Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation

Abstract
:Introduction

Results and Discussion


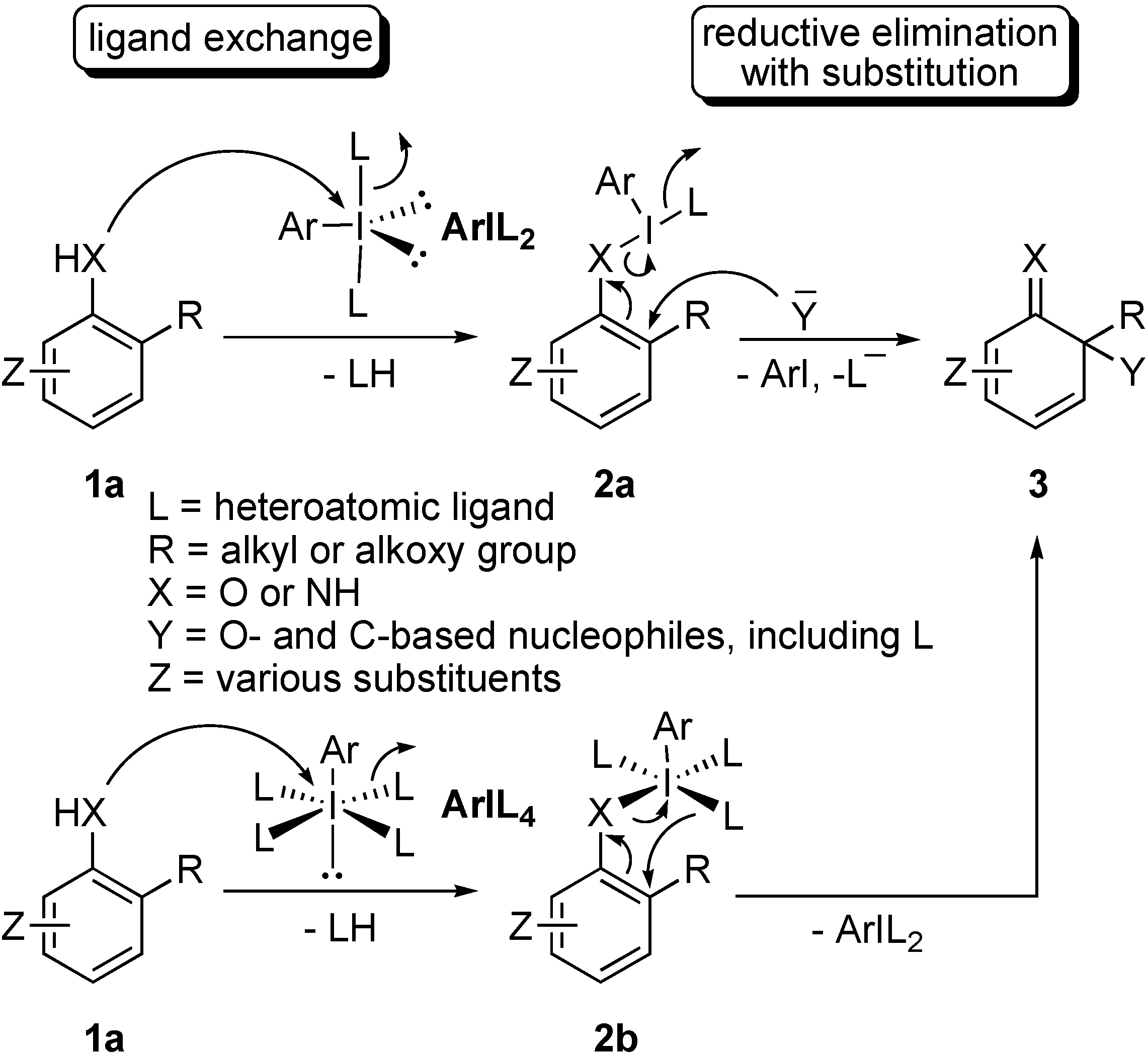{kind=link}
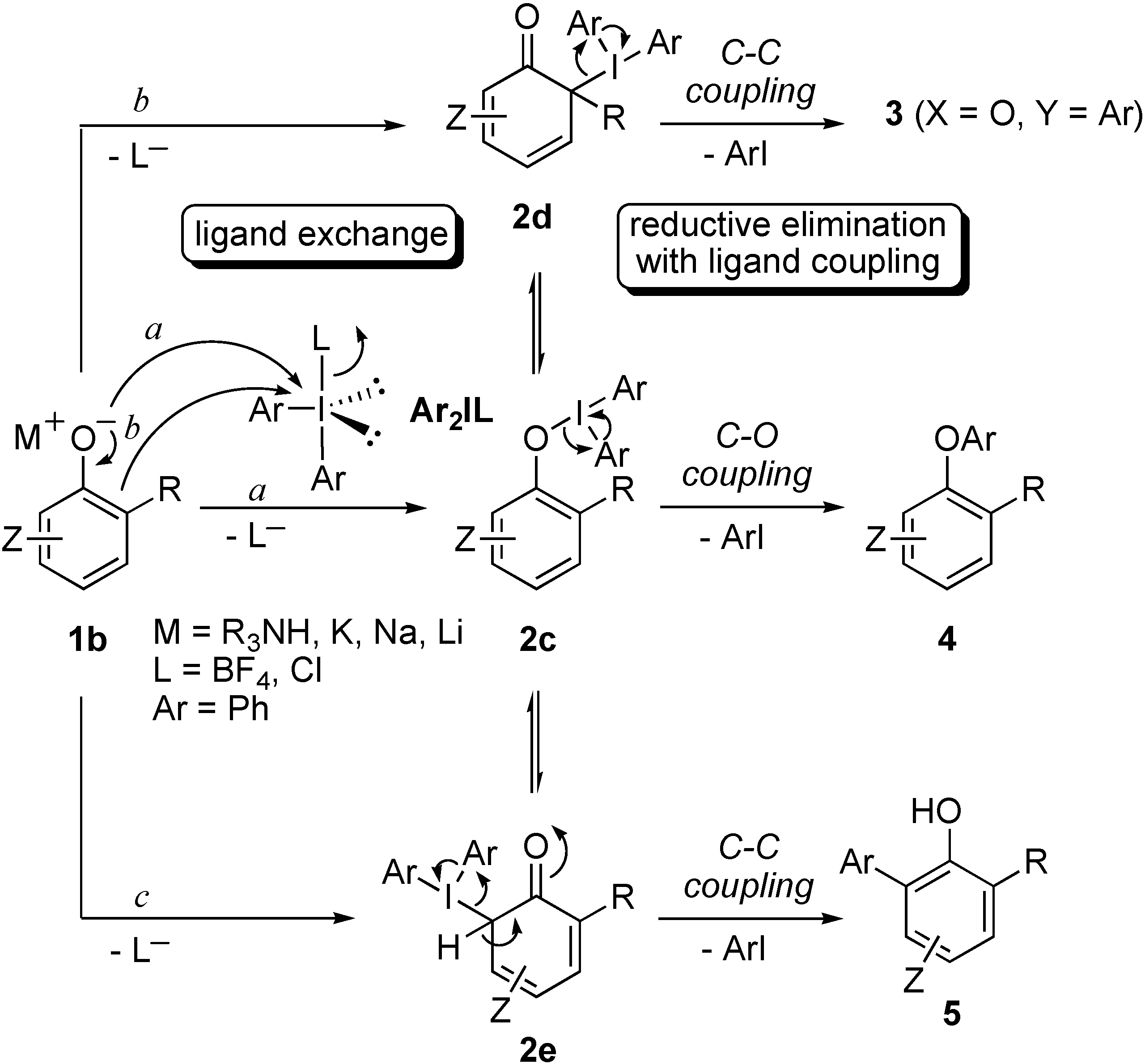{kind=link}
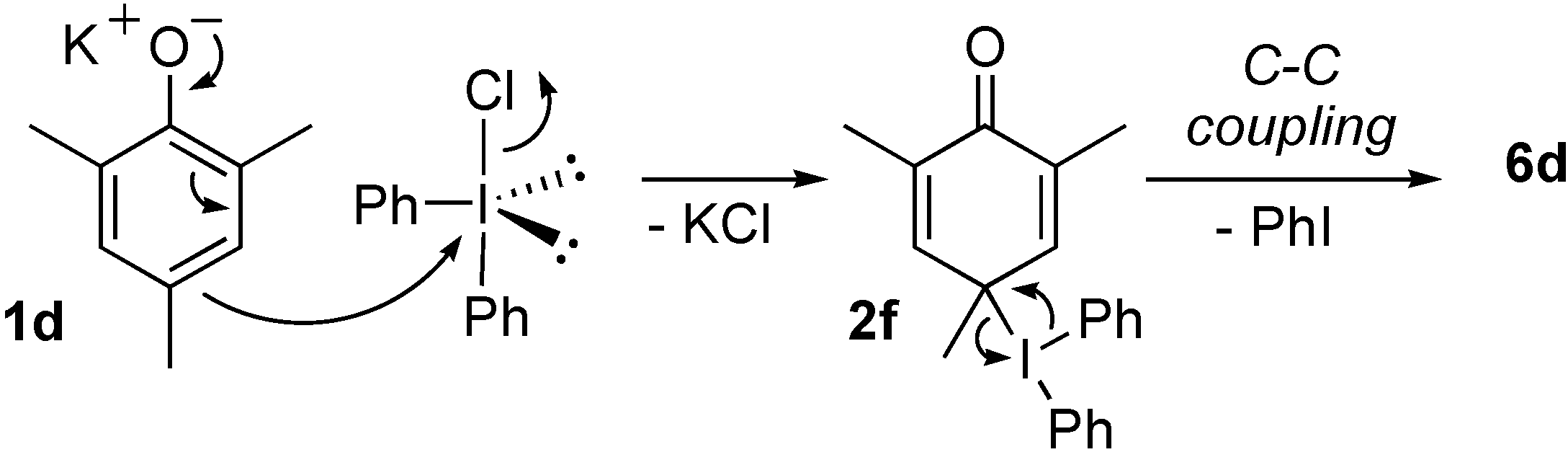{kind=link}
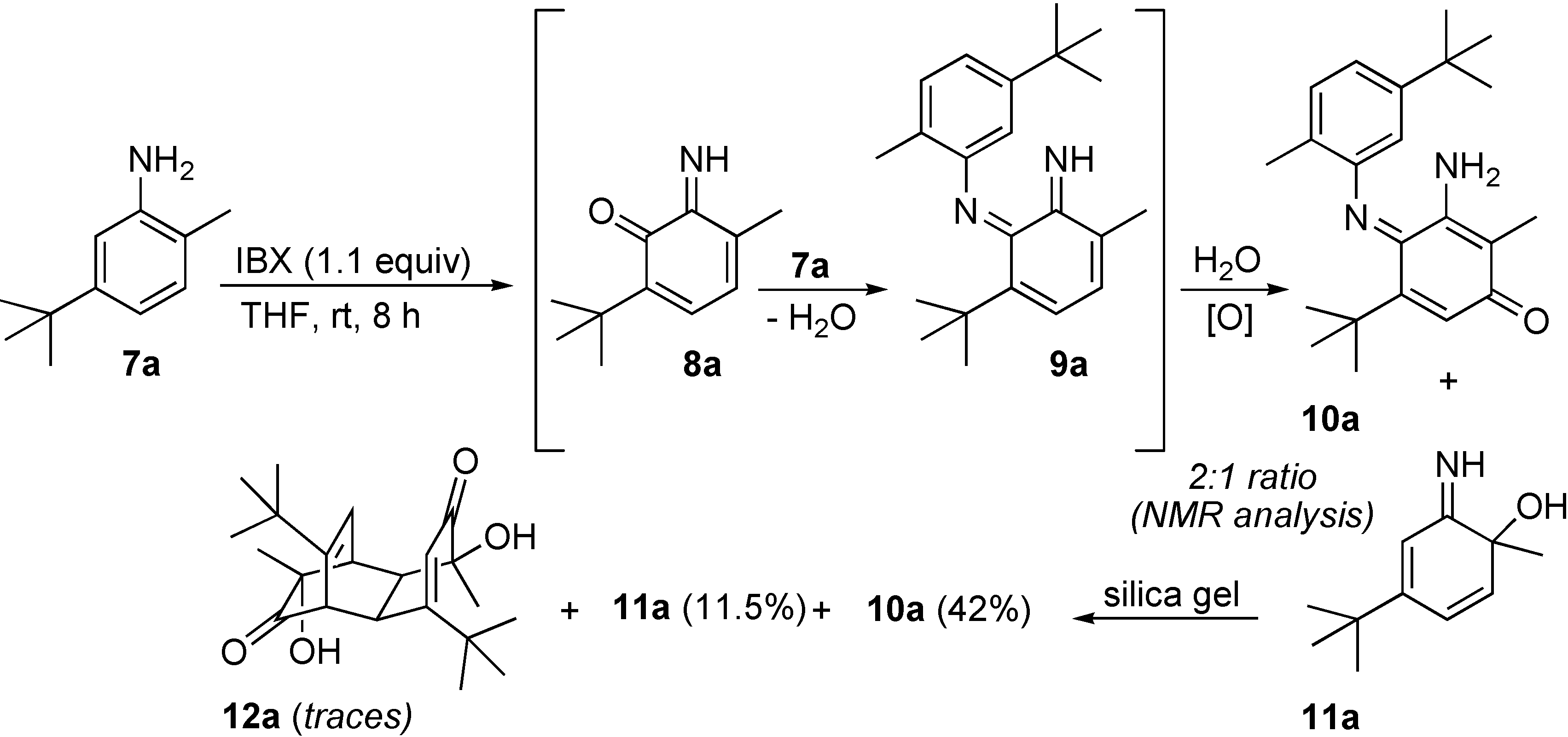{kind=link}
{kind=link}
{kind=link}
{kind=link}







