General
All commercially available reagents were used without purification and solvents were dried according to standard procedures. Product purification was carried out using flash chromatography on silica gel (Merck silica gel 60 (0.040-0.063 mm). TLC was run on Merck silica gel 60 F254 analytical plates; detection was carried out with either UV, iodine and spraying with a solution of KMnO4, with subsequent heating. The melting points were determined on the Kofler block, and are uncorrected. Optical rotations were measured in chloroform, using a P3002 Krüss polarimeter and reported as follows: [α]d25 (c in g/100 mL, solvent). NMR spectra were recorded at room temperature on a FT NMR spectrometer Varian Mercury Plus 400 (1H at 400.13 MHz and 13C at 100.6 MHz) using CDCl3 as the solvent and TMS as internal reference. For 1H δ are given in parts per million relative to TMS (0 ppm), for 13C relative to CDCl3 (77 ppm). 13C-NMR multiplicities were determined by a DEPT pulse sequence. IR spectra were recorded on a Perkin-Elmer 599 IR spectrometer in CHCl3. All reactions were performed under nitrogen atmosphere when anhydrous solvents were used. Microwave experiments were conducted using a focused microwave system (CEM Discover). All experiments were performed in glass vessels (10 mL) sealed with a septum. At the end of reaction, the vessels and contents were cooled rapidly using a stream of compressed air.
Methyl 3,6-anhydro-2-deoxy-4,5:7,8-di-O-isopropylidene-d-glycero-d-galacto-octanoate (
1) and
Methyl 3,6-anhydro-2-deoxy-4,5:7,8-di-O-isopropylidene-d-glycero-d-talo-octanoate (
2).
1:
1H-NMR: δ 1.33 (3H, s, CH
3), 1.37 (3H, s, CH
3), 1.44 (3H, s, CH
3), 1.46 (3H, s, CH
3), 2.73 (1H, dd,
J2,2=16.7 Hz,
J3,2=6.4 Hz, H
2), 2.81 (1H, dd,
J2,2=16.7 Hz,
J3,2=7.3 Hz, H
2), 3.52 (1H, m, H
6), 3.70 (3H, s, OCH
3), 3.94 (1H, m, H
3), 4.04 (1H, dd,
J8,8=8.7 Hz,
J8,7=4.7 Hz, H
8), 4.07 (1H, dd,
J8,8=8.7 Hz,
J8,7=6.1 Hz, H
8), 4.38 (1H, ddd,
J7,6=7.5 Hz,
J8,7=6.1 Hz,
J8,7=4.7 Hz, H
7), 4.76 (2H, m, H
4, H
5);
13C- NMR: δ 24.6, 25.2, 25.7, 26.9, 33.3, 51.8, 66.9, 73.1, 77.7, 80.7, 81.0, 81.6, 109.1, 112.6, 171.4. The procedure and [α]
D were consistent with those reported [
11].
2:
1H-NMR: δ 1.34 (3H, s, CH
3), 1.37 (3H, s, CH
3), 1.45 (3H, s, CH
3), 1.51 (3H, s, CH
3), 2.47 (1H, dd,
J2,2=15.2 Hz,
J3,2=7.1 Hz, H
2), 2.54 (1H, dd,
J2,2=15.2 Hz,
J3,2=7.7 Hz, H
2), 3.71 (3H, s, OCH
3), 3.79 (1H, dd,
J7,6=7.7 Hz,
J6,5=3.7 Hz, H
6), 4.00 (1H, dd,
J8,8=8.7 Hz,
J8,7=4.4 Hz, H
8), 4.08 (1H, dd,
J8,8=8.7 Hz,
J8,7=6.3 Hz, H
8), 4.39 (1H, ddd,
J7,6=7.7 Hz,
J8,7=6.3 Hz,
J8,7=4.4, H
7), 4.49 (1H, dd,
J3,2=7.7 Hz,
J3,2=7.1 Hz, H
3), 4.64 (1H, d,
J5,4=6.0 Hz, H
4), 4.81 (1H, dd,
J5,4=6.0 Hz,
J6,5=3.7 Hz, H
5);
13C-NMR: δ 24.7, 25.2, 26.1, 27.0, 36.2, 51.9, 66.9, 73.3, 80.8, 80.9, 80.9, 84.9, 109.2, 112.9, 170.6. The procedure, m.p. and [α]
d were consistent with those reported [
11].
