Neuroprotection by Radical Avoidance: Search for Suitable Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
The Multiple Levels of Enhanced Radical Formation and Radical Avoidance
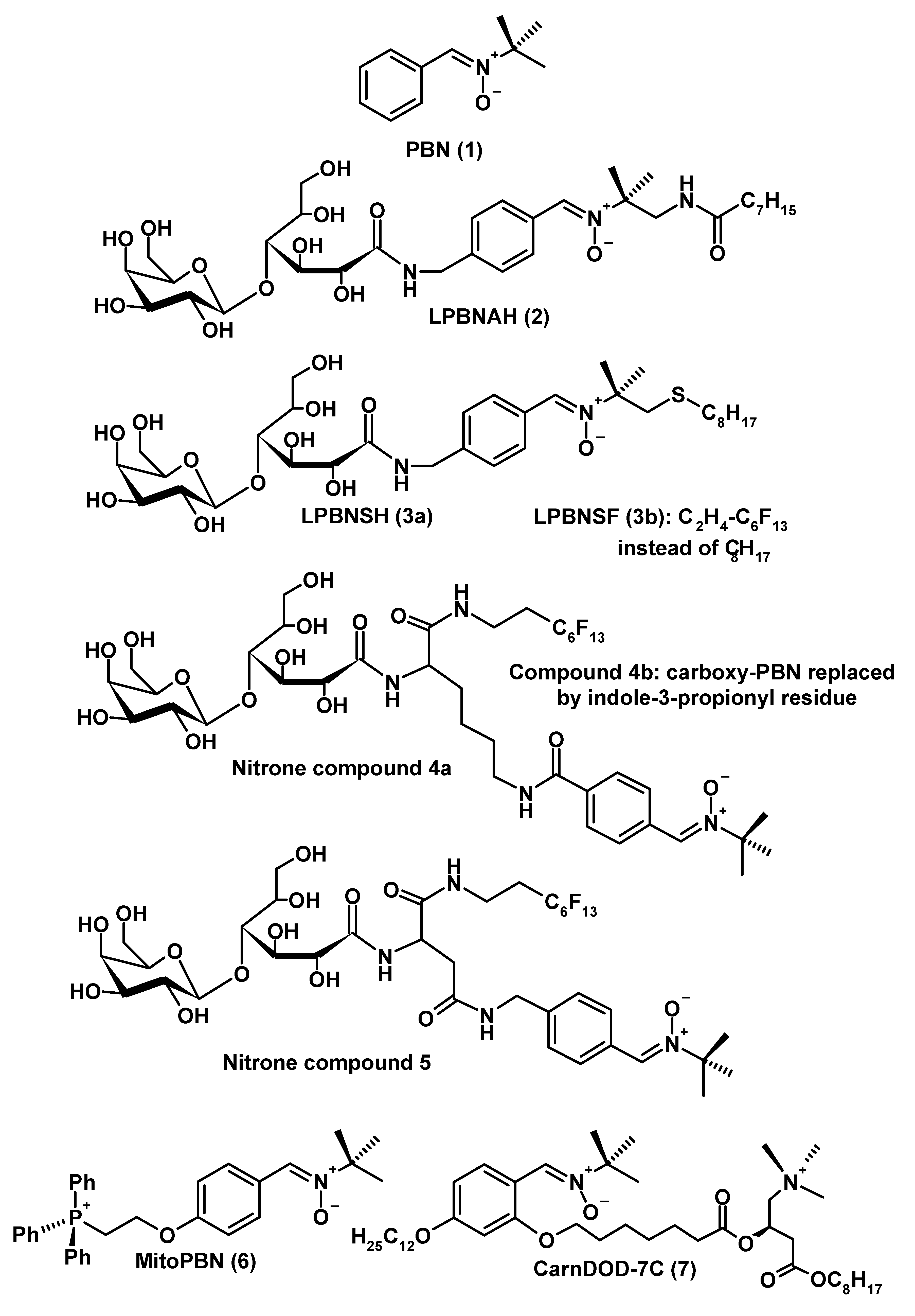
Nitrones

Leptin
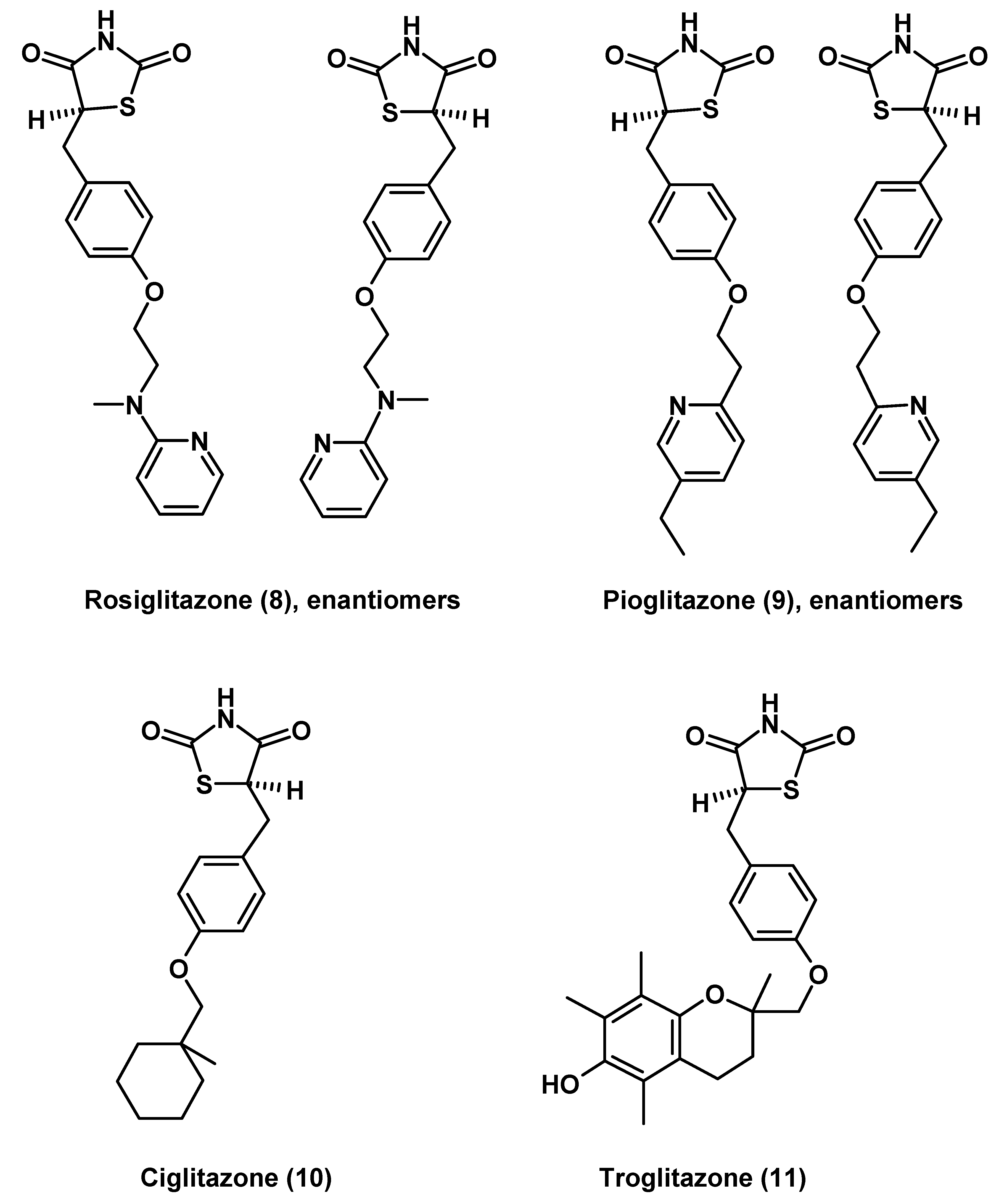