Acknowledgements
Experimental
General
General Procedure for λ3-Iodane-Mediated Phenylation of Phenols
3,5,6-Trimethyl-6-phenylcyclohexa-2,4-dienone (3a) and 1,2,5-trimethyl-3-phenoxybenzene (4a)
Phenoxybenzene (4b)
2,6-Dimethyl-6-phenylcyclohexa-2,4-dienone (3c) and 1,3-dimethyl-2-phenoxybenzene (4c)
2,4,6-Trimethyl-6-phenylcyclohexa-2,4-dienone (3d), 1,3,5-trimethyl-2-phenoxybenzene (4d) and 2,4,6-trimethyl-4-phenylcyclohexa-2,5-dienone (6d)
4-tert-Butyl-2-methylaniline (7a)
N-acetyl-5-tert-butyl-2-methylaniline (7b)
2,4,6-trimethylaniline (7c)
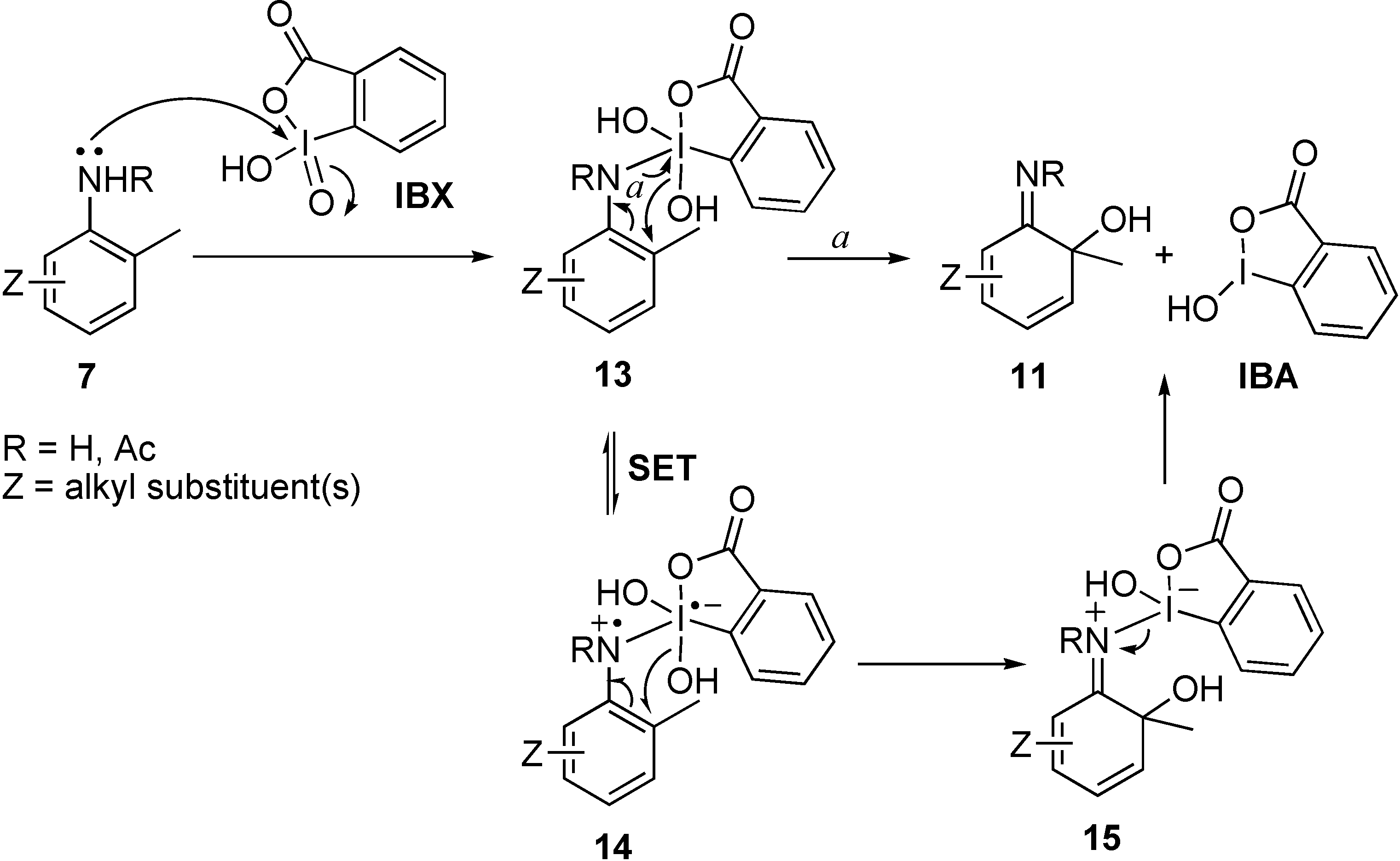
3-Amino-5-tert-butyl-4-[5-tert-butyl-2-methylphenylimino]-2-methylcyclohexa-2,5-dienone (10a) and 4-tert-butyl-6-imino-1-methylcyclohexa-2,4-dienol (11a).