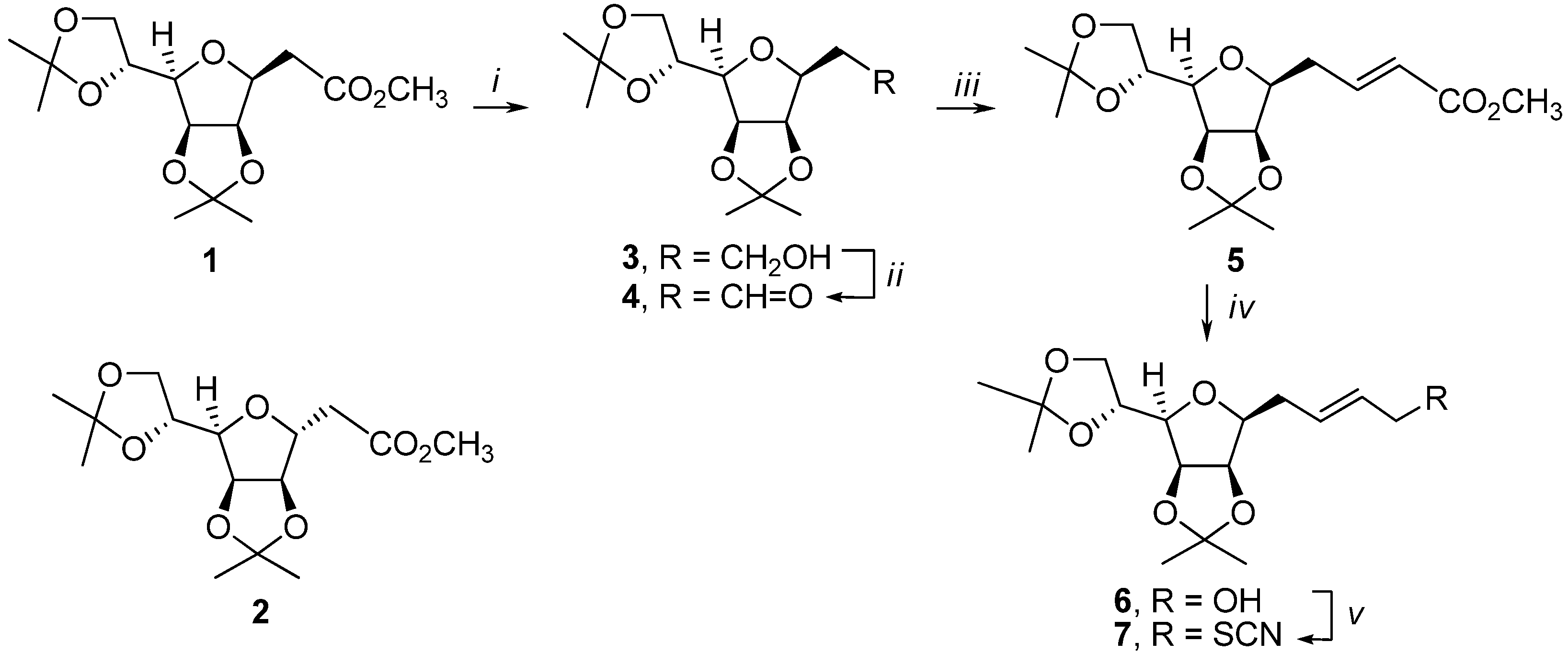
3,6-Anhydro-2-deoxy-4,5:7,8-di-O-isopropylidene-d-glycero-d-galacto-octitol (3): LiAlH4 (0.87 g, 23.0 mmol) was added at 0 oC to a solution of ester 1 (3.83 g, 12.1 mmol) in dry Et2O (70 mL). The reaction mixture was stirred at 0 oC for 15 min and then for 45 min at room temperature. The reaction was quenched by careful addition of water (3 mL) and the precipitate was removed by filtration. The filtrate was dried (Na2SO4) and concentrated under reduced pressure. The chromatography of the residue on silica gel (hexane-ethyl acetate, 2:1) afforded 3.03 g (88%) of alcohol 3 as a colorless oil; [α]d25 = -23 (c 0.49, CHCl3); 1H-NMR: δ 1.34 (3H, s, CH3), 1.38 (3H, s, CH3), 1.44 (3H, s, CH3), 1.48 (3H, s, CH3), 1.93 (1H, m, H2), 2.05 (1H, m, H2), 3.54 (1H, dd, J7,6=7.2 Hz, J6,5=3.7 Hz, H6), 3.70 (1H, ddd, J3,2=8.4 Hz, J3,2=5.1 Hz, J4,3=3.7 Hz, H3), 3.80 (2H, m, H1), 4.05 (1H, dd, J8,8=8.7 Hz, J8,7=4.8 Hz, H8), 4.09 (1H, dd, J8,8=8.7 Hz, J8,7=6.1 Hz, H8), 4.40 (1H, m, H7), 4.66 (1H, dd, J5,4=6.1 Hz, J4,3=3.7 Hz, H4), 4.76 (1H, dd, J5,4=6.1 Hz, J6,5=3.7 Hz, H5); 13C-NMR: δ 24.6, 25.3, 25.7, 26.9, 31.1, 60.4, 66.8, 73.1, 80.5, 80.6, 81.7, 81.7, 109.0, 112.4; Anal. Calcd for C14H24O6 (288.34): C 58.32, H 8.39; found C 58.54, H 8.66.
