Introduction
Synthetic access to the β-benzocycloalkenones and their ring-substituted derivatives,
1, was facilitated by the 1977 publication of a two-step protocol involving Wittig olefination of α-benzo-cycloalkenones,
2, and oxidative ring expansion of the resulting alkenes with thallium(III) nitrate in methanol [
1]. This procedure, exemplified by equation 1, enables the regiospecific placement of alkyl groups at the α-carbon atoms of the β-cycloalkenone ring, and affords dimethyl ketals when trimethyl orthoformate is employed as a co-solvent. It was later demonstrated that thallium(III) nitrate can be replaced with the Prevost combination, AgNO
3 and I
2, for two-step conversions of α-tetralones (n = 2) to β-benzosuberones [
2], although the experimental procedure is a bit less convenient than the thallium nitrate method.
![Molecules 10 00217 i001]()
We have recently reported that the treatment of arylalkenes with [hydroxy(tosyloxy)iodo]benzene (
3, HTIB) in 95% methanol provides a general, regiospecific synthesis of α-aryl ketones (equation 2) [
3]. This oxidative rearrangement is fundamentally equivalent to the ring-expansion step depicted in eq. 1, and we now report that HTIB is an excellent alternative to Tl(NO
3)
3·3H
2O or AgNO
3/I
2 for the two-step synthesis of β-benzocycloalkenones from their lower α-benzocycloalkenone homologs. An obvious advantage of HTIB is its relatively benign character in comparison to Tl(NO
3)
3·3H
2O and AgNO
3[
4].
![Molecules 10 00217 i002]()
Results and Discussion
Syntheses of the exocyclic alkenes and β-benzocycloalkenones shown in
Table 1 were accomplished as indicated in equation 3. The olefination procedure was adapted from the Fitjer-Quabeck approach, wherein potassium
tert-butoxide is utilized as the Wittig base in Et
2O or benzene [
5]. More specifically, the α-benzocycloalkenones were added to a pre-stirred mixture of potassium
tert-butoxide and the appropriate alkyltriphenylphosphonium iodide in Et
2O at room temperature. After approximately 4 h, the reaction mixtures were filtered through Celite® and concentrated. Passage of the residual material through silica gel with hexanes gave the exocyclic alkenes (characterized by
1H‑NMR) which were used without further purification.
The oxidative ring-expansions were effected by the addition of crystalline HTIB (10 mmol) to a small excess of the alkene in 95% MeOH (mildly exothermic) at room temperature. After about 20 minutes and a preliminary aqueous workup, the resulting β-benzocycloalkenones were isolated by column chromatography in yields (from the alkenes) of 80 to 99%.
![Molecules 10 00217 i003]()
Table 1.
Oxidative ring expansions of alkylidenebenzocycloalkenes to β-benzocycloalkenones with HTIB in 95% MeOH
As indicated in
Table 1 (entries 1-3), conversions of unsubstituted α-benzocycloalkenones to homologous β-benzocycloalkenones containing six, seven and eight-membered rings,
via the Wittig-HTIB sequence, were demonstrated. Furthermore, as with the Tl(III)-induced rearrangements, the proper selection of substrates enables syntheses of regioisomeric pairs of methyl-substituted β-benzocycloalkenones (cf. entries 4-7). For example, the treatment of 2-methyl-1-methylideneindan with HTIB gave 3-methyl-2-tetralone (entry 4), while similar treatment of 1-ethylideneindan gave 1-methyl-2-tetralone (entry 5).
The Wittig-HTIB sequence was also employed for incorporation of a 13C-label into the β-tetralone nucleus (entry 8). To this end, 13C-methyltriphenylphosphonium iodide was prepared from 13C-labeled iodomethane and utilized for the olefination of 1-indanone. Exposure of 1-(13C-methylidene)indan (5.0 mmol) to HTIB (4.54 mmol) in 95% MeOH (25 mL) gave 1-13C-2-tetralone in 91% isolated yield. The location of the label at C-1 in the β-tetralone ring was clearly revealed by NMR analysis. Thus, the singlet at δ 3.58 in the 1H-spectrum of unlabeled β-tetralone appears as a doublet at δ 3.62 (JCH = 128.6 Hz) in the 1H spectrum of the 13C-isotopomer. A 13C-NMR spectrum of the labeled compound, recorded after only 16 scans, exhibits a markedly enhanced singlet at δ 45.03, while the resonances of the remaining carbons are either very weak in comparison or not perceptible.