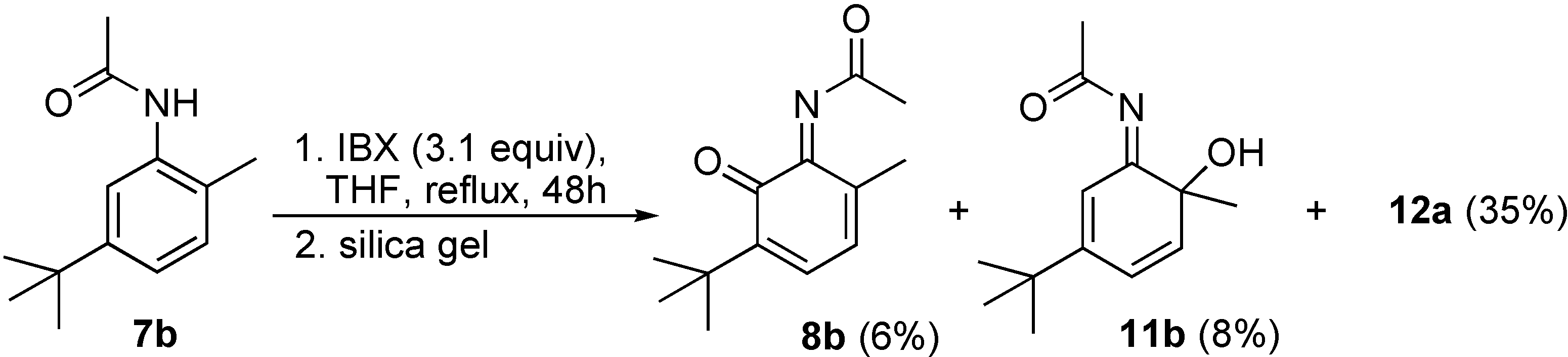
6-tert-Butyl-3-methyl-1,2-benzoquinone-2-(N-acetyl)imine (8b) and 6-(N-acetylimino)-4-tert-butyl-1-methyl-cyclohexa-2,4-dienol (11b)
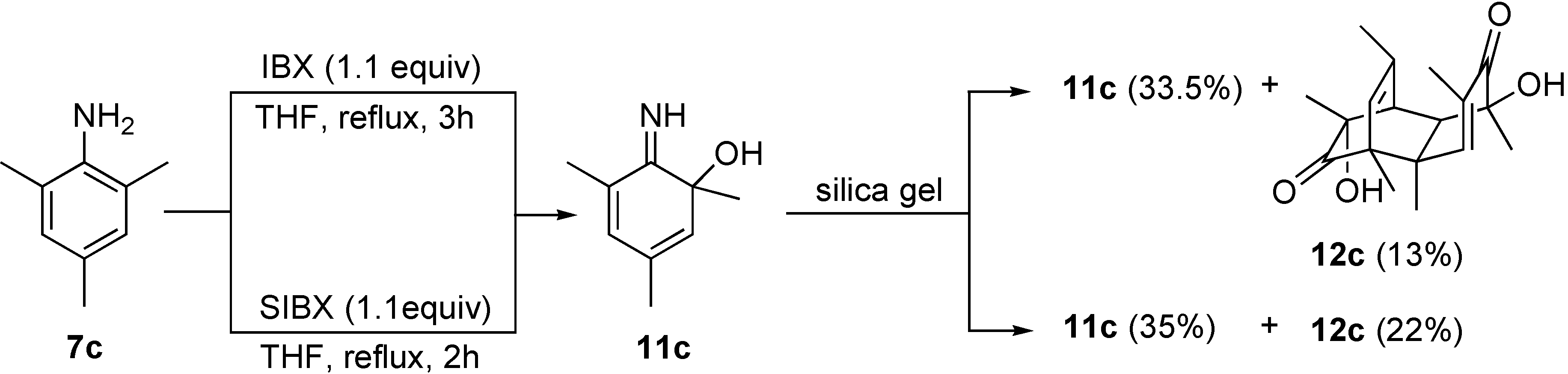
6-Imino-1,3,5-trimethyl-cyclohexa-2,4-dienol (11c) and 3,10-dihydroxy-3,5,7,8,10,12-hexamethyl-tricyclo[6.2.2.02,7]dodeca-5,11-diene-4,9-dione (12c).
References and Notes
- Quideau, S. Modern Arene Chemistry; Astruc, D., Ed.; Wiley-VCH: Weinheim, 2002; pp. 539–573. [Google Scholar]
- Quideau, S.; Pouységu, L.; Deffieux, D. Curr. Org. Chem. 2004, 8, 113–148.
- Quideau, S.; Pouységu, L. Org. Prep. Proc. Int. 1999, 31, 617–680.
- Ozanne, A.; Pouységu, L.; Depernet, D.; François, B.; Quideau, S. Org. Lett. 2003, 5, 2903–2906.
- Quideau, S.; Pouységu, L.; Deffieux, D.; Ozanne, A.; Gagnepain, J.; Fabre, I.; Oxoby, M. Arkivoc 2003, 6, 106–119.
- Quideau, S.; Lebon, M.; Lamidey, A.-M. Org. Lett. 2002, 22, 3975–3978.
- Pouységu, L.; Avellan, A.-V.; Quideau, S. J. Org. Chem. 2002, 67, 3425–3436.
- Quideau, S.; Pouységu, L.; Oxoby, M.; Looney, M.A. Tetrahedron 2001, 57, 319–329.
