. [Google Scholar] [CrossRef]
- Jolly, A.; Fanti, A.K.; Gräßer, I.; Becker, N.B.; Höfer, T. CycleFlow quantifies cell-cycle heterogeneity in vivo. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, A.D.; Mayer, C.T.; Oliveira, T.Y.; Shulman, Z.; Jones, M.J.; Koren, A.; Nussenzweig, M.C. T cell help controls the speed of the cell cycle in germinal center B cells. Science 2015, 349, 643–646. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, L.; Flossdorf, M.; Mir, J.; Cho, Y.L.; Plambeck, M.; Treise, I.; Toska, A.; Heinzel, S.; Schiemann, M.; Busch, D.H.; et al. Differential expansion of T central memory precursor and effector subsets is regulated by division speed. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.S.; Jaehnert, I.; Schichor, C.; Or-Guil, M.; Carneiro, J. Quantifying the length and variance of the eukaryotic cell cycle phases by a stochastic model and dual nucleoside pulse labelling. PLoS Comput. Biol. 2014, 10, e1003616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilman, A.; Ganusov, V.V.; Perelson, A.S. Stochastic models of lymphocyte proliferation and death. PLoS ONE 2010, 5, e12775. [Google Scholar] [CrossRef] [PubMed]
- Trucco, E. Mathematical models for cellular systems. The von Foerster equation. Part II. Bull. Math. Biophys. 1965, 27, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Wellard, C.; Markham, J.F.; Hawkins, E.D.; Hodgkin, P.D. The cyton model for lymphocyte proliferation and differentiation. In Mathematical Models and Immune Cell Biology; Springer: New York, NY, USA, 2011; pp. 107–120. [Google Scholar]
- Miles, J. The Laplace transform of the lognormal distribution. arXiv 2018, arXiv:1803.05878. [Google Scholar]
- Bernard, D.; Mondesert, O.; Gomes, A.; Duthen, Y.; Lobjois, V.; Cussat-Blanc, S.; Ducommun, B. A checkpoint-oriented cell cycle simulation model. Cell Cycle 2019, 18, 795–808. [Google Scholar] [CrossRef]
- Dowling, M.R.; Kan, A.; Heinzel, S.; Zhou, J.H.; Marchingo, J.M.; Wellard, C.J.; Markham, J.F.; Hodgkin, P.D. Stretched cell cycle model for proliferating lymphocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 6377–6382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 2018, 24, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambon, A.C. Use of the Ki67 promoter to label cell cycle entry in living cells. Cytom. Part A 2010, 77, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bains, I.; Thiébaut, R.; Yates, A.J.; Callard, R. Quantifying thymic export: Combining models of naive T cell proliferation and TCR excision circle dynamics gives an explicit measure of thymic output. J. Immunol. 2009, 183, 4329–4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahoz-Beneytez, J.; Schaller, S.; Macallan, D.; Eissing, T.; Niederalt, C.; Asquith, B. Physiologically based simulations of deuterated glucose for quantifying cell turnover in humans. Front. Immunol. 2017, 8, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwata, N.; Igarashi, H.; Ohmura, T.; Aizawa, S.; Sakaguchi, N. Cutting edge: Absence of expression of RAG1 in peritoneal B-1 cells detected by knocking into RAG1 locus with green fluorescent protein gene. J. Immunol. 1999, 163, 6355–6359. [Google Scholar] [PubMed]
- Winter, S.J.; Krueger, A. Development of unconventional T cells controlled by MicroRNA. Front. Immunol. 2019, 10, 2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernitz, J.M.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic stem cells count and remember self-renewal divisions. Cell 2016, 167, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Morcos, M.N.; Zerjatke, T.; Glauche, I.; Munz, C.M.; Ge, Y.; Petzold, A.; Reinhardt, S.; Dahl, A.; Anstee, N.; Bogeska, R.; et al. Proliferative behavior of hematopoietic stem cells revisited: No evidence for mitotic memory. bioRxiv 2019, 745729. [Google Scholar] [CrossRef]
- Prinz, I.; Sansoni, A.; Kissenpfennig, A.; Ardouin, L.; Malissen, M.; Malissen, B. Visualization of the earliest steps of γδ T cell development in the adult thymus. Nat. Immunol. 2006, 7, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Yo, M.; Komatsu, N.; Hiratsuka, T.; Kogure, T.; Hoshida, T.; Goshima, N.; Matsuda, M.; Miyoshi, H.; Miyawaki, A. Genetically encoded tools for optical dissection of the mammalian cell cycle. Mol. Cell 2017, 68, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Kurd, N.S.; Lutes, L.K.; Yoon, J.; Chan, S.W.; Dzhagalov, I.L.; Hoover, A.R.; Robey, E.A. A role for phagocytosis in inducing cell death during thymocyte negative selection. ELife 2019, 8, e48097. [Google Scholar] [CrossRef] [PubMed]
- Surh, C.D.; Sprent, J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature 1994, 372, 100–103. [Google Scholar] [CrossRef]
- Laufer, T.M.; DeKoning, J.; Markowitz, J.S.; Lo, D.; Glimcher, L.H. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature 1996, 383, 81–85. [Google Scholar] [CrossRef]
- van Meerwijk, J.P.; Marguerat, S.; Lees, R.K.; Germain, R.N.; Fowlkes, B.; MacDonald, H.R. Quantitative impact of thymic clonal deletion on the T cell repertoire. J. Exp. Med. 1997, 185, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.; Partington, K.M.; Jenkinson, E.J. Differential effects of peptide diversity and stromal cell type in positive and negative selection in the thymus. J. Immunol. 1998, 161, 6599–6603. [Google Scholar]
- Merkenschlager, M.; Graf, D.; Lovatt, M.; Bommhardt, U.; Zamoyska, R.; Fisher, A.G. How many thymocytes audition for selection? J. Exp. Med. 1997, 186, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Itano, A.; Robey, E. Highly efficient selection of CD4 and CD8 lineage thymocytes supports an instructive model of lineage commitment. Immunity 2000, 12, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Daley, S.R.; Hu, D.Y.; Goodnow, C.C. Helios marks strongly autoreactive CD4+ T cells in two major waves of thymic deletion distinguished by induction of PD-1 or NF-κB. J. Exp. Med. 2013, 210, 269–285. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.D.; Bunker, J.J.; Erickson, S.A.; Oh-Hora, M.; Bendelac, A. Crossreactive αβ T cell receptors are the predominant targets of thymocyte negative selection. Immunity 2015, 43, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Hogquist, K.A.; Jameson, S.C. The self-obsession of T cells: How TCR signaling thresholds affect fate ’decisions’ and effector function. Nat. Immunol. 2014, 15, 815. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Singer, A. Tuning of activation thresholds explains flexibility in the selection and development of T cells in the thymus. Proc. Natl. Acad. Sci. USA 1996, 93, 14747–14752. [Google Scholar] [CrossRef] [Green Version]
- Bhakta, N.R.; Oh, D.Y.; Lewis, R.S. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat. Immunol. 2005, 6, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Melichar, H.J.; Ross, J.O.; Herzmark, P.; Hogquist, K.A.; Robey, E.A. Distinct temporal patterns of T cell receptor signaling during positive versus negative selection in situ. Sci. Signal. 2013, 6, ra92. [Google Scholar] [CrossRef] [Green Version]
- Melichar, H.J.; Ross, J.O.; Taylor, K.T.; Robey, E.A. Stable interactions and sustained TCR signaling characterize thymocyte–thymocyte interactions that support negative selection. J. Immunol. 2015, 194, 1057–1061. [Google Scholar] [CrossRef]
- Ross, J.O.; Melichar, H.J.; Au-Yeung, B.B.; Herzmark, P.; Weiss, A.; Robey, E.A. Distinct phases in the positive selection of CD8+ T cells distinguished by intrathymic migration and T-cell receptor signaling patterns. Proc. Natl. Acad. Sci. USA 2014, 111, E2550–E2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurd, N.; Robey, E.A. T-cell selection in the thymus: A spatial and temporal perspective. Immunol. Rev. 2016, 271, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khailaie, S.; Robert, P.A.; Toker, A.; Huehn, J.; Meyer-Hermann, M. A signal integration model of thymic selection and natural regulatory T cell commitment. J. Immunol. 2014, 193, 5983–5996. [Google Scholar] [CrossRef] [PubMed]
- Sant’Angelo, D.B.; Waterbury, P.G.; Cohen, B.E.; Martin, W.D.; Van Kaer, L.; Hayday, A.C.; Janeway, C.A., Jr. The imprint of intrathymic self-peptides on the mature T cell receptor repertoire. Immunity 1997, 7, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.J.; Jiang, S.; Xie, J.; Li, Q.J.; Davis, M.M. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat. Immunol. 2009, 10, 1162. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.L.; Felix, N.J.; Walters, J.J.; Rohrs, H.; Gross, M.L.; Allen, P.M. An endogenous peptide positively selects and augments the activation and survival of peripheral CD4+ T cells. Nat. Immunol. 2009, 10, 1155. [Google Scholar] [CrossRef] [PubMed]