Thiazolidinediones

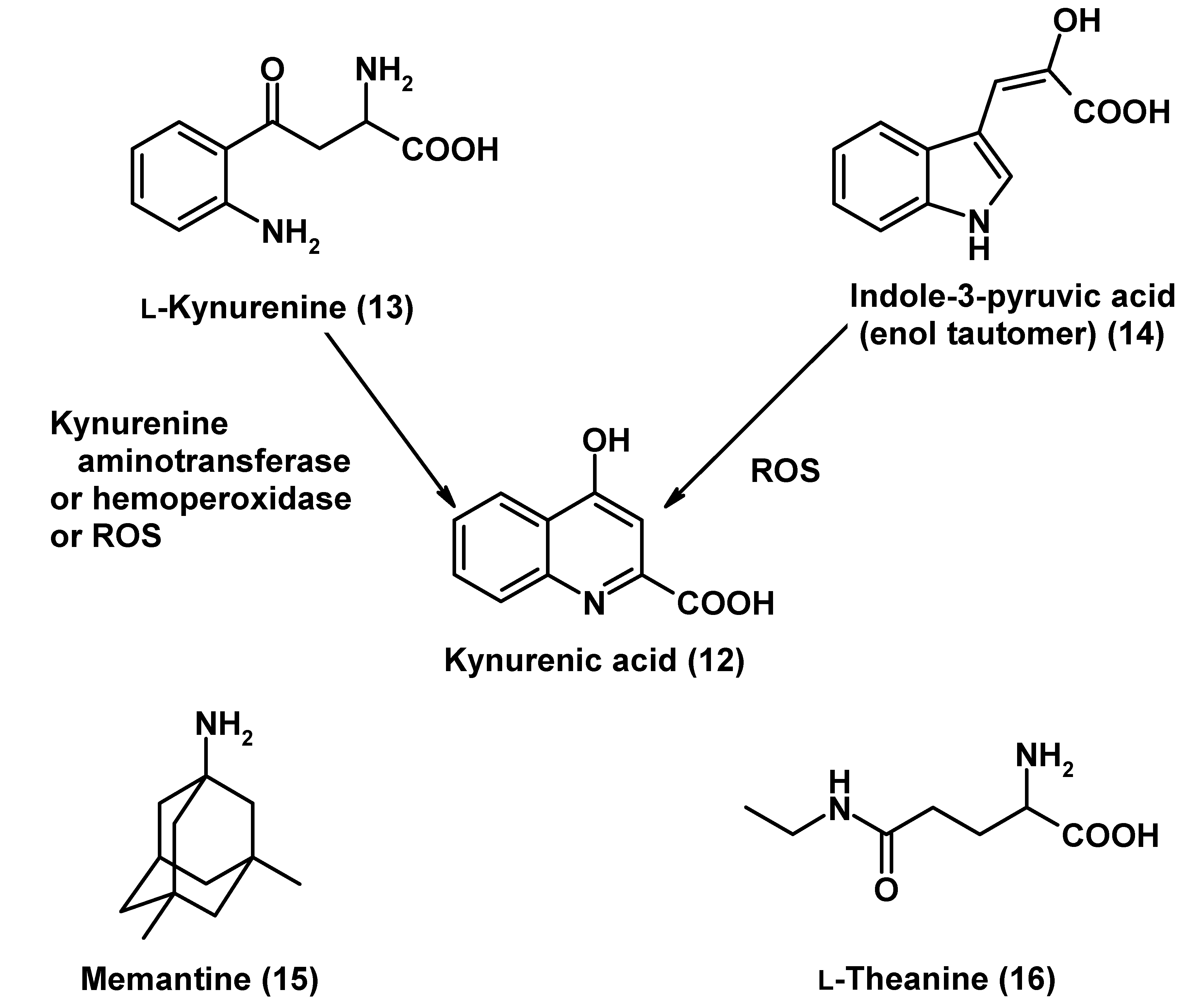
Glutamatergic Modulators

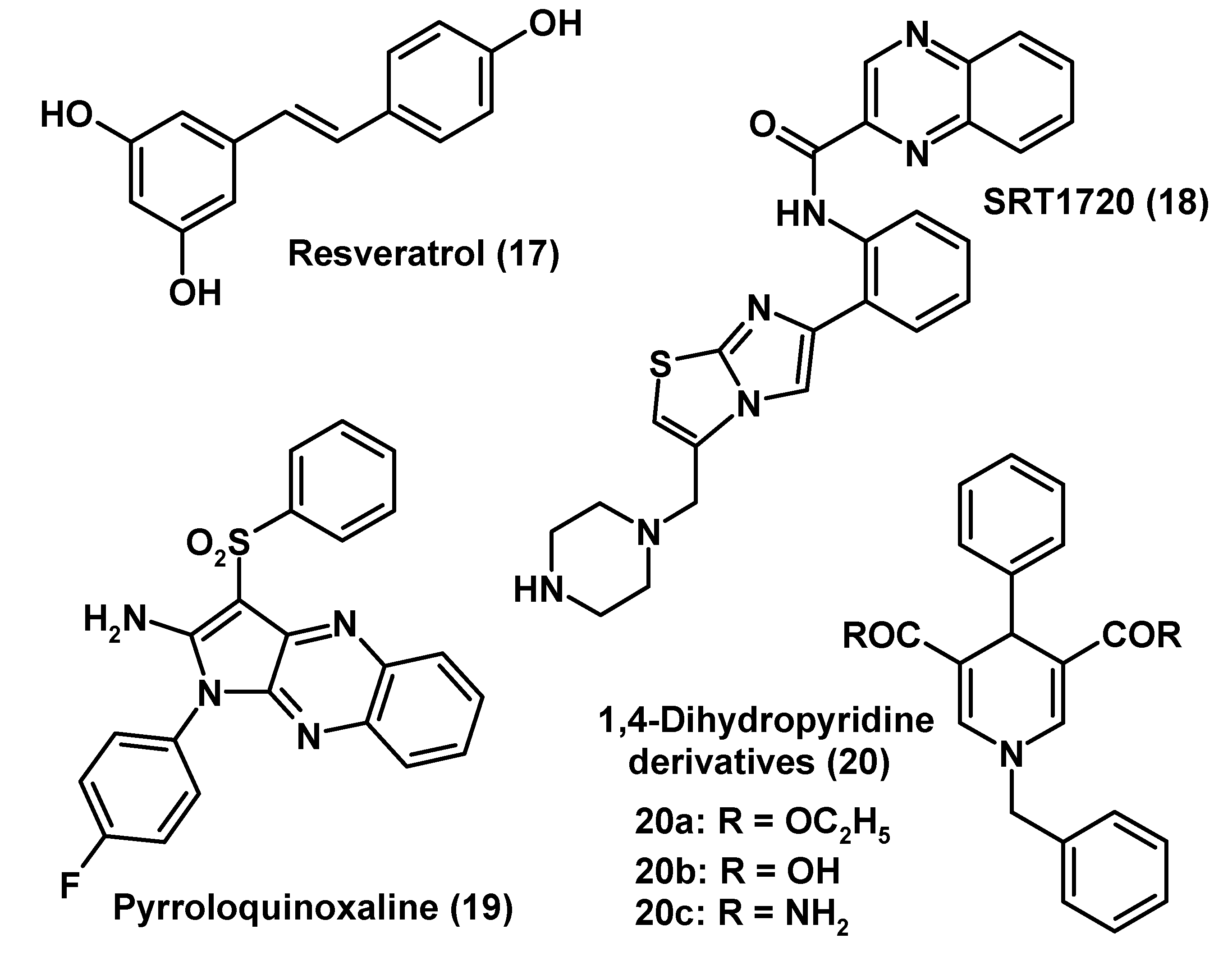
Resveratrol and Other Sirtuin Activators