- Quideau, S.; Pouységu, L.; Avellan, A.-V.; Whelligan, D.K.; Looney, M.A. Tetrahedron Lett. 2001, 42, 7393–7396.
- Quideau, S.; Looney, M.A.; Pouységu, L. Org. Lett. 1999, 1, 1651–1654.
- Quideau, S.; Pouységu, L.; Looney, M.A. J. Org. Chem. 1998, 63, 9597–9600.
- Magdziak, D.; Rodriguez, A.A.; Van De Water, R.W.; Pettus, T.R.R. Org. Lett. 2002, 4, 285–288.
- Bell, H.C.; Pinhey, J.T.; Sternhell, S. Aus. J. Chem. 1979, 32, 1551–1560.
- Bell, H.C.; May, G.L.; Pinhey, J.T.; Sternhell, S. Tetrahedron Lett. 1976, 47, 4303–4306.
- Barton, D.H.R.; Yadav-Bhatnagar, N.; Finet, J.-P.; Khamsi, J.; Motherwell, W.B.; Stanforth, S.P. Tetrahedron 1987, 43, 323–332.
- Barton, D.H.R.; Bhatnagar, N.Y.; Blazejewski, J.-C.; Charpiot, B.; Finet, J.-P.; Lester, D.J.; Motherwell, W.B.; Papoula, M.T.B.; Stanforth, S.P. J. Chem. Soc., Perkin Trans. 1 1985, 2657–2665.
- Ochiai, M. Top. Curr. Chem. 2003, 224, 5–68.
- Finet, J.-P. Ligand Coupling Reactions With Heteroatomic Compounds; Elsevier Science Ltd, 1998; Vol. 18. [Google Scholar]
- Ochiai, M.; Kitagawa, Y.; Toyonari, M. Arkivoc 2003, 6, 43–48.
- Ochiai, M.; Kitagawa, Y.; Takayama, N.; Takaoka, Y.; Shiro, M. J. Am. Chem. Soc. 1999, 121, 9233–9234.
- Ochiai, M.; Shu, T.; Nagaoka, T.; Kitagawa, Y. J. Org. Chem. 1997, 62, 2130–2138.
- Andersson, G. Acta Chem. Scand. 1976, B 30, 403–406.
- Yen, C.-F.; Peddinti, R.K.; Liao, C.-C. Org. Lett. 2000, 2, 2909–2912.
- Ochiai, M.; Sumi, K.; Takaoka, Y.; Kunishima, M.; Nagao, Y.; Shiro, M.; Fujita, E. Tetrahedron 1988, 44, 4095–4112.
- Beringer, F.M.; Forgione, P.S.; Yudis, M.D. Tetrahedron 1960, 8, 49–63.
- Beringer, F.M.; Daniel, W.J.; Galton, S.A.; Rubin, G. J. Org. Chem. 1966, 31, 4315–4318.
- For a previous preparation of a diaryl ether using a diphenyl-λ3-iodane reagent, see: Scherrer, R.A.; Beatty, H.R. J. Org. Chem. 1980, 45, 2127–2131.
- See also: Crowder, J.R.; Glover, E.E.; Grundon, M.F.; Kaempfen, H.X. J. Chem. Soc. 1963, 4578–4585.
- Nicolaou, K.C.; Sugita, K.; Baran, P.S.; Zhong, Y.-L. Angew. Chem. Int. Ed. 2001, 40, 207–210.
- Coutts, I.G.C.; Culbert, N.J.; Edwards, M.; Hadfield, J.A.; Musto, D.R.; Pavlidis, V.H.; Richards, D.J. J. Chem. Soc., Perkin Trans. 1 1985, 1829–1836.
- Coutts, I.G.C.; Pavlidis, V.H.; Reza, K.; Southcott, M.R.; Wiley, G. Tetrahedron Lett. 1997, 38, 5563–5566.
- For a preparation of para-quinone imide ketals using iodosybenzene, see: Barret, R.; Daudon, M. Tetrahedron Lett. 1991, 32, 2133–2134.
- Kokil, P.B.; Patil, S.D.; Ravindranathan, T.; Nair, P.M. Tetrahedron Lett. 1979, 989–992.