3,6-Anhydro-2-deoxy-4,5:7,8-di-O-isopropylidene-d-glycero-d-galacto-octose (4): To a solution of alcohol 3 (3.0 g, 10.5 mmol) in CH3CN (55 mL) was added IBX (4.41 g, 15.7 mmol). The resulting suspension was heated under reflux for 40 min. Then the reaction was cooled to room temperature and filtered through a medium glass frit. The filter cake was washed with further portions of acetonitrile (2 x 15 mL). The combined filtrates were concentrated under reduced pressure. The chromatography of the residue on silica gel (hexane-ethyl acetate, 3:1) afforded 2.73 g (93%) of aldehyde 4 as a colorless oil; [α]d25 = -12 (c 0.68, CHCl3); 1H-NMR: δ 1.32 (3H, s, CH3), 1.38 (3H, s, CH3), 1.45 (3H, s, CH3), 1.46 (3H, s, CH3), 2.87-2.89 (2H, m, H2), 3.55 (1H, dd, J7,6=7.4 Hz, J6,5=3.3 Hz, H6), 3.99 (1H, ddd, J3,2=6.4 Hz, J3,2=6.4 Hz, J4,3=3.3 Hz, H3), 4.03 (1H, dd, J8,8=8.7 Hz, J8,7=4.7 Hz, H8), 4.08 (1H, dd, J8,8=8.7 Hz, J8,7=6.2 Hz, H8), 4.39 (1H, ddd, J7,6=7.4 Hz, J8,7=6.2 Hz, J8,7=4.7 Hz, H7), 4.76 (1H, dd, J5,4=6.1 Hz, J4,3=3.3 Hz, H4), 4.79 (1H, dd, J5,4=6.1 Hz, J6,5=3.3 Hz, H5), 9.81 (1H, t, J=1.3 Hz, CHO); 13C-NMR: δ 24.5, 25.2, 25.6, 26.9, 42.8, 66.8, 73.0, 76.6, 80.6, 81.0, 81.6, 109.1, 112.6, 199.9; Anal. Calcd for C14H22O6 (286.33): C 58.73, H 7.74; found C 58.49, H 7.51.
Methyl 5,8-anhydro-2,3,4-trideoxy-6,7:9,10-di-O-isopropylidene-d-glycero-d-galacto-dec-2(E)-enoate (5): [(Methoxycarbonyl)methylidene]triphenylphosphorane (3.82 g, 11.4 mmol) was added to a solution of aldehyde 4 (2.73 g, 9.5 mmol) in dry CH2Cl2 (25 mL). The reaction mixture was stirred for 1.5 h at room temperature. The solvent was removed under reduced pressure and the residue was purified by chromatography on silica gel (hexane-ethyl acetate, 5:1) to afford 2.79 g (87%) of (E)-5 as a colorless oil; [α]d25 = -12 (c 0.34, CHCl3); 1H-NMR: δ 1.33 (3H, s, CH3), 1.38 (3H, s, CH3), 1.44 (3H, s, CH3), 1.48 (3H, s, CH3), 2.61 (2H, m, H4), 3.50 (1H, dd, J9,8=7.5 Hz, J8,7=3.6 Hz, H8), 3.59 (1H, ddd, J5,4=6.8 Hz, J5,4=6.8 Hz, J6,5=3.6 Hz, H5), 3.73 (3H, s, OCH3), 4.05 (1H, dd, J10,10=8.7 Hz, J10,9=4.8 Hz, H10), 4.09 (1H, dd, J10,10=8.7 Hz, J10,9=6.1 Hz, H10), 4.39 (1H, ddd, J9,8=7.5 Hz, J10,9=6.1 Hz, J10,9=4.8 Hz, H9), 4.63 (1H, dd, J7,6=6.1 Hz, J6,5=3.6 Hz, H6), 4.76 (1H, dd, J7,6=6.1 Hz, J8,7=3.6 Hz, H7), 5.94 (1H, ddd, J3,2=15.7 Hz, J4,2=1.5 Hz, J4,2=1.5 Hz, H2), 6.99 (1H, ddd, J3,2=15.7 Hz, J4,3=7.1 Hz, J4,3=7.1 Hz, H3); 13C-NMR: δ 24.6, 25.3, 25.7, 26.9, 31.4, 51.5, 66.9, 73.1, 80.3, 80.7, 81.1, 81.7, 109.1, 112.6, 123.0, 144.9, 166.8; Anal. Calcd for C17H26O7 (342.39): C 59.64, H 7.65; found C 59.73, H 7.79.
5,8-Anhydro-2,3,4-trideoxy-6,7:9,10-di-O-isopropylidene-d-glycero-d-galacto-dec-2(E)-enitol (6): To a solution of ester 5 (2.79 g, 8.15 mmol) in dry CH2Cl2 (37 mL) diisobutylaluminum hydride (24.6 mL of 1.2 M toluene solution) was added dropwise at -15 °C. The resulting mixture was stirred for 45 min at the same temperature and then quenched with methanol (6.2 mL). The mixture was allowed to warm to room temperature and poured into 30% aqueous K/Na tartrate (123 mL). After stirring for 30 min, the product was extracted with CH2Cl2 (3 x 37 mL). The combined organic layers were dried (Na2SO4) and the solvent evaporated under reduced pressure. Chromatography of the residue on silica gel (hexane-ethyl acetate, 1:1) afforded 2.34 g (91%) of allylic alcohol 6 as a colorless oil; [α]d25 = +22 (c 0.28, CHCl3); 1H-NMR: δ 1.34 (3H, s, CH3), 1.38 (3H, s, CH3), 1.45 (3H, s, CH3), 1.48 (3H, s, CH3), 2.47 (2H, m, H4), 3.48 (1H, m, H8), 3.52 (1H, m, H5), 4.05 (1H, dd, J10,10=8.7 Hz, J10,9=4.8 Hz, H10), 4.08 (1H, dd, J10,10=8.7 Hz, J10,9=6.0 Hz, H10), 4.11 (2H, m, H1), 4.40 (1H, ddd, J9,8=7.4 Hz, J10,9=6.0 Hz, J10,9=4.8 Hz, H9), 4.62 (1H, dd, J7,6=6.1 Hz, J6,5=3.6 Hz, H6), 4.74 (1H, dd, J7,6=6.1 Hz, J8,7=3.7 Hz, H7), 5.76 (2H, m, H2, H3); 13C-NMR: δ 24.7, 25.3, 25.8, 26.9, 31.2, 63.6, 66.9, 73.2, 80.7, 81.1, 81.5, 81.6, 109.0, 112.4, 128.2, 131.5; Anal. Calcd for C16H26O6 (314.38): C 61.13, H 8.34; found C 61.32, H 8.50.