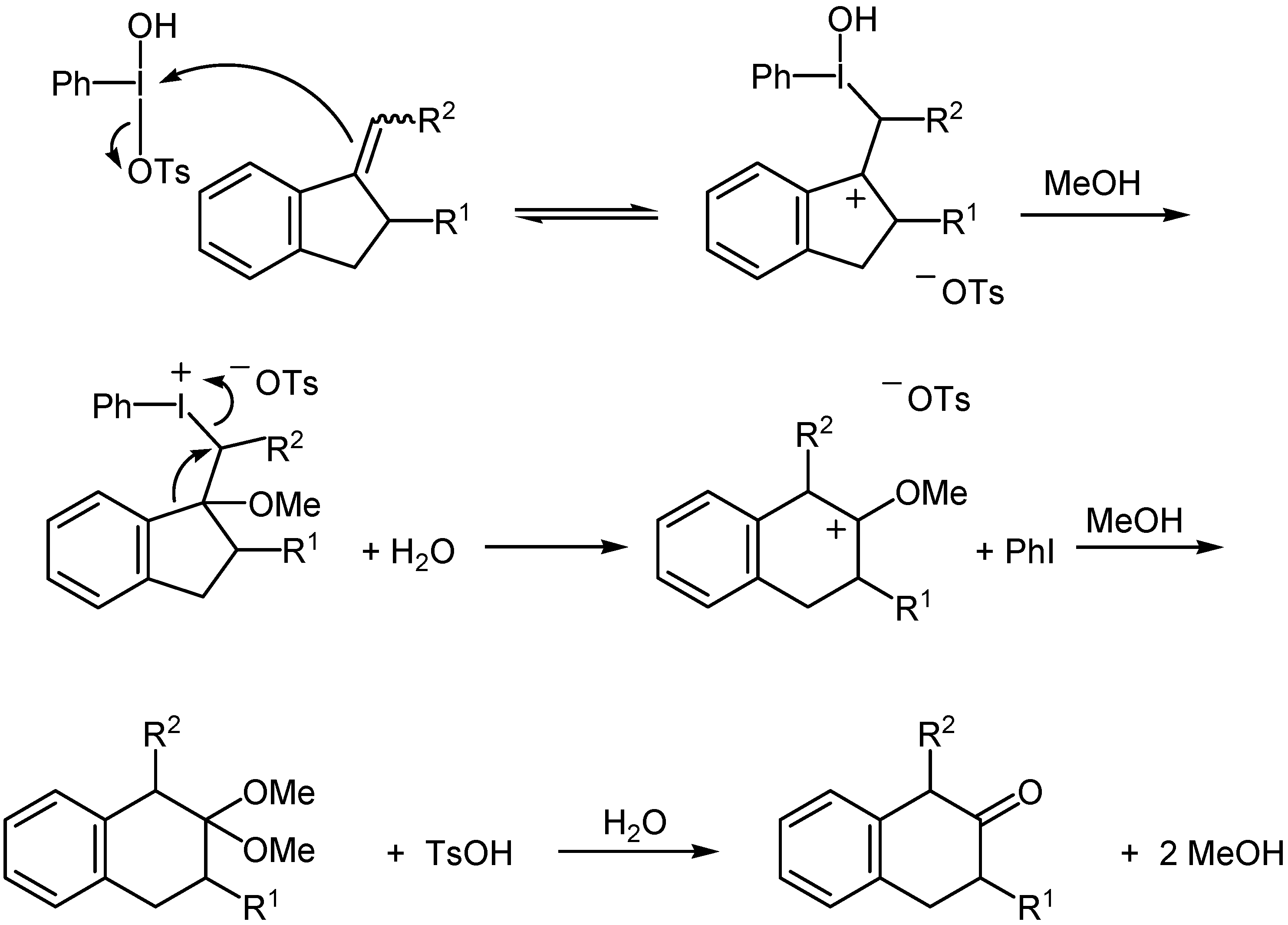
A plausible mechanism for the HTIB-induced ring-expansions reported herein is presented in
Scheme 1. It is analogous to that proposed for the oxidative rearrangement of arylalkenes to α-aryl ketones [
3], and accounts for the regiochemistry of β-benzocycloalkenone formation. The similarity between iodine(III) and thallium(III) reagents in the context of oxidative rearrangements has been reviewed by Prakash [
6] and almost certainly originates from their electrophilic character and capacity for reduction to the respective iodine(I) and thallium(I) oxidation states.