- For an elegant synthesis of dynemicin A using a λ3-iodane-mediated preparation of a key para-quinone imine monoketal intermediate, see: Myers, A.G.; Tom, N.J.; Fraley, M.E.; Cohen, S.B.; Madar, D.J. J. Am. Chem. Soc. 1997, 119, 6072–6094.
- For an example of a preparation of o-benzoquinone monosulfonimides using MnO2, see: Fujita, S. J. Org. Chem. 1983, 48, 177–183.
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L.; Sugita, K. J. Am. Chem. Soc. 2002, 124, 2212–2220.
- Nicolaou, K.C.; Sugita, K.; Baran, P.S.; Zhong, Y.-L. J. Am. Chem. Soc. 2002, 124, 2221–2232.
- More, J.D.; Finney, N.S. Org. Lett. 2002, 4, 3001–3003.
- Nicolaou, K.C.; Baran, P.S.; Kranich, R.; Zhong, Y.-L.; Sugita, K.; Zou, N. Angew. Chem. Int. Ed. 2001, 40, 202–206.
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. J. Am. Chem. Soc. 2002, 124, 2233–2244.
- Adler, E.; Dahlen, J.; Westin, G. Acta Chem. Scand. 1960, 14, 1580–1596. [CrossRef] [Green Version]
- Barton, D.H.R.; Ley, S.V.; Magnus, P.D.; Rosenfeld, M.N. J. Chem. Soc., Perkin Trans. 1 1977, 567–572.
- Nicolaou, K.C.; Mathison, C.J.N.; Montagnon, T. J. Am. Chem. Soc. 2004, 126, 5192–5201.
- Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537–4538.
- Bates, R.B.; Janda, K.D. J. Org. Chem. 1982, 47, 4374–4376.
- Factor, A.; Finkbeiner, H.; Jerussi, R.A.; White, D.M. J. Org. Chem. 1970, 35, 57–62.
- Mazzocchi, P.H.; Ammon, H.L.; Colicelli, S.E. Org. Magn. Reson. 1978, 11, 143–149.
- Tashiro, M.; Yamato, T. J. Org. Chem. 1979, 44, 3037–3041.
- Carpenter, M.S.; Easter, W.M.; Wood, T.F. J. Org. Chem. 1951, 16, 586–616.
- Fischer, A.; Röderer, R. Can. J. Chem. 1976, 54, 3978–3985.
- Tashiro, M.; Fukuda, Y.; Yamato, T. J. Org. Chem. 1983, 48, 1927–1928.
- Glatzhofer, D.T.; Roy, R.R.; Cossey, K.N. Org. Lett. 2002, 4, 2349–2352.
- Masnovi, J.M.; Sankararaman, S.; Kochi, J.K. J. Am. Chem. Soc. 1989, 111, 2263–2276.
- Evans, D.; Smith, C.E.; Williamson, W.R.N. J. Med. Chem. 1977, 20, 169–171.
- Johnson, T.W.; Corey, E.J. J. Am. Chem. Soc. 2001, 123, 4475–4479.
- Sprecher, M.; Kost, D. J. Am. Chem. Soc. 1994, 116, 1016–1026.
- †Ar2IL-type compounds are usually referred to as diaryliodonium salts despite the fact that their geometry is not tetrahedral, but pseudotrigonal bipyramidal, as expected for hypervalent iodine(III) species. The lamba iodane terminology is thus used here to name hypervalent iodine compounds [17].
- Sample Availability: Available from the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Quideau, S.; Pouységu, L.; Ozanne, A.; Gagnepain, J. Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation. Molecules 2005, 10, 201-216. https://doi.org/10.3390/10010201
Quideau S, Pouységu L, Ozanne A, Gagnepain J. Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation. Molecules. 2005; 10(1):201-216. https://doi.org/10.3390/10010201
Chicago/Turabian StyleQuideau, S., L. Pouységu, A. Ozanne, and J. Gagnepain. 2005. "Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation" Molecules 10, no. 1: 201-216. https://doi.org/10.3390/10010201
APA StyleQuideau, S., Pouységu, L., Ozanne, A., & Gagnepain, J. (2005). Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation. Molecules, 10(1), 201-216. https://doi.org/10.3390/10010201