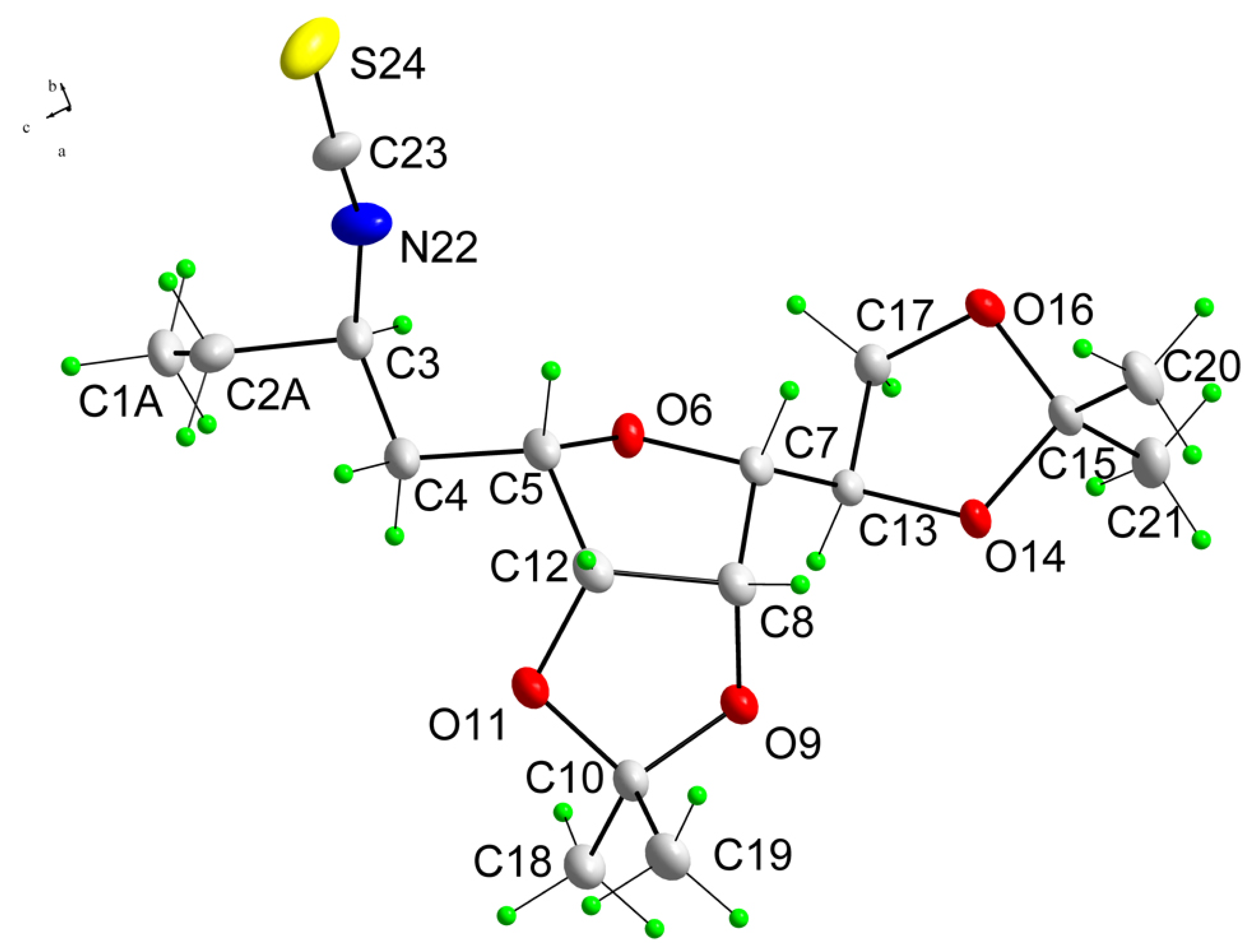
5,8-Anhydro-1,2,3,4-tetradeoxy-6,7:9,10-di-O-isopropylidene-1-thiocyanato-d-glycero-d-galacto-dec-2(E)-enitol (7): To a solution of alcohol 6 (2.34 g, 7.44 mmol) in dry dichloromethane (26 mL) were added triethylamine (1.55 mL, 11.17 mmol) and CH3SO2Cl (0.69 mL, 8.93 mmol) at 0 oC. The mixture was stirred at 0 oC for 15 min and then further 45 min at room temperature. The solvent was evaporated under reduced pressure. The residue was diluted with diethyl ether (40 mL) and the solid was removed by filtration. The solvent was evaporated to afford the crude mesylate which was used in the subsequent reaction directly without further purification. To the crude mesylate dissolved in CH3CN (26 mL), KSCN (1.09 g, 11.17 mmol) was added. After stirring at room temperature for 1 h, the solvent was evaporated. The residue was diluted with diethyl ether (40 mL) and the solid was removed by filtration. Evaporation of the solvent and chromatography of the residue (hexane-ethyl acetate, 5:1) afforded 2.0 g (76%) of thiocyanate 7 as white crystals; m.p. 81–82 °C; [α]d25 = +23 (c 0.28, CHCl3); 1H-NMR: δ 1.34 (3H, s, CH3), 1.38 (3H, s, CH3), 1.44 (3H, s, CH3), 1.48 (3H, s, CH3), 2.50-2.54 (2H, m, H4), 3.49 (1H, dd, J9,8=7.5 Hz, J8,7=3.7 Hz, H8), 3.52-3.55 (3H, m, 2 x H1, H5), 4.05 (1H, dd, J10,10=8.7 Hz, J10,9=4.8 Hz, H10), 4.09 (1H, dd, J10,10=8.7 Hz, J10,9=6.1 Hz, H10), 4.39 (1H, ddd, J9,8=7.5 Hz, J10,9= 6.1 Hz, J10,9=4.8 Hz, H9), 4.67 (1H, dd, J7,6=6.1 Hz, J6,5=3.6 Hz, H6), 4.74 (1H, dd, J7,6=6.1 Hz, J8,7=3.7 Hz, H7), 5.70 (1H, m, H2), 5.87 (1H, m, H3); 13C-NMR: δ 24.6, 25.3, 25.8, 26.9, 31.4, 36.3, 66.9, 73.1, 80.7 81.0, 81.2, 81.6, 109.0, 111.9, 112.4, 124.9, 134.0; Anal. Calcd for C17H25NO5S (355.46): C 57.44, H 7.09, N 3.94; found C 57.61, H 7.28, N 4.04.
5,8-Anhydro-1,2,3,4-tetradeoxy-6,7:9,10-di-O-isopropylidene-3(S)-isothiocyanato-d-glycero-d-galacto-dec-1-enitol (8a) and 5,8-Anhydro-1,2,3,4-tetradeoxy-6,7:9,10-di-O-isopropylidene-3(R)-isothiocyanato-d-glycero-d-galacto-dec-1-enitol (8b): Conventional method for the preparation of 8a, 8b: A solution of thiocyanate 7 (1.80 g, 5.06 mmol) in dry n-heptane (30 mL) was heated at 90 oC for 6 h under nitrogen atmosphere. The solvent was evaporated under reduced pressure. The chromatography of the residue on silica gel (hexane-ethyl acetate, 9:1) afforded isothiocyanates 8a (0.75 g, 42%) and 8b (0.73 g, 41%).
Microwave-assisted synthesis of 8a, 8b: The (E)-thiocyanate 7 (20 mg, 0.056 mmol) was weighed in a 10 ml glass pressure microwave tube equipped with a magnetic stirrer bar. Dry n-heptane (0.4 mL) was added, the tube was closed with a silicon septum and the reaction mixture was subjected to microwave irradiation for 2 h (power: 150 W, temperature: 90 oC, pressure: 12 bar). The reaction mixture was allowed to cool to room temperature and transferred into a round bottom flask. The solvent was evaporated under reduced pressure. The chromatography of the residue on silica gel (hexane-ethyl acetate, 9:1) gave 0.16 mg (86%) of isothiocyanates 8a, 8b. Compound 8a: white crystals; m.p. 54-56 oC; [α]d25 = -19 (c 0.27, CHCl3); υmax (liquid film) 2033 (NCS) cm-1; 1H-NMR: δ 1.33 (3H, s, CH3), 1.38 (3H, s, CH3), 1.45 (3H, s, CH3), 1.46 (3H, s, CH3), 1.93 (1H, m, H4), 2.07 (1H, ddd, J4,4=14.3 Hz, J5,4=9.3 Hz, J4,3=3.7 Hz, H4), 3.54 (1H, dd, J9,8=7.6 Hz, J8,7=3.7 Hz, H8), 3.71 (1H, ddd, J5,4=9.3 Hz, J6,5=3.7 Hz, J5,4=3.5 Hz, H5), 4.06 (1H, dd, J10,10=8.7 Hz, J10,9=4.8 Hz, H10), 4.09 (1H, dd, J10,10=8.7 Hz, J10,9=6.0 Hz, H10), 4.39 (1H, ddd, J9,8=7.6 Hz, J10,9=6.0 Hz, J10,9=4.8 Hz, H9), 4.45 (1H, m, H3), 4.64 (1H, dd, J7,6=6.1 Hz, J6,5=3.7 Hz, H6), 4.78 (1H, dd, J7,6=6.1 Hz, J8,7=3.7 Hz, H7), 5.24 (1H, dd, J2,1cis=10.2 Hz, J3,1cis=1.4 Hz, H1cis), 5.39 (1H, dd, J2,1trans=16.9 Hz, J3,1trans=1.6 Hz, H1trans), 5.83 (1H, ddd, J2,1trans=16.9 Hz, J2,1cis=10.2 Hz, J3,2=5.4 Hz, H2); 13C-NMR: δ 24.5, 25.3, 25.7, 26.9, 35.3, 57.5, 66.9, 73.0, 78.1, 80.8, 81.5, 81.7, 109.2, 112.5, 116.5, 132.9, 135.2; Anal. Calcd for C17H25NO5S (355.46): C 57.44, H 7.09, N 3.94; found C 57.25, H 7.20, N 4.11. Compound 8b: a colorless oil; [α]d25 = -10 (c 0.41, CHCl3); υmax (liquid film) 2020 (NCS) cm-1; 1H-NMR: δ 1.33 (3H, s, CH3), 1.38 (3H, s, CH3), 1.44 (3H, s, CH3), 1.47 (3H, s, CH3), 2.03 (1H, m, H4), 2.16 (1H, m, H4), 3.49 (1H, dd, J9,8=7.6 Hz, J8,7=3.6 Hz, H8), 3.61 (1H, ddd, J5,4=7.4 Hz, J5,4=6.1 Hz, J6,5=3.7 Hz, H5), 4.05 (1H, dd, J10,10=8.8 Hz, J10,9=4.6 Hz, H10), 4.09 (1H, dd, J10,10=8.8 Hz, J10,9=6.2 Hz, H10), 4.36–4.41 (2H, m, H3, H9), 4.64 (1H, dd, J7,6=6.1 Hz, J6,5=3.7 Hz, H6), 4.77 (1H, dd, J7,6=6.1 Hz, J8,7=3.6 Hz, H7), 5.25 (1H, ddd, J2,1´cis=10.2 Hz, J3,1cis=1.1 Hz, J1cis,1trans=0.5 Hz, H1cis), 5.36 (1H, ddd, J2,1trans=16.8 Hz, J3,1trans=1.3 Hz, J1cis,1trans=0.5 Hz, H1trans), 5.81 (1H, ddd, J2,1trans=16.8 Hz, J2,1cis=10.2 Hz, J3,2=6.0 Hz, H2); 13C-NMR: δ 24.6, 25.2, 25.7, 27.0, 34.8, 57.3, 66.9, 73.0, 78.4, 80.7, 81.0, 81.7, 109.1, 112.6, 117.2, 132.7, 134.8; Anal. Calcd for C17H25NO5S (355.46): C 57.44, H 7.09, N 3.94; found C 57.62, H 6.94, N 3.88.
5,8-Anhydro-1,2,3,4-tetradeoxy-6,7:9,10-di-O-isopropylidene-3(S)-(methoxycarbonylamino)-d-glycero-d-galacto-dec-1-enitol (10a): To a solution of isothiocyanate 8a (0.54 g, 1.52 mmol) in dry methanol (15 mL) was added sodium methoxide (90 mg, 1.67 mmol). The reaction mixture was stirred for 3 h at room temperature under nitrogen atmosphere. The solvent was evaporated and the residue was partitioned between CH2Cl2 (25 mL) and water (7 mL). The organic layer was dried (Na2SO4) and the solvent was evaporated under reduced pressure to provide the crude thiourethane 9a which was used in the subsequent reaction directly without further purification. To a solution of 9a (436 mg, 1.12 mmol) in dry acetonitrile (10.8 mL) was added mesitonitrile oxide (218 mg, 1.35 mmol). The mixture was stirred at room temperature for 2 h under nitrogen atmosphere. The solvent was evaporated under reduced pressure. The chromatography of the residue (hexane-ethyl acetate, 3:1) gave 0.35 g (85%) of 10a as a colorless oil; 1H-NMR: δ 1.33 (3H, s, CH3), 1.38 (3H, s, CH3), 1.45 (3H, s, CH3), 1.47 (3H, s, CH3), 1.88 (1H, m, H4), 2.04 (1H, m, H4), 3.51 (1H, dd, J9,8=6.7 Hz, J8,7=3.7 Hz, H8), 3.63 (1H, m, H5), 3.66 (3H, s, CH3O), 4.05 (1H, dd, J10,10=8.7 Hz, J10,9=4.8 Hz, H10), 4.10 (1H, dd, J10,10=8.7 Hz, J10,9=6.2 Hz, H10), 4.36-4.41 (2H, m, H3, H9), 4.59 (1H, dd, J7,6=6.1 Hz, J6,5=3.7 Hz, H6), 4.72 (1H, dd, J7,6=6.1 Hz, J8,7=3.7 Hz, H7), 5.13 (1H, d, J2,1cis=10.4 Hz, H1cis), 5.20 (1H, d, J2,1trans=17.1 Hz, H1trans), 5.38 (1H, d, J3,NH=6.4 Hz, NH), 5.80 (1H, ddd, J2,1trans=17.1 Hz, J2,1cis=10.4 Hz, J3,2=5.1 Hz, H2); 13C- NMR: δ 24.5, 25.3, 25.7, 26.9, 32.6, 51.0, 52.0, 66.8, 73.1, 79.1, 80.5, 81.7, 81.8, 109.1, 112.5, 114.7, 138.2, 156.4; Anal. Calcd for C18H29NO7 (371.43): C 58.21, H 7.87, N 3.77; found C 58.46, H 7.61, N 3.92.