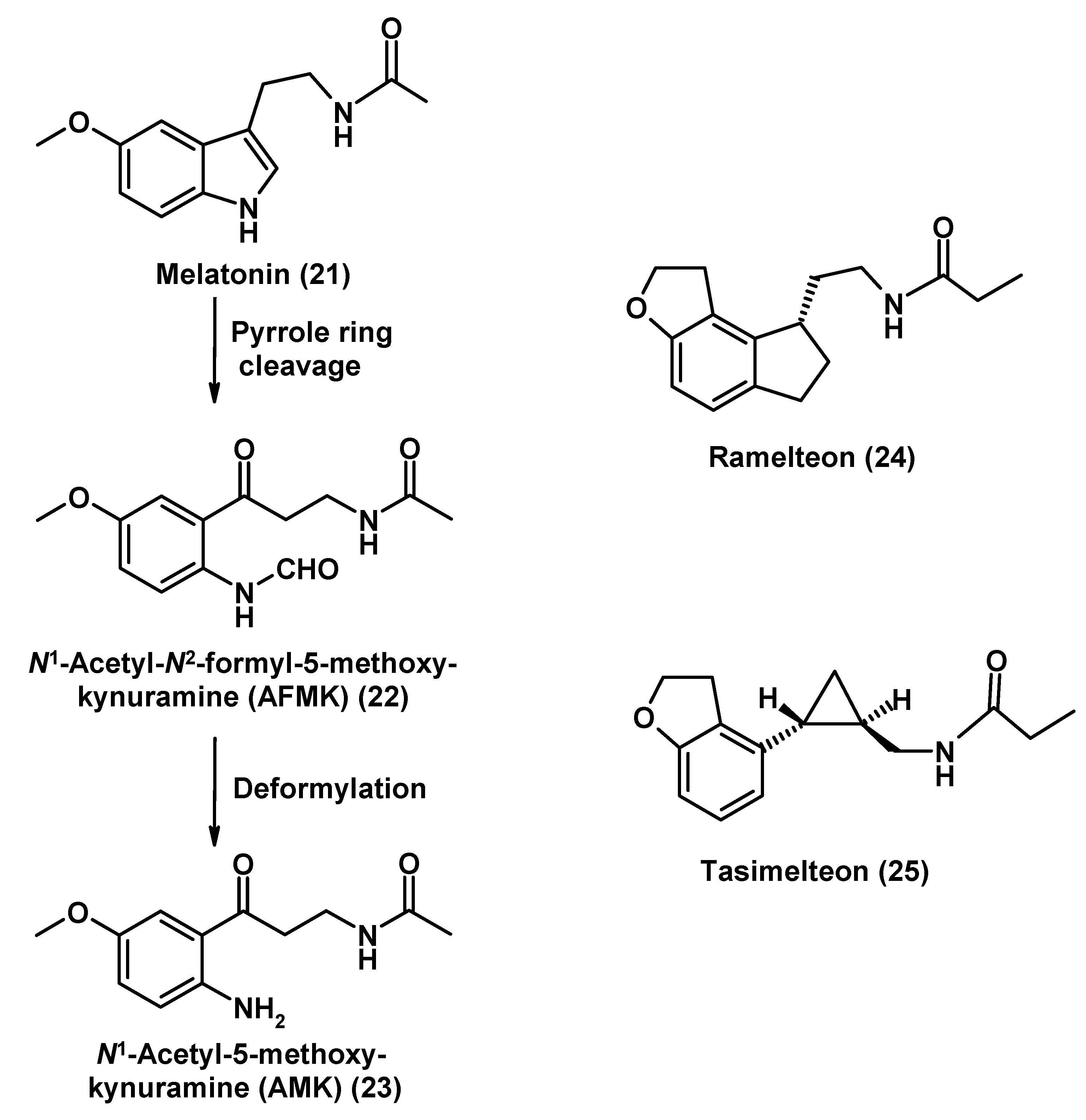
Melatonin, Its Analogs and Metabolites

Conclusions
- Samples Availability: Not available.
References and Notes
- Grundman, M.; Grundman, M.; Delaney, P. Antioxidant strategies for Alzheimer's disease. Proc. Nutr. Soc. 2002, 61, 191–202. [Google Scholar] [CrossRef]
- Frank, B.; Gupta, S. A review of antioxidants and Alzheimer's disease. Ann. Clin. Psychiatry 2005, 17, 269–286. [Google Scholar] [CrossRef]
- Boothby, L.A.; Doering, P.L. Vitamin C and vitamin E for Alzheimer's disease. Ann. Pharmacother. 2005, 39, 2073–2080. [Google Scholar] [CrossRef]
- Bhat, V.; Weiner, W.J. Parkinson's disease. Diagnosis and the initiation of therapy. Minerva Med. 2005, 96, 145–154. [Google Scholar]
- Isaac, M.G.E.K.N.; Quinn, R.; Tabet, N. Vitamin E for Alzheimer's disease and mild cognitive impairment. Cochrane Database Syst. Rev. 2008, 3, CD002854. [Google Scholar]
- Poeggeler, B.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C. Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis. J. Pineal Res. 1993, 14, 151–168. [Google Scholar] [CrossRef]
- Poeggeler, B. Melatonin, aging, and age-related diseases: Perspectives for prevention, intervention, and therapy. Endocrine 2005, 27, 201–212. [Google Scholar] [CrossRef]
- Dröge, W.; Schipper, H.M. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 2007, 6, 361–370. [Google Scholar] [CrossRef]
- Figueiredo, P.A.; Mota, M.P.; Appell, H.J.; Duarte, J.A. The role of mitochondria in aging of skeletal muscle. Biogerontology 2008, 9, 67–84. [Google Scholar] [CrossRef]
- Rattan, S.I. Increased molecular damage and heterogeneity as the basis of aging. Biol. Chem. 2008, 389, 267–272. [Google Scholar]
- Wei, Y.H.; Wu, S.B.; Ma, Y.S.; Lee, H.C. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang Gung Med. J. 2009, 32, 113–132. [Google Scholar]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Pennisi, G.; Calafato, S.; Bellia, F.; Bates, T.E.; Giuffrida Stella, A.M.; Schapira, T.; Dinkova Kostova, A.T.; Rizzarelli, E. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem. Res. 2008, 33, 2444–2471. [Google Scholar]
- Chigurupati, S.; Wie, Z.; Belal, C.; Vandermey, M.; Kyriazis, G.A.; Arumugam, T.V.; Chan, S.L. The homocysteine-inducible endoplasmic reticulum stress protein counteracts calcium store depletion and induction of CCAAT enhancer-binding protein homologous protein in a neurotoxin model of Parkinson disease. J. Biol. Chem. 2009, 284, 18323–18333. [Google Scholar]
- Hardeland, R. Antioxidative protection by melatonin: Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef]
- Halliwell, B.B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Kanki, T.; Nakayama, H.; Sasaki, N.; Takio, K.; Alam, T.I.; Hamasaki, N.; Kang, D. Mitochondrial nucleoid and transcription factor A. Ann. N.Y. Acad. Sci. 2004, 1011, 61–68. [Google Scholar]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. N.Y. Acad. Sci. 2005, 1042, 101–108. [Google Scholar]
- Kienhöfer, J.; Häussler, D.J.; Ruckelshausen, F.; Muessig, E.; Weber, K.; Pimentel, D.; Ullrich, V.; Bürkle, A.; Bachschmid, M.M. Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents. FASEB J. 2009, 23, 2034–2044. [Google Scholar] [CrossRef]
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci. 2008, 31, 251–256. [Google Scholar] [CrossRef]
- Thompson, L.V. Oxidative stress, mitochondria and mtDNA-mutator mice. Exp. Gerontol. 2006, 41, 1220–1222. [Google Scholar] [CrossRef]
- Reilly, T.; Waterhouse, J.; Atkinson, G. Aging, rhythms of physical performance, and adjustment to changes in the sleep-activity cycle. Occup. Environ. Med. 1997, 54, 812–816. [Google Scholar] [CrossRef]
- Touitou, Y.; Bogdan, A.; Haus, E.; Touitou, C. Modifications of circadian and circannual rhythms with aging. Exp. Gerontol. 1997, 32, 603–614. [Google Scholar] [CrossRef]
- Weinert, D. Age-dependent changes of the circadian system. Chronobiol. Int. 2000, 17, 261–283. [Google Scholar] [CrossRef]
- Van Someren, E.J. Circadian rhythms and sleep in human aging. Chronobiol. Int. 2000, 17, 233–243. [Google Scholar] [CrossRef]
- Yamazaki, S.; Straume, M.; Tei, H.; Sakaki, Y.; Menaker, M.; Block, G.D. Effects of aging on central and peripheral mammalian clocks. Proc. Natl. Acad. Sci. USA 2002, 99, 10801–10806. [Google Scholar]
- Weinert, D. The temporal order of mammals. Evidence for multiple central and peripheral control mechanisms and for endogenous and exogenous components: some implications for research on aging. Biol. Rhythm Res. 2005, 36, 293–308. [Google Scholar]
- Morin, L.P. Age-related changes in hamster circadian period, entrainment and rhythm splitting. Biol. Rhythm Res. 1988, 3, 237–248. [Google Scholar] [CrossRef]
- Penev, P.D.; Zee, P.C.; Turek, F.W. Quantitative analysis of the age-related fragmentation of hamster 24-h activity rhythms. Am. J. Physiol. 1997, 273, R2132–R2137. [Google Scholar]
- Weinert, H.; Weinert, D.; Waterhouse, J. The circadian activity and body temperature rhythms of mice during their last days of life. Biol. Rhythm Res. 2002, 33, 199–212. [Google Scholar] [CrossRef]
- Humbert, W.; Pévet, P. The decrease of pineal melatonin production with age. Causes and consequences. Ann. N.Y. Acad. Sci. 1994, 719, 43–63. [Google Scholar] [CrossRef]
- Magri, F.; Locatelli, M.; Balza, G.; Molla, G.; Cuzzoni, G.; Fioravanti, M.; Solerte, S.B.; Ferrari, E. Changes in endocrine circadian rhythms as markers of physiological and pathological brain aging. Chronobiol. Int. 1997, 14, 385–396. [Google Scholar] [CrossRef]
- Touitou, Y. Human aging and melatonin. Clinical relevance. Exp. Gerontol. 2001, 36, 1083–1100. [Google Scholar] [CrossRef]
- Karasek, M.; Reiter, R.J. Melatonin and aging. Neuroendocrinol. Lett. 2002, 23 (Suppl. 1), 14–16. [Google Scholar]
- Karasek, M. Melatonin, human aging, and age-related diseases. Exp. Gerontol. 2004, 39, 1723–1729. [Google Scholar] [CrossRef]
- Coto-Montes, A.; Hardeland, R. Diurnal rhythm of protein carbonyl as an indicator of oxidative damage in Drosophila melanogaster: Influence of clock gene alleles and deficiencies in the formation of free-radical scavengers. Biol. Rhythm Res. 1999, 30, 383–391. [Google Scholar] [CrossRef]
- Coto-Montes, A.; Tomás-Zapico, C.; Rodríguez-Colunga, M.J.; Tolivia-Cadrecha, D.; Martínez-Fraga, J.; Hardeland, R.; Tolivia, D. Effects of the circadian mutation 'tau' on the Harderian glands of Syrian hamsters. J. Cell. Biochem. 2001, 83, 426–434. [Google Scholar] [CrossRef]
- Hardeland, R.; Coto-Montes, A.; Poeggeler, B. Circadian rhythms, oxidative stress and antioxidative defense mechanisms. Chronobiol. Int. 2003, 20, 921–962. [Google Scholar] [CrossRef]
- Krishnan, N.; Davis, A.J.; Giebultowicz, J.M. Circadian regulation of response to oxidative stress in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2008, 374, 299–303. [Google Scholar] [CrossRef]
- Coto-Montes, A.; Boga, J.A.; Tomás-Zapico, C.; Rodríguez-Colunga, M.J.; Martínez-Fraga, J.; Tolivia-Cadrecha, D.; Menéndez, G.; Hardeland, R.; Tolivia, D. Physiological oxidative stress model: Syrian hamster Harderian gland—Sex differences in antioxidant enzymes. Free Radic. Biol. Med. 2001, 30, 785–792. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003, 34, 325–337. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004, 36, 287–294. [Google Scholar] [CrossRef]
- Gardoni, F.; di Luca, M. New targets for pharmacological intervention in the glutamatergic synapse. Eur. J. Pharmacol. 2006, 545, 2–10. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Hilditch-Maguire, P.; Trettel, F.; Passani, L.A.; Auerbach, A.; Persichetti, F.; MacDonald, M.E. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 2000, 9, 2789–2797. [Google Scholar] [CrossRef]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are direct effects of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar]
- Ellerby, L.M. Hunting for excitement: NMDA receptors in Huntington’s disease. Neuron 2002, 33, 841–842. [Google Scholar] [CrossRef]
- Panov, A.V.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: Relevance to Huntington’s disease. Arch. Biochem. Biophys. 2003, 410, 1–6. [Google Scholar] [CrossRef]
- Estrada Sánchez, A.M.; Mejía-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington's disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Jin, Y.N.; Fuenzalida, K.; Bronfman, M.; Johnson, G.V. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: Possible role of peroxisome proliferator-activated receptor-γ (PPARγ) in the pathogenesis of Huntington disease. J. Biol. Chem. 2008, 283, 25628–25637. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington's disease. Biochim. Biophys. Acta. 2009. [Google Scholar] [CrossRef]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar]
- Srinivasan, V.; Pandi-Perumal, S.R.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin in Alzheimer’s disease and other neurodegenerative disorders. Behav. Brain Funct. 2006, 2. [Google Scholar] [CrossRef]
- Rogers, J.; Webster, S.; Lue, L.F.; Brachova, W.; Civin, W.H.; Emmerling, M.; Shivers, B.; Walker, D.; McGeer, P. Inflammation and Alzheimer's disease pathogenesis. Neurobiol. Aging 1996, 17, 681–686. [Google Scholar] [CrossRef]
- Pereira, H.A.; Kumar, P.; Gramma, P. Expression of CAP37, a novel inflammatory mediator, in Alzheimer's disease. Neurobiol. Aging 1996, 17, 753–759. [Google Scholar]
- Sheng, J.G.; Ito, K.; Skinner, R.D.; Mrak, R.E.; Rovnaghi, C.R.; van Eldik, L.J.; Griffin, W.S.T. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol. Aging 1996, 17, 761–766. [Google Scholar] [CrossRef]
- Yan, S.D.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; Schmidt, A.M. Amyloid-β peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: A proinflammatory pathway in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar]
- Carpenter, A.F.; Carpenter, P.W.; Markesbery, W.R. Morphometric analysis of microglia in Alzheimer's disease. J. Neuropathol. Exp. Neurol. 1993, 52, 601–608. [Google Scholar] [CrossRef]
- MacKenzie, I.R.A.; Hao, C.; Muñoz, D.G. Role of microglia in senile plaque formation. Neurobiol. Aging 1995, 16, 797–804. [Google Scholar] [CrossRef]
- McGeer, E.G.; McGeer, P.L. Neuroinflammation in Alzheimer's disease and mild cognitive impairment: A field in its infancy. J. Alzheimers Dis. 2009. [Google Scholar] [CrossRef]
- Landreth, G.E. Microglia in central nervous dystem diseases. J. Neuroimmune Pharmacol. 2009. [Google Scholar] [CrossRef]
- Alberati-Giani, D.; Ricciardi-Castagnoli, P.; Köhler, C.; Cesura, A.M. Regulation of the kynurenine metabolic pathway by interferon-γ in murine cloned macrophages and microglial cells. J. Neurochem. 1996, 66, 996–1004. [Google Scholar]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320, 595–597. [Google Scholar]
- Guillemin, G.J.; Smythe, G.A.; Veas, L.A.; Takikawa, O.; Brew, B.J. Aβ1-42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport 2003, 14, 2311–2315. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.; Kovac, A.D.; Ullrich, O.; Zipp, F.; Nitsch, R.; Bechmann, I. IDO (indolamine 2,3-dioxygenase) expression and function in the CNS. Adv. Exp. Med. Biol. 2003, 527, 113–118. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Williams, K.R.; Smith, D.G.; Smythe, G.A.; Croitoru-Lamoury, J.; Brew, B.J. Quinolinic acid in the pathogenesis of Alzheimer's disease. Adv. Exp. Med. Biol. 2003, 527, 167–176. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Yadav, M.C.; Burudi, E.M.; Alirezaei, M.; Flynn, C.C.; Watry, D.D.; Lanigan, C.M.; Fox, H.S. IFN-γ-induced IDO and WRS expression in microglia is differentially regulated by IL-4. Glia 2007, 55, 1385–1396. [Google Scholar] [CrossRef]
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology 1983, 22, 1331–1342. [Google Scholar] [CrossRef]
- Schurr, A.; West, C.A.; Rigor, B.M. Neurotoxicity of quinolinic acid and its derivatives in hypoxic rat hippocampal slices. Brain Res. 1991, 568, 199–204. [Google Scholar] [CrossRef]
- Misztal, M.; Skangiel-Kramska, J.; Niewiadomska, G.; Danysz, W. Subchronic intraventricular infusion of quinolinic acid produces working memory impairment—A model of progressive excitotoxicity. Neuropharmacology 1996, 35, 449–458. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington's disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef]
- Park, L.; Anrather, J.; Girouard, H.; Zhou, P.; Iadecola, C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 2007, 27, 1908–1918. [Google Scholar] [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J. Neurosci. Res. 2008, 86, 2524–2534. [Google Scholar] [CrossRef]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef]
- Collins-Underwood, J.R.; Zhao, W.; Sharpe, J.G.; Robbins, M.E. NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic. Biol. Med. 2008, 45, 929–938. [Google Scholar] [CrossRef]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef]
- Schiavone, S.; Sorce, S.; Dubois-Dauphin, M.; Jaquet, V.; Colaianna, M.; Zotti, M.; Cuomo, V.; Trabace, L.; Krause, K.H. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol. Psychiatry 2009, 66, 384–392. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef]
- Hardeland, R.; Poeggeler, B.; Pappolla, M.A. New vistas on mitochondrial electron flux rates and aging. Cell. 2008. Available online: http://www.cell.com/content/article/comments?uid= PIIS0092867408000627/ accessed on 4 December 2009.
- Hardeland, R. Melatonin, mitochondrial electron flux and leakage: Recent findings and resolution of contradictory results. Adv. Stud. Biol. 2009, 1, 207–230. [Google Scholar]
- Hardeland, R.; Poeggeler, B.; Pappolla, M.A. Mitochondrial actions of melatonin — an endeavor to identify their adaptive and cytoprotective mechanisms. Abh. Sächs. Akad. Wiss. Math. Nat. Kl. 2009, 65(4), 14–31. [Google Scholar]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; Yin, J.; Wang, W.; Mattson, M.P.; Kao, J.P.; Lakatta, E.G.; Sheu, S.S.; Ouyang, K.; Chen, J.; Dirksen, R.T.; Cheng, H. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef]
- Sheu, S.S.; Wang, W.; Cheng, H.; Dirksen, R.T. Superoxide flashes: Illuminating new insights into cardiac ischemia/reperfusion injury. Future Cardiol. 2008, 4, 551–554. [Google Scholar] [CrossRef]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef]
- Genova, M.L.; Merlo Pich, M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N.Y. Acad. Sci. 2004, 1011, 86–100. [Google Scholar]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Merlo Pich, M.; Paolucci, U.; Parenti Castelli, G.; Ventura, B. Role of mitochondria in oxidative stress and aging. Ann. N.Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar]
- Ohnishi, S.T.; Ohnishi, T.; Muranaka, S.; Fujita, H.; Kimura, H.; Uemura, K.; Yoshida, K.; Utsumi, K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J. Bioenerg. Biomembr. 2005, 37, 1–15. [Google Scholar] [CrossRef]
- Lenaz, G.; Fato, R.; Genova, M.L.; Bergamini, C.; Bianchi, C.; Biondi, A. Mitochondrial complex I: Structural and functional aspects. Biochim. Biophys. Acta 2006, 1757, 1406–1420. [Google Scholar] [CrossRef]
- Genova, M.L.; Merlo Pich, M.; Biondi, A.; Bernacchia, A.; Falasca, A.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. Mitochondrial production of oxygen radical species and the role of coenzyme Q as an antioxidant. Exp. Biol. Med. Maywood 2003, 228, 506–513. [Google Scholar]
- Staniek, K.; Gille, L.; Kozlov, A.V.; Nohl, H. Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Radic. Res. 2002, 36, 381–387. [Google Scholar] [CrossRef]
- Gong, X.; Yu, L.; Xia, D.; Yu, C.A. Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex. J. Biol. Chem. 2005, 280, 9251–9257. [Google Scholar]
- Miwa, S.; Brand, M.D. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim. Biophys. Acta 2005, 1709, 214–219. [Google Scholar]
- Tyagi, N.; Moshal, K.S.; Sen, U.; Lominadze, D.; Ovechkin, A.V.; Tyagi, S.C. Ciglitazone ameliorates homocysteine-mediated mitochondrial translocation and matrix metalloproteinase-9 activation in endothelial cells by inducing peroxisome proliferator activated receptor-γ activity. Cell. Mol. Biol. (Noisy-le-grand) 2006, 52, 21–27. [Google Scholar]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159. [Google Scholar]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2009. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar]
- Uo, T.; Dworzak, J.; Kinoshita, C.; Inman, D.M.; Kinoshita, Y.; Horner, P.J.; Morrison, R.S. Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Exp. Neurol. 2009, 218, 274–285. [Google Scholar] [CrossRef]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef]
- Radak, Z.; