
A Drive towards Thermodynamic Efficiency for Dissipative Structures in Chemical Reaction Networks



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Stochastic Chemical Reaction Network Simulations
2.2. Quantification of Minimum Work-Rate Required to Maintain Nonequilibrium Steady States
2.3. Quantification of the Thermodynamic Efficiency of Bifurcation-Based Work-Harvesting Processes
3. Results
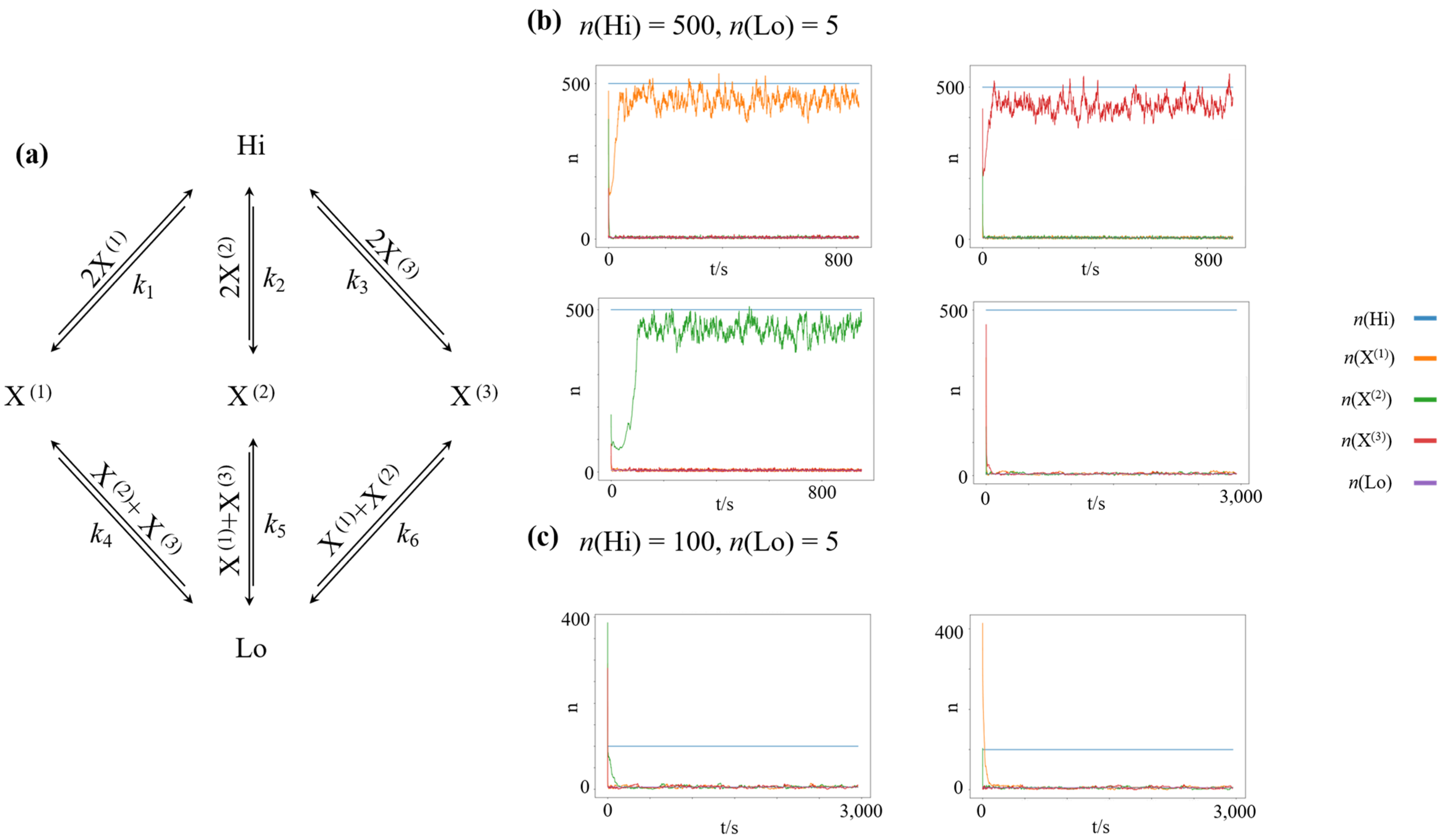
3.1. Chemical Winner-Take-All Dynamics
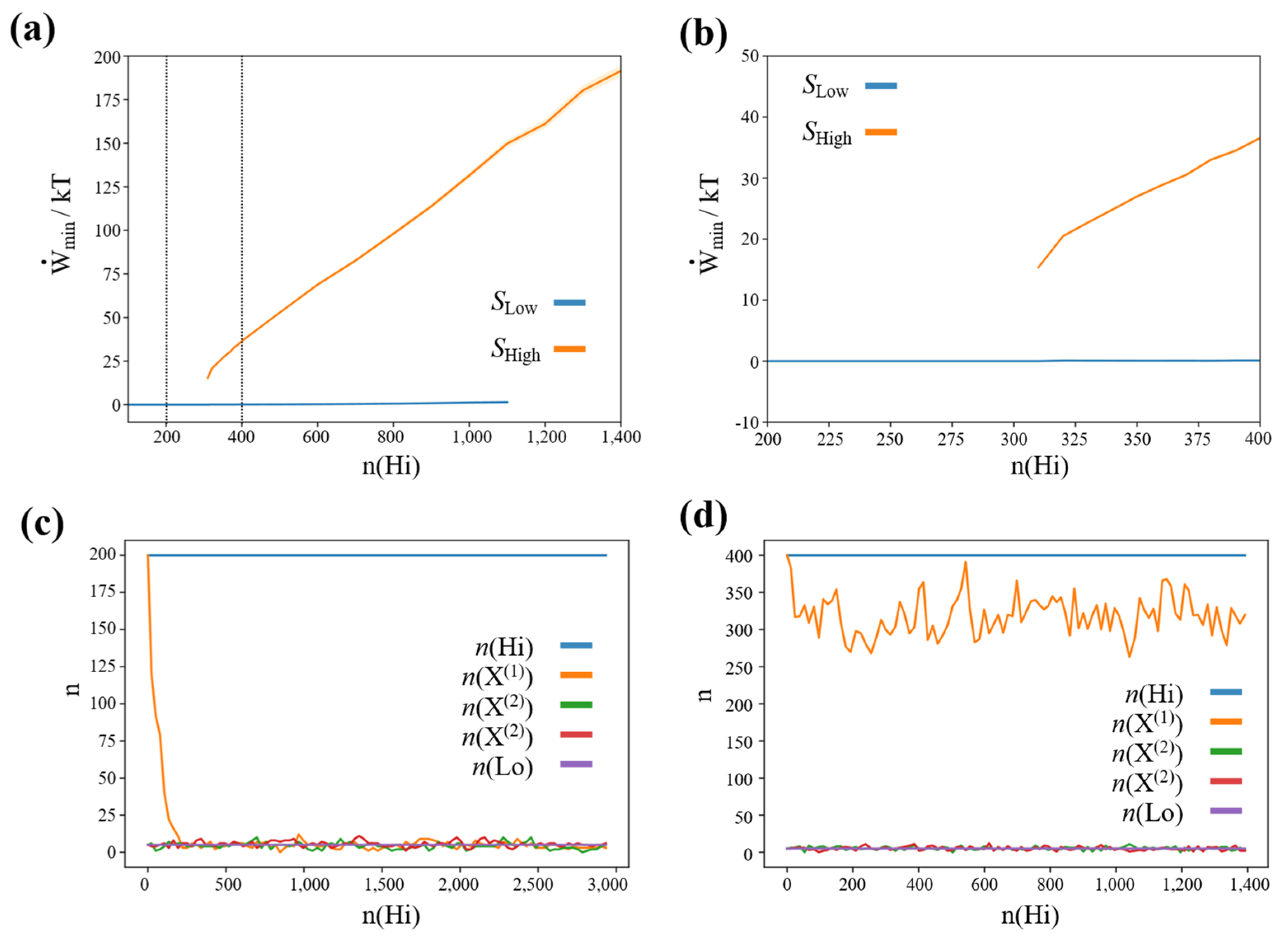
3.2. Finite Minimum Work-Rate Required to Maintain High-Concentration States
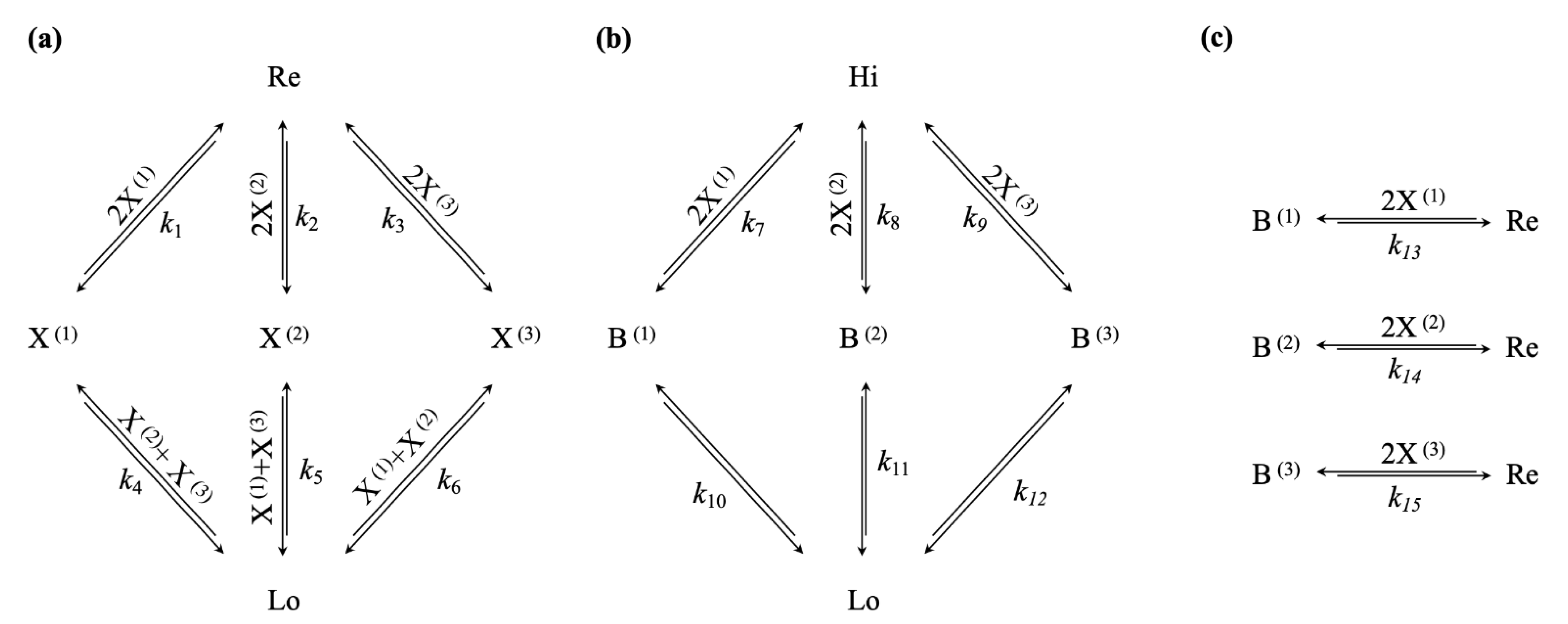
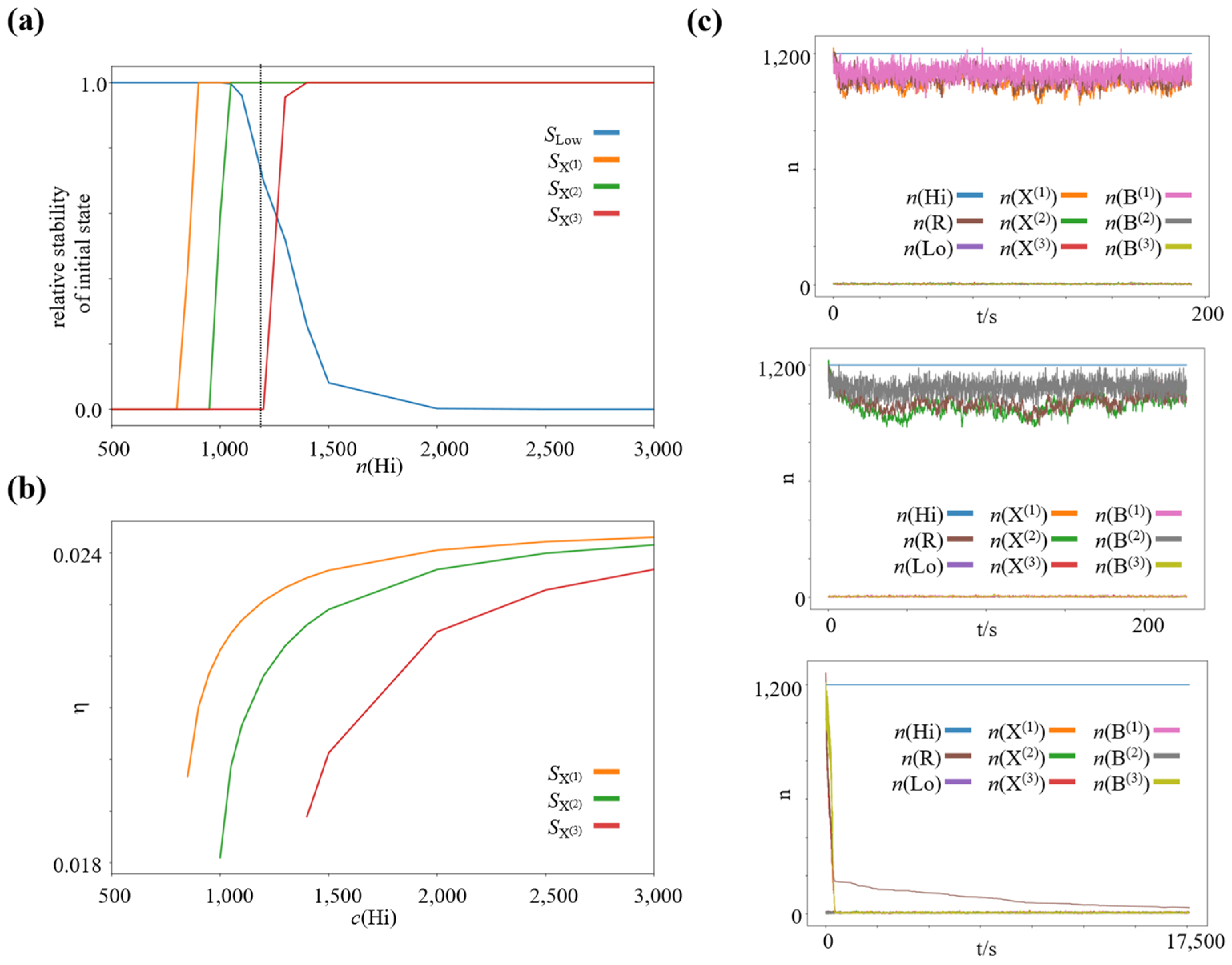
3.3. Decreasing the Driving Chemical Potential Difference Leads to Self-Selection of Nonequilibrium Steady States with High-Efficiency Bifurcation Processes
4. Discussion
4.1. A Simple Selection Mechanism for Thermodynamic Efficiency in Dissipative Nonequilibrium Steady States of Chemical Reaction Networks
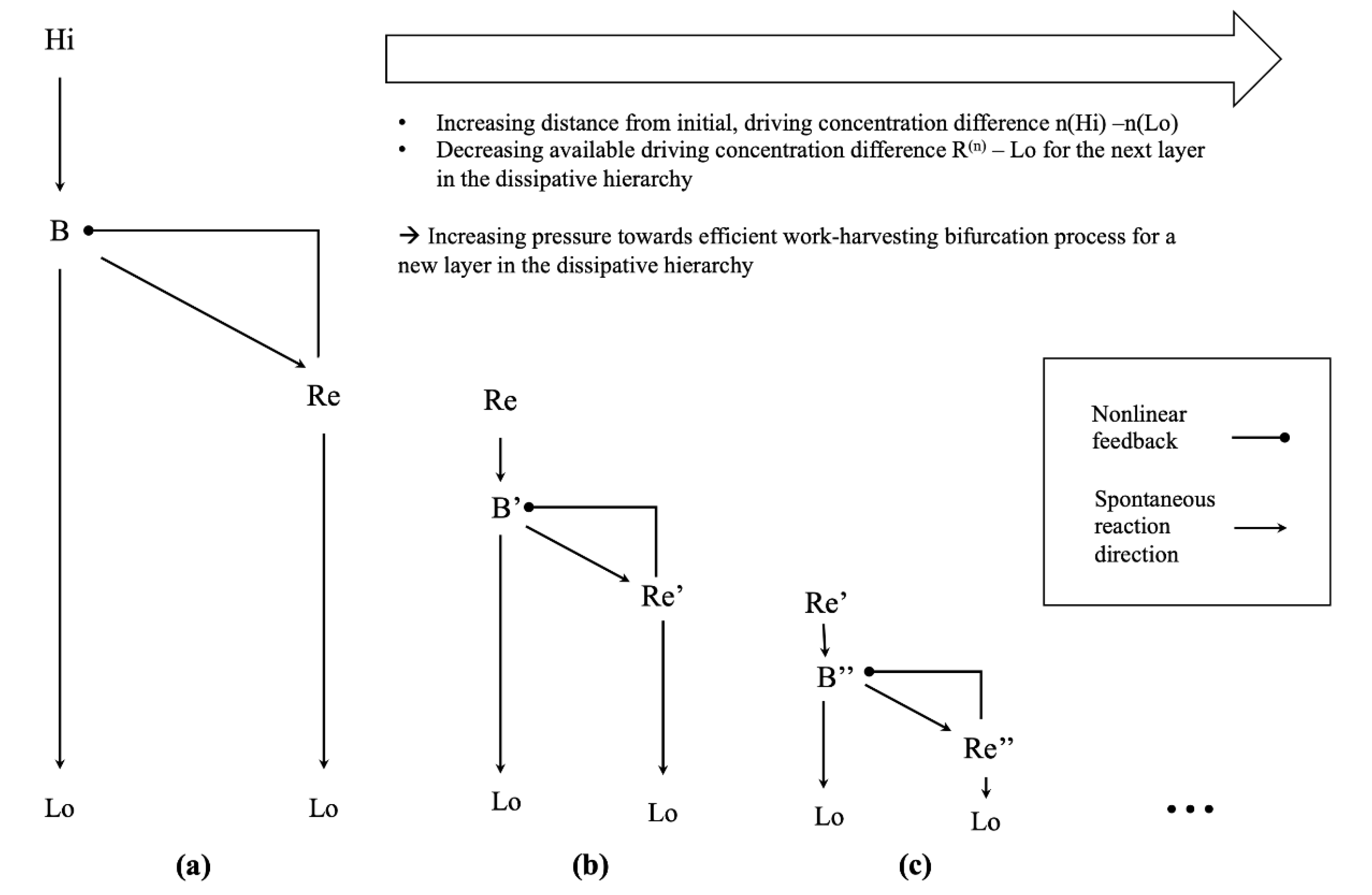
4.2. Emergent Pressure towards Thermodynamic Efficiency in a Hierarchy of Dissipative Structures
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branscomb, E.; Russell, M.J. Turnstiles and bifurcators: The disequilibrium converting engines that put metabolism on the road. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 62–78. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.J.; Nitschke, W.; Branscomb, E. The inevitable journey to being. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120254. [Google Scholar] [CrossRef] [PubMed]
- Morowitz, H.; Smith, E. Energy flow and the organization of life. Complexity 2007, 13, 51–59. [Google Scholar] [CrossRef]
- Goldenfeld, N.; Biancalani, T.; Jafarpour, F. Universal biology and the statistical mechanics of early life. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2017, 375, 20160341. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Morowitz, H.J. The Origin and Nature of Life on Earth: The Emergence of the Fourth Geosphere; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Jeffery, K.; Pollack, R.; Rovelli, C. On the Statistical Mechanics of Life: Schrödinger Revisited. Entropy 2019, 21, 1211. [Google Scholar] [CrossRef] [Green Version]
- Lotka, A.J. Natural Selection as a Physical Principle. Proc. Natl. Acad. Sci. USA 1922, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotka, A.J. Contribution to the Energetics of Evolution. Proc. Natl. Acad. Sci. USA 1922, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigogine, I.; Nicolis, G. Biological order, structure and instabilities. Q. Rev. Biophys. 1971, 4, 107–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottrell, A. The natural philosophy of engines. Contemp. Phys. 1979, 20, 1–10. [Google Scholar] [CrossRef]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E 1999, 60, 2721–2726. [Google Scholar] [CrossRef] [Green Version]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef] [Green Version]
- Van den Broeck, C.; Esposito, M. Ensemble and trajectory thermodynamics: A brief introduction. Phys. A Stat. Mech. Its Appl. 2015, 418, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U. Stochastic thermodynamics: From principles to the cost of precision. Phys. A Stat. Mech. Its Appl. 2018, 504, 176–191. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U. Stochastic thermodynamics, fluctuation theorems and molecular machines. Rep. Prog. Phys. 2012, 75, 126001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perunov, N.; Marsland, R.A.; England, J.L. Statistical Physics of Adaptation. Phys. Rev. X 2016, 6, 021036. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.I.; Kim, H.; Davies, P.C.W. The informational architecture of the cell. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150057. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.I.; Davies, P.C.W. The algorithmic origins of life. J. R. Soc. Interface 2013, 10, 20120869. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.C.W.; Walker, S.I. The hidden simplicity of biology. Rep. Prog. Phys. 2016, 79, 102601. [Google Scholar] [CrossRef] [PubMed]
- Still, S.; Sivak, D.A.; Bell, A.J.; Crooks, G.E. Thermodynamics of Prediction. Phys. Rev. Lett. 2012, 109, 120604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishby, N.; Pereira, F.C.; Bialek, W. The information bottleneck method. In Proceedings of the 37th Annual Allerton Conference on Communication, Control and Computing; Hajek, B., Sreenivas, R.S., Eds.; University of Illinois: Urbana, IL, USA, 1999; pp. 368–377. [Google Scholar]
- Still, S. Information-theoretic approach to interactive learning. EPL 2009, 85, 28005. [Google Scholar] [CrossRef] [Green Version]
- Still, S.; Precup, D. An information-theoretic approach to curiosity-driven reinforcement learning. Theory Biosci. 2012, 131, 139–148. [Google Scholar] [CrossRef]
- Still, S. Information Bottleneck Approach to Predictive Inference. Entropy 2014, 16, 968–989. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, V.; Kimber, D.; Ii, M.J.B. Metabolically Efficient Information Processing. Neural Comput. 2001, 13, 799–815. [Google Scholar] [CrossRef]
- Krieg, D.; Triesch, J. A unifying theory of synaptic long-term plasticity based on a sparse distribution of synaptic strength. Front. Synaptic Neurosci. 2014, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Perge, J.A.; Koch, K.; Miller, R.; Sterling, P.; Balasubramanian, V. How the Optic Nerve Allocates Space, Energy Capacity, and Information. J. Neurosci. 2009, 29, 7917. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; McLean, J.; Segev, R.; Freed, M.A.; Berry, M.J., II; Balasubramanian, V.; Sterling, P. How Much the Eye Tells the Brain. Curr. Biol. 2006, 16, 1428–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, W.B.; Baxter, R.A. Energy-Efficient Neuronal Computation via Quantal Synaptic Failures. J. Neurosci. 2002, 22, 4746. [Google Scholar] [CrossRef] [PubMed]
- Levy, W.B.; Baxter, R.A. Energy Efficient Neural Codes. Neural Comput. 1996, 8, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Sarpeshkar, R. Analog Versus Digital: Extrapolating from Electronics to Neurobiology. Neural Comput. 1998, 10, 1601–1638. [Google Scholar] [CrossRef]
- Balasubramanian, V. Heterogeneity and Efficiency in the Brain. Proc. IEEE 2015, 103, 1346–1358. [Google Scholar] [CrossRef]
- Schmiedl, T.; Seifert, U. Stochastic thermodynamics of chemical reaction networks. J. Chem. Phys. 2007, 126, 044101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedl, T.; Speck, T.; Seifert, U. Entropy Production for Mechanically or Chemically Driven Biomolecules. J. Stat. Phys. 2007, 128, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Gaspard, P. Fluctuation theorem for nonequilibrium reactions. J. Chem. Phys. 2004, 120, 8898–8905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieux, D.; Gaspard, P. Fluctuation theorem and Onsager reciprocity relations. J. Chem. Phys. 2004, 121, 6167–6174. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M. Open questions on nonequilibrium thermodynamics of chemical reaction networks. Commun. Chem. 2020, 3, 107. [Google Scholar] [CrossRef]
- Penocchio, E.; Rao, R.; Esposito, M. Thermodynamic efficiency in dissipative chemistry. Nat. Commun. 2019, 10, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzini, F.; Falasco, G.; Esposito, M. Thermodynamics of chemical waves. J. Chem. Phys. 2019, 151, 234103. [Google Scholar] [CrossRef]
- Falasco, G.; Rao, R.; Esposito, M. Information Thermodynamics of Turing Patterns. Phys. Rev. Lett. 2018, 121, 108301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzini, F.; Penocchio, E.; Falasco, G.; Esposito, M. Nonequilibrium thermodynamics of non-ideal chemical reaction networks. J. Chem. Phys. 2021, 154, 094114. [Google Scholar] [CrossRef] [PubMed]
- Avanzini, F.; Falasco, G.; Esposito, M. Thermodynamics of non-elementary chemical reaction networks. New J. Phys. 2020, 22, 093040. [Google Scholar] [CrossRef]
- Wachtel, A.; Rao, R.; Esposito, M. Thermodynamically consistent coarse graining of biocatalysts beyond Michaelis–Menten. New J. Phys. 2018, 20, 042002. [Google Scholar] [CrossRef]