5,8-Anhydro-1,2,3,4-tetradeoxy-6,7:9,10-di-O-isopropylidene-3(R)-(methoxycarbonylamino)-d-glycero-d-galacto-dec-1-enitol (10b): To a solution of isothiocyanate 8b (416 mg, 1.17 mmol) in dry methanol (11.6 mL) was added sodium methoxide (69.5 mg, 1.29 mmol). The reaction mixture was stirred for 4 h at room temperature under nitrogen atmosphere. The solvent was evaporated and the residue was partitioned between CH2Cl2 (20 mL) and water (6 mL). The organic layer was dried (Na2SO4). The solvent was evaporated under reduced pressure to give the crude thiourethane 9b which was used in the subsequent reaction directly without further purification. To a solution of 9b (288 mg, 0.74 mmol) in dry acetonitrile (7 mL) was added mesitonitrile oxide (144 mg, 0.89 mmol). The mixture was stirred at room temperature for 2 h under nitrogen atmosphere. The solvent was evaporated under reduced pressure and the chromatography of the residue (hexane-ethyl acetate, 3:1) afforded 0.25 g (92%) of 10b as a colorless oil; 1H-NMR: δ 1.33 (3H, s, CH3), 1.37 (3H, s, CH3), 1.44 (3H, s, CH3), 1.47 (3H, s, CH3), 1.84 (1H, ddd, J4,4=14.2 Hz, J4,3=9.5 Hz, J5,4=6.5 Hz, H4), 2.00 (1H, m, H4), 3.47 (1H, dd, J9,8=7.5 Hz, J8,7=3.6 Hz, H8), 3.59 (1H, ddd, J5,4=6.5 Hz, J5,4=6.5 Hz, J6,5=3.6, H5), 3.67 (3H, s, CH3O), 4.03 (1H, dd, J10,10=8.7 Hz, J10,9=4.6 Hz, H10), 4.08 (1H, dd, J10,10=8.7 Hz, J10,9=6.2 Hz, H10), 4.29 (1H, m, H3), 4.38 (1H, ddd, J9,8=7.5 Hz, J10,9=6.2 Hz, J10,9=4.6 Hz, H9), 4.67 (1H, dd, J7,6=6.1 Hz, J6,5=3.6 Hz, H6), 4.73 (1H, dd, J7,6=6.1 Hz, J8,7=3.6 Hz, H7), 4.93 (1H, m, NH), 5.12 (1H, dd, J2,1cis=10.4 Hz, J3,1cis=1.3 Hz, H1cis), 5.21 (1H, dd, J2,1trans=17.0 Hz, J3,1trans=1.2 Hz, H1trans), 5.80 (1H, ddd, J2,1trans=17.0 Hz, J2,1cis=10.4 Hz, J3,2=5.6 Hz, H2); 13C-NMR: δ 24.6, 25.2, 25.8, 26.9, 33.7, 51.4, 52.1, 66.9, 73.1, 79.5, 80.6, 81.4, 81.7, 109.1, 112.4, 114.8, 138.5, 156.6; Anal. Calcd for C18H29NO7 (371.43): C 58.21, H 7.87, N 3.77; found C 58.05, H 7.54, N 3.53.
4,7-Anhydro-2,3-dideoxy-5,6:8,9-di-O-isopropylidene-2(S)-(methoxycarbonylamino)-d-glycero-d-galacto-nononic acid (12a): A solution of 10a (0.28 g, 0.76 mmol) in methanol (28 mL) was cooled to -78 oC. Ozone was then passed through the solution under vigorous stirring. The maximum time for the ozone treatment was 30 min. This resulted in the formation of a bluish solution. Dry nitrogen was passed through the cold solution in order to remove excess ozone. Ph3P (0.20 g, 0.76 mmol) and CH2Cl2 (11 mL) were added and the solution was allowed to warm up to room temperature while stirring was continued for 1.5 h. The solvent was removed under reduced pressure and the chromatography of the residue (hexane-ethyl acetate, 2:1) afforded 0.25 g (87%) of 11a as a colorless oil which was used immediately in the next step. 1H-NMR: δ 1.32 (3H, s, CH3), 1.38 (3H, s, CH3), 1.44 (3H, s, CH3), 1.47 (3H, s, CH3), 2.07-2.17 (1H, m, H3), 2.24 (1H, m, H3), 3.54 (1H, dd, J8,7=6.8 Hz, J7,6=3.7 Hz, H7), 3.62-3.68 (1H, m, H4), 3.70 (3H, s, CH3O), 4.03 (1H, dd, J9,9=8.7 Hz, J9,8=4.8 Hz, H9), 4.08 (1H, dd, J9,9=8.7 Hz, J9,8=6.3 Hz, H9), 4.32-4.40 (2H, m, H2, H8), 4.60 (1H, dd, J6,5=6.1 Hz, J5,4=3.7 Hz, H5), 4.74 (1H, dd, J6,5=6.1 Hz, J7,6=3.7 Hz, H6), 5.77 (1H, m, NH), 9.64 (1H, bs, CH=O). A solution of NaClO2 (80%, 0.57 g, 6.3 mmol) and NaH2PO4 (0.71 g, 4.5 mmol) in 3.8 mL of water was added dropwise to a solution of aldehyde 11a (0.25 g, 0.68 mmol) in acetonitrile/tert-butyl alcohol/2-methyl-2-butene (15 mL, 4:4:1) at 0 oC over 5 min and stirred at the same temperature for 35 min. The reaction mixture was poured into brine (12 mL) and extracted with ethyl acetate (2 x 25 mL). The combined organic layers were dried (Na2SO4). The solvent was removed under reduced pressure and chromatography of the residue on silica gel (hexane-ethyl acetate, 1:2) afforded 0.20 g (74%) of carboxylic acid 12a as a colorless oil; [α]d25 = -33 (c 0.12, CHCl3); 1H-NMR: δ 1.31 (3H, s, CH3), 1.37 (3H, s, CH3), 1.44 (3H, s, CH3), 1.45 (1H, s, CH3), 2.18-2.26 (2H, m, H3), 3.51 (1H, dd, J8,7=6.8 Hz, J7,6=3.6 Hz, H7), 3.62-3.66 (4H, m, H4, CH3O), 4.02-4.08 (2H, m, H9), 4.37 (1H, ddd, J8,7=6.8 Hz, J9,8=6.1 Hz, J9,8=4.8 Hz, H8), 4.48 (1H, m, H2), 4.59 (1H, dd, J6,5=6.1 Hz, J5,4=3.7 Hz, H5), 4.70 (1H, dd, J6,5=6.1 Hz, J7,6=3.6 Hz, H6), 5.76 (1H, d, J2,NH=7.9 Hz, NH); 13C-NMR: δ 24.6, 25.3, 25.7, 26.9, 30.9, 52.1, 52.3, 66.7, 73.1, 80.0, 2 x 81.0, 81.6, 109.2, 112.5, 156.7, 171.2; Anal. Calcd for C17H27NO9 (389.40): C 52.44, H 6.99, N 3.60; found C 52.10, H 7.00, N 3.41.
4,7-Anhydro-2,3-dideoxy-5,6:8,9-di-O-isopropylidene-2(R)-(methoxycarbonylamino)-d-glycero-d-galacto-nononic acid (12b): A solution of 10b (185 mg, 0.498 mmol) in methanol (18 mL) was cooled to -78 oC. Ozone was then passed through the solution under vigorous stirring. The maximum time for the ozone treatment was 30 min. This resulted in the formation of a bluish solution. Dry nitrogen was passed through the cold solution in order to remove excess ozone. Ph3P (0.13 g, 0.498 mmol) and CH2Cl2 (7 mL) were added and the solution was allowed to warm up to room temperature while stirring was continued for 1.5 h. The solvent was removed under reduced pressure and the chromatography of the residue (hexane-ethyl acetate, 2:1) gave 0.16 g (88%) of aldehyde 11b as a colorless oil which was used immediately in the next step. 1H-NMR: δ 1.33 (3H, s, CH3), 1.36 (3H, s, CH3), 1.43 (3H, s, CH3), 1.48 (3H, s, CH3), 2.11-2.21 (1H, m, H3), 2.26-2.36 (1H, m, H3), 3.48 (1H, dd, J8,7=7.0 Hz, J7,6=3.6 Hz, H7), 3.62-3.67 (1H, m, H4), 3.70 (3H, s, CH3O), 3.94 (1H, dd, J9,9=8.7 Hz, J9,8=4.8 Hz, H9), 4.04 (1H, dd, J9,9=8.7 Hz, J9,8=6.2 Hz, H9), 4.32-4.36 (2H, m, H2, H8), 4.68 (1H, dd, J6,5=6.1 Hz, J5,4= 3.5 Hz, H5), 4.74 (1H, dd, J6,5=6.1 Hz, J7,6=3.6 Hz, H6), 5.63 (1H, m, NH), 9.52 (1H, bs, CH=O). A solution of NaClO2 (80%, 0.37 g, 4.1 mmol) and NaH2PO4 (0.46 g, 2.9 mmol) in 2.5 mL of water was added dropwise to a solution of aldehyde 11b (0.16 g, 0.43 mmol) in acetonitrile/tert-butyl alcohol/2-methyl-2-butene (10 mL, 4:4:1) at 0 oC over 5 min and stirred at the same temperature for 45 min. The reaction mixture was poured into brine (8 mL) and extracted with ethyl acetate (2 x 16 mL). The combined organic layers were dried (Na2SO4). The solvent was removed under reduced pressure and the chromatography of the residue on silica gel (hexane-ethyl acetate, 1:2) gave 0.12 g (73%) of carboxylic acid 12b as a white viscous oil; [α]d25 = -7 (c 0.49, CHCl3); 1H-NMR: δ 1.32 (3H, s, CH3), 1.35 (3H, s, CH3), 1.42 (3H, s, CH3), 1.45 (3H, s, CH3), 2.05-2.13 (1H, m, H3), 2.29-2.36 (1H, m, H3), 3.44 (1H, m, H7), 3.60-3.63 (1H, m, H4), 3.65 (3H, s, CH3O), 3.98-4.04 (2H, m, H9), 4.34 (1H, ddd, J8,7=7.3 Hz, J9,8=6.2 Hz, J9,8=5.4 Hz, H8), 4.39-4.40 (1H, m, H2), 4.70 (2H, m, H5, H6), 5.61 (1H, d, J2,NH=7.1 Hz, NH); 13C-NMR: δ 24.7, 25.3, 25.8, 26.9, 31.4, 51.9, 52.1, 66.8, 73.1, 80.7, 2 x 81.4, 81.6, 109.0, 112.4, 156.7, 173.4; Anal. Calcd for C17H27NO9 (389.40): C 52.44, H 6.99, N 3.60; found C 52.68, H 7.03, N 3.82.





{kind=link}
{kind=link}
{kind=link}