Experimental
General
NMR spectra were recorded on a Varian model Gemini-300 spectrometer at resonance frequencies of 300 (
1H-) and 75 (
13C-) MHz. The NMR solvent in all cases was CDCl
3; chemical shifts are expressed relative to residual CHCl
3 (
1H spectra) or to CDCl
3 (
13C- spectra). Multiplets in
1H-NMR spectra are sometimes specified with a range of chemical shifts corresponding to the highest and lowest lines in the multiplet. IR spectra were recorded on a Bomem MB-100 FT-IR spectrophotometer. IR samples were neat films or a Nujol® mull (Entry 4). The elemental analysis was performed by Midwest Microlab, LTD (Indianapolis, IN). Melting points were recorded on a Thomas-Hoover Unimelt melting point apparatus and are uncorrected. 2-Methyl-1-indanone and 2-methyl-1-tetralone (used for preparation of the alkenes in entries 4 and 6) were prepared by adaptation of literature methods [
7,
8]. All other solvents and chemical reagents were obtained from commercial sources and used as received. Flash column chromatography was performed on a 42 mm internal diameter column packed with Kieselgel (230-400 mesh) silica gel purchased from the Aldrich Chemical Company. Thin layer chromatography (TLC) was done with glass backed 250 micron silica gel plates containing a fluorescent indicator and purchased from Alltech. The 95% methanol used as the solvent for the treatment of the substrate alkenes with [hydroxy(tosyloxy)iodo]benzene (HTIB), refers to a 95:5 (v/v) mixture of methanol and water. Consumption of the oxidant was verified prior to work-up by adding a drop of the reaction mixture to 10% KI (aq.) solution. Yields are based on the material used for the spectroscopic data presented in this paper.
Preparation of Hypervalent Iodine Reagents
Typical Synthesis of (Diacetoxyiodo)benzene
A solution of 32% (w/v) peracetic acid (100 mL, 421 mmol) in acetic acid was added dropwise with mechanical stirring to a cooled flask (15
oC) containing iodobenzene (65.28 g, 320.0 mmol), over a period of 1 hour. The rate of addition was adjusted to keep the temperature of the reaction mixture between 25-30
oC. Mechanical stirring was continued for 4 hours during which time a white precipitate separated. Water (100 mL) was added to facilitate precipitation and to dilute any remaining oxidant. The solid was isolated by vacuum filtration, washed with water (2 x 100 mL) and ether (150 mL), dried over P
2O
5 in a vacuum dessicator overnight and identified as (diacetoxyiodo)benzene; yield 94.86 g (92%); mp 160-161 °C (lit. [
9] mp 159-161 °C).
Typical Synthesis of [Hydroxy(tosyloxy)iodo]benzene (HTIB, 3)
A solution of (diacetoxyiodo)benzene (40.26 g 125.0 mmol) in boiling acetonitrile (120 mL) was added at once to a solution of
p-toluenesulfonic acid monohydrate (24.72 g, 130.0 mmol) in boiling acetonitrile (80 mL), with magnetic stirring, to give a yellow-green solution. The solution was allowed to cool to room temperature, and the crystalline [hydroxy(tosyloxy)iodo]benzene that separated was isolated by vacuum filtration and washed with acetonitrile (100 mL) and ether (100 mL); yield, 45.39 g (93%); mp 136-7
oC, dec. (lit.[
10] mp 136-138.5
oC).
Representative Procedure - Reaction of HTIB with 1-Methylideneindan (4) (Entry 1).
Crystalline HTIB (3.92 g, 10.0 mmol) was added to a stirred solution of 1-methylideneindan (1.43 g, 11.0 mmol) in 95% methanol (40 mL). The solid dissolved rapidly (~15 sec) with the evolution of heat (41 oC) to give a colorless solution. The solution was stirred at room temperature for 20 minutes and the solvent was removed in vacuo to give an oily mixture. This mixture was partitioned between CH2Cl2 (40 mL) and H2O (25 mL) and transferred to a separatory funnel. The organic layer was separated, washed with H2O (2 x 25 mL) and brine (1 x 30 mL), dried over MgSO4, and concentrated in vacuo to a bright yellow oil (1.51 g), which was subjected to flash column chromatography on silica gel (hexanes; 5 % ethyl acetate/hexanes) to give β-tetralone (5) as a light yellow oil (Rf = 0.29, 5 % ethyl acetate/hexanes); yield, 1.38 g (94 %); 1H-NMR δ 2.54 (t, J = 6.6, 2H), 3.06 (t, J = 6.6, 2H), 3.58 (s, 2H), 7.11-7.15 (m, 1H), 7.20-7.24 (m, 3H); 13C-NMR δ 28.13, 37.95, 44.91, 126.80, 126.87, 127.58, 128.19, 133.28, 136.71, 210.71; IR (C=O) 1719 cm-1.
Summary of Purification, Yield and Spectral Data for β-benzocycloalkenones (Entries 2-7)
Conversion of Alkene 6 to Ketone 7 (Entry 2)
Flash column chromatography of the oil on silica gel (petroleum ether; 10% ethyl acetate/petroleum ether) gave
2-benzosuberone (
7) as a colorless oil; yield, 1.58 g (99%);
1H-NMR δ 1.99 (m, 2H), 2.57 (dd,
J = 6.9, 6.9, 2H), 2.95 (m, 2H), 3.73 (s, 2H), 7.14-7.22 (m, 4H);
13C-NMR δ 26.11, 32.85, 43.56, 50.04, 127.14, 127.59, 129.24, 129.60, 133.61, 140.51, 208.90; IR (C=O) 1710 cm
-1; 2,4-dinitro- phenylhydrazone, mp 174-176 °C (lit. [
11] mp 169-170 °C).
Conversion of Alkene 8 to Ketone 9 (Entry 3)
Flash column chromatography of the oil on silica gel (hexanes; 5% ethyl acetate/hexanes) gave
7,8,9,10-tetrahydro-5H-benzocyclooctan-6-one (
9) as a colorless oil (R
f = 0.32, 5% ethyl acetate/ hexanes); yield, 1.51 g (87%);
1H-NMR δ 1.76 (m, 2H), 1.87 (m, 2H), 2.35 (m, 2H), δ 2.84 (m, 2H), 3.82 (s, 2H), 7.14-7.30 (m, 3H) [
12];
13C-NMR δ 24.63, 31.14, 32.98, 40.87, 48.58, 126.73, 127.99, 129.98, 130.27, 133.66, 141.13, 212.05; IR (C=O) 1700 cm
-1; oxime, mp 128-129 °C (lit.[
13] mp 130-131 °C).
Conversion of Alkene 10 to Ketone 11 (Entry 4)
Flash column chromatography on silica gel (5% ethyl acetate/hexanes; 10% ethyl acetate/hexanes) gave a yellow oil (R
f = 0.78, 10% ethyl acetate in hexanes). Kugelrohr distillation of this oil gave 3-methyl-2-tetralone (
11) as white rosettes; yield, 1.31 g (82%); mp 38-40
oC (lit. [
14] mp 37-40 °C);
1H‑NMR δ 1.20 (d, J = 6.9, 3H), 2.54 (m, 1H), 2.84 (dd, J = 15.4, 11.0, 1H), 3.08 (dd, J = 15.4, 5.8, 1H), 3.61 (s, 2H), 7.12-7.26 (m, 4H);
13C-NMR δ 14.89, δ 36.73, δ 42.31, δ 44.07, δ 126.76, δ 126.87, δ 127.80, δ 128.13, δ 133.51, δ 136.24, δ 212.28; IR (C=O) 1722 cm
-1.
Conversion of Alkene 12 to Ketone 13 (Entry 5)
Flash column chromatography of the oil on silica gel (hexanes; 5% ethyl acetate/hexanes) gave 1‑methyl-2-tetralone (13) as a colorless oil (Rf = 0.24, 5% ethyl acetate/hexanes); yield, 1.48 g (92%); 1H‑ NMR δ 1.48 (d, J = 6.9, 3H), 2.43-2.69 (overlapping m’s, 2H), 3.08 (m, 2H), 3.54 (quartet, J = 6.9, 1H), 7.21-7.29 (m, 4H); 13C-NMR δ 13.74, 27.70, 36.82, 47.12, 125.72, 126.36, 126.73, 127.18, 136.56, 137.63, 211.83; IR (C=O) 1714 cm-1; Anal. Calcd. for C11H12O: C: 82.46%, H: 7.55 %, Found: C 82.35 %, H 7.60%.
Conversion of Alkene 14 to Ketone 15 (Entry 6)
Flash column chromatography of the oil on silica gel (hexanes; 5% ethyl acetate/hexanes) gave 3‑methyl-2-benzosuberone (15) as a colorless oil (Rf = 0.27, 5% ethyl acetate/hexanes); yield, 1.40 g (80%); 1H-NMR δ 1.03 (d, J = 6.3, 3H), 1.56 (m, 1H), 2.13 (m, 1H), 2.80 (m, 1H), 2.92 (m, 2H), 3.72 (AB doublet pair, J = 54.9, 15.4, 2H), 7.14-7.26 (m, 4H); 13C-NMR δ 10.80, 27.94, 30.75, 41.69, 45.15, 122.57, 122.98, 124.46, 125.00, 129.39, 136.17, 206.11; IR (C=O) 1708 cm-1.
Conversion of Alkene 16 to Ketone 17 (Entry 7)
Flash column chromatography of the oil on silica gel (hexanes; 5% ethyl acetate/hexanes; 10% ethyl acetate/hexanes) gave
1-methyl-2-benzosuberone (
17) as a colorless oil (R
f = 0.26, 5% ethyl acetate/hexanes); yield, 1.47 g (85%);
1H-NMR δ 1.45 (d,
J = 7.1, 3H), 1.92 (m, 1H), 2.08 (m, 1H), 2.47 (m, 1H), 2.66 (m, 1H), 2.79 (m, 1H), 2.95 (m, 1H), 3.88 (quartet, 6.9, 1H), 7.12-7.26 (m, 4H);
13C‑NMR δ 14.50, 27.07, 32.33, 42.21, 50.80, 127.12, 127.16, 127.21, 129.40, 138.61, 139.65, 211.89; IR (C=O) 1709 cm
-1; 2,4-dinitrophenylhydrazone, mp 144-146 °C (lit. [
15] mp 150-151 °C).
Preparation of 13C-Methyltriphenylphosphonium Iodide
A solution of
13C-labeled iodomethane [
16] (5.00 g, 35.0 mmol) in benzene (20 mL) was added dropwise to a cooled (-4 to 0 °C) solution of triphenylphosphine (8.39 g, 32.0 mmol) in benzene (50 mL) over one hour. A white solid began to separate after 40 minutes. This mixture was allowed to warm to room temperature and stirred for a period of 4 hours. The white solid was collected by vacuum filtration and identified by melting point as
13C-methyltriphenylphosphonium iodide; yield, 12.60 (97%); melting point, 182-184 °C (lit. [
17] mp 183-184 °C).
Preparation of 13C-Methylideneindan (18)
Potassium tert-butoxide (1.35 g, 12.0 mmol) was added at once under argon to a mechanically stirred mixture of 13C-methyltriphenylphosphonium iodide (4.86 g, 12.0 mmol) in dry ether (50 mL) to give a canary yellow mixture. This mixture was vigorously stirred for 30 minutes. A solution of 1‑indanone (1.32 g, 10.0 mmol) in dry ether (20 mL) was added to the mixture over a period of 5 minutes. The color of the mixture gradually became a vivid blue as it was stirred overnight (14 hours). The resulting mixture was filtered through Celite® (10 g), and the filtrate was concentrated in vacuo to give a light yellow oil. The oil was eluted with hexanes through a pad of silica gel (30 g) on a sintered glass funnel under aspirator vacuum. The eluant was concentrated in vacuo to give 1-(13C-methylidene)indan (18) as a colorless oil; yield, 0.91 g (69%); 1H-NMR δ 2.97 (m, 2H), 3.14 (m, 2H), 5.16 (dt, JHH = 2.1, JHC = 127.20, 1H), 5.68 (dt, JHH = 2.1, JHC = 125.4, 1H), 7.34-7.44 (m, 3H), 7.66 (d, J = 6.9, 1H); 13C-NMR δ 12.93, 30.01, 31.10, 102.73 (enhanced), 120.50, 120.53, 125.24, 126.31, 128.13, 128.15; IR (12C=13C) 1715 cm-1.
Reaction of 13C Labeled Methylideneindan with HTIB (Entry 8)
Crystalline HTIB (1.78 g, 4.54 mmol) was added to a stirred solution of 1-(13C-methylidene)indan (18, 0.66 g, 5.0 mmol) in 95% methanol (25 mL). The solid dissolved rapidly with the evolution of heat to give a colorless solution. The solution was stirred at room temperature for 20 minutes, and the solvent was removed in vacuo to give an oily mixture. This mixture was partitioned between CH2Cl2 (25 mL) and H2O (25 mL). The organic layer was washed with H2O (2 x 25 mL) and brine (1 x 20 mL), dried over MgSO4, and concentrated in vacuo to give a bright yellow oil (0.79 g). Flash column chromatography of the oil on silica gel (hexanes; 5 % ethyl acetate/ hexanes) gave 1-13C-β-tetralone (19) as a light yellow oil (Rf = 0.30, 5 % ethyl acetate/hexanes); yield, 0.61 g (91 %); 1H-NMR δ 2.58 (t, J = 6.6, 2H), δ 3.10 (t, J = 6.6, 2H), δ 3.62 (d, JHC = 128.6, 2H), δ 7.15-7.19 (m, 4H); 13C-NMR, (16 transients) δ 45.03; IR (C=O) 1718 cm-1.





{kind=link}
