- Rao, R.; Esposito, M. Nonequilibrium Thermodynamics of Chemical Reaction Networks: Wisdom from Stochastic Thermodynamics. Phys. Rev. X 2016, 6, 041064. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Esposito, M. Conservation laws and work fluctuation relations in chemical reaction networks. J. Chem. Phys. 2018, 149, 245101. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, D.T. Stochastic Simulation of Chemical Kinetics. Annu. Rev. Phys. Chem. 2007, 58, 35–55. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. Annu. Conf. Neural Inf. Process. Syst. 2019, 32, 8026–8037. [Google Scholar]
- Horowitz, J.M.; Zhou, K.; England, J.L. Minimum energetic cost to maintain a target nonequilibrium state. Phys. Rev. E 2017, 95, 042102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlögl, F. On thermodynamics near a steady state. Z. Für Phys. A Hadron. Nucl. 1971, 248, 446–458. [Google Scholar] [CrossRef]
- Branscomb, E.; Biancalani, T.; Goldenfeld, N.; Russell, M. Escapement mechanisms and the conversion of disequilibria; the engines of creation. Phys. Rep. 2017, 677, 1–60. [Google Scholar] [CrossRef]
- Kinosita, K.; Yasuda, R.; Noji, H.; Adachi, K. A rotary molecular motor that can work at near 100% efficiency. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2000, 355, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, K.S.; Rooney, N. The more food webs change, the more they stay the same. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Kachman, T.; Owen, J.A.; England, J.L. Self-Organized Resonance during Search of a Diverse Chemical Space. Phys. Rev. Lett. 2017, 119, 038001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.; Endres, R.G. Thermodynamics of switching in multistable non-equilibrium systems. J. Chem. Phys. 2020, 152, 054108. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Smith, H.B.; Mathis, C.; Raymond, J.; Walker, S.I. Universal scaling across biochemical networks on Earth. Sci. Adv. 2019, 5, eaau0149. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.M.; Mathis, C.; Carrick, E.; Keenan, G.; Cooper, G.J.T.; Graham, H.; Craven, M.; Gromski, P.S.; Moore, D.G.; Walker, S.I.; et al. Identifying molecules as biosignatures with assembly theory and mass spectrometry. Nat. Commun. 2021, 12, 3033. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueltzhöffer, K.; Da Costa, L.; Cialfi, D.; Friston, K. A Drive towards Thermodynamic Efficiency for Dissipative Structures in Chemical Reaction Networks. Entropy 2021, 23, 1115. https://doi.org/10.3390/e23091115
Ueltzhöffer K, Da Costa L, Cialfi D, Friston K. A Drive towards Thermodynamic Efficiency for Dissipative Structures in Chemical Reaction Networks. Entropy. 2021; 23(9):1115. https://doi.org/10.3390/e23091115
Chicago/Turabian StyleUeltzhöffer, Kai, Lancelot Da Costa, Daniela Cialfi, and Karl Friston. 2021. "A Drive towards Thermodynamic Efficiency for Dissipative Structures in Chemical Reaction Networks" Entropy 23, no. 9: 1115. https://doi.org/10.3390/e23091115
APA StyleUeltzhöffer, K., Da Costa, L., Cialfi, D., & Friston, K. (2021). A Drive towards Thermodynamic Efficiency for Dissipative Structures in Chemical Reaction Networks. Entropy, 23(9), 1115. https://doi.org/10.3390/e23091115