- Vrisekoop, N.; Monteiro, J.P.; Mandl, J.N.; Germain, R.N. Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity 2014, 41, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burroughs, N.J.; de Boer, R.J.; Keşmir, C. Discriminating self from nonself with short peptides from large proteomes. Immunogenetics 2004, 56, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganti, R.S.; Lo, W.L.; McAffee, D.B.; Groves, J.T.; Weiss, A.; Chakraborty, A.K. How the T cell signaling network processes information to discriminate between self and agonist ligands. Proc. Natl. Acad. Sci. USA 2020, 117, 26020–26030. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, G.P.; Bugaj, L.J.; Anderson, W.; Daniels, K.G.; Rawlings, D.J.; Lim, W.A. T cells selectively filter oscillatory signals on the minutes timescale. Proc. Natl. Acad. Sci. USA 2021, 118, e2019285118. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Tsutsumi, M.; Nitta, S.; Muro, R.; Suzuki, E.C.; Nakano, K.; Tomofuji, Y.; Sawa, S.; Okamura, T.; Penninger, J.M.; et al. Fibroblasts as a source of self-antigens for central immune tolerance. Nat. Immunol. 2020, 21, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Rudensky, A.Y. Conceiving the inconceivable: The function of Aire in immune tolerance to peripheral tissue-restricted antigens in the thymus. J. Immunol. 2021, 206, 245–247. [Google Scholar] [CrossRef]
- Breed, E.R.; Lee, S.T.; Hogquist, K.A. Directing T cell fate: How thymic antigen presenting cells coordinate thymocyte selection. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 84, pp. 2–10. [Google Scholar]
- Cosway, E.J.; Ohigashi, I.; Schauble, K.; Parnell, S.M.; Jenkinson, W.E.; Luther, S.; Takahama, Y.; Anderson, G. Formation of the intrathymic dendritic cell pool requires CCL21-mediated recruitment of CCR7+ progenitors to the thymus. J. Immunol. 2018, 201, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Li, Y.; Van Nieuwenhuijze, A.; Selden, H.J.; Jarrett, A.M.; Sorace, A.G.; Yankeelov, T.E.; Liston, A.; Ehrlich, L.I. CCR7 modulates the generation of thymic regulatory T cells by altering the composition of the thymic dendritic cell compartment. Cell Rep. 2017, 21, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Davalos-Misslitz, A.C.; Worbs, T.; Willenzon, S.; Bernhardt, G.; Förster, R. Impaired responsiveness to T-cell receptor stimulation and defective negative selection of thymocytes in CCR7-deficient mice. Blood J. Am. Soc. Hematol. 2007, 110, 4351–4359. [Google Scholar] [CrossRef]
- Garg, G.; Nikolouli, E.; Hardtke-Wolenski, M.; Toker, A.; Ohkura, N.; Beckstette, M.; Miyao, T.; Geffers, R.; Floess, S.; Gerdes, N.; et al. Unique properties of thymic antigen-presenting cells promote epigenetic imprinting of alloantigen-specific regulatory T cells. Oncotarget 2017, 8, 35542. [Google Scholar] [CrossRef] [Green Version]
- Efroni, S.; Harel, D.; Cohen, I.R. Toward rigorous comprehension of biological complexity: Modeling, execution, and visualization of thymic T-cell maturation. Genome Res. 2003, 13, 2485–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: A user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efroni, S.; Harel, D.; Cohen, I.R. Emergent dynamics of thymocyte development and lineage determination. PLoS Comput. Biol. 2007, 3, e13. [Google Scholar] [CrossRef] [PubMed]
- Souza-e Silva, H.; Savino, W.; Feijóo, R.A.; Vasconcelos, A.T.R. A cellular automata-based mathematical model for thymocyte development. PLoS ONE 2009, 4, e8233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Textor, J.; Henrickson, S.E.; Mandl, J.N.; Von Andrian, U.H.; Westermann, J.; De Boer, R.J.; Beltman, J.B. Random migration and signal integration promote rapid and robust T cell recruitment. PLoS Comput. Biol. 2014, 10, e1003752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, A.; Robert, P.A.; Halle, S.; Meyer-Hermann, M. Evaluation of CD8 T cell killing models with computer simulations of 2-photon imaging experiments. PLoS Comput. Biol. 2020, 16, e1008428. [Google Scholar] [CrossRef]
- Brown, A.J.; Snapkov, I.; Akbar, R.; Pavlović, M.; Miho, E.; Sandve, G.K.; Greiff, V. Augmenting adaptive immunity: Progress and challenges in the quantitative engineering and analysis of adaptive immune receptor repertoires. Mol. Syst. Des. Eng. 2019, 4, 701–736. [Google Scholar] [CrossRef]
- Marcou, Q.; Mora, T.; Walczak, A.M. High-throughput immune repertoire analysis with IGoR. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Elhanati, Y.; Murugan, A.; Callan, C.G.; Mora, T.; Walczak, A.M. Quantifying selection in immune receptor repertoires. Proc. Natl. Acad. Sci. USA 2014, 111, 9875–9880. [Google Scholar] [CrossRef] [Green Version]
- Pei, W.; Feyerabend, T.B.; Rössler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Botting, R.A.; Conde, C.D.; Popescu, D.M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 2020, 367, eaay3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, H.; Rothenberg, E.V. How transcription factors drive choice of the T cell fate. Nat. Rev. Immunol. 2020, 21, 162–176. [Google Scholar] [CrossRef] [PubMed]
- yszkiewicz, M.; Ziętara, N.; Föhse, L.; Puchałka, J.; Diestelhorst, J.; Witzlau, K.; Prinz, I.; Schambach, A.; Krueger, A. Limited niche availability suppresses murine intrathymic dendritic-cell development from noncommitted progenitors. Blood J. Am. Soc. Hematol. 2015, 125, 457–464. [Google Scholar]
- Ishikawa, T.; Akiyama, N.; Akiyama, T. In Pursuit of Adult Progenitors of Thymic Epithelial Cells. Front. Immunol. 2021, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- James, K.D.; Jenkinson, W.E.; Anderson, G. Non-epithelial stromal cells in thymus development and function. Front. Immunol. 2021, 12, 634367. [Google Scholar] [CrossRef]
- Albinsson, S.; Lingblom, C.; Lundqvist, C.; Telemo, E.; Ekwall, O.; Wennerås, C. Eosinophils interact with thymocytes and proliferate in the human thymus. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Santamaria, J.; Darrigues, J.; van Meerwijk, J.P.; Romagnoli, P. Antigen-presenting cells and T-lymphocytes homing to the thymus shape T cell development. Immunol. Lett. 2018, 204, 9–15. [Google Scholar] [CrossRef]
- Yamano, T.; Nedjic, J.; Hinterberger, M.; Steinert, M.; Koser, S.; Pinto, S.; Gerdes, N.; Lutgens, E.; Ishimaru, N.; Busslinger, M.; et al. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity 2015, 42, 1048–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S.; Cervantes, M.; Sassone-Corsi, P. The circadian clock and cell cycle: Interconnected biological circuits. Curr. Opin. Cell Biol. 2013, 25, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamy, M.; Beck-Garcia, K.; Beck-Garcia, E.; Hartl, F.A.; Morath, A.; Yousefi, O.S.; Dopfer, E.P.; Molnár, E.; Schulze, A.K.; Blanco, R.; et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity 2016, 44, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombs, D.; Dushek, O.; van der Merwe, P.A. A review of mathematical models for T cell receptor triggering and antigen discrimination. Math. Model. Immune Cell Biol. 2011, 25–45. [Google Scholar] [CrossRef]
- Altan-Bonnet, G.; Germain, R.N. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005, 3, e356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzella, T.S.; Barreto, V.M.; Carneiro, J. Partitioning stable and unstable expression level variation in cell populations: A theoretical framework and its application to the T cell receptor. PLoS Comput. Biol. 2020, 16, e1007910. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robert, P.A.; Kunze-Schumacher, H.; Greiff, V.; Krueger, A. Modeling the Dynamics of T-Cell Development in the Thymus. Entropy 2021, 23, 437. https://doi.org/10.3390/e23040437
Robert PA, Kunze-Schumacher H, Greiff V, Krueger A. Modeling the Dynamics of T-Cell Development in the Thymus. Entropy. 2021; 23(4):437. https://doi.org/10.3390/e23040437
Chicago/Turabian StyleRobert, Philippe A., Heike Kunze-Schumacher, Victor Greiff, and Andreas Krueger. 2021. "Modeling the Dynamics of T-Cell Development in the Thymus" Entropy 23, no. 4: 437. https://doi.org/10.3390/e23040437
APA StyleRobert, P. A., Kunze-Schumacher, H., Greiff, V., & Krueger, A. (2021). Modeling the Dynamics of T-Cell Development in the Thymus. Entropy, 23(4), 437. https://doi.org/10.3390/e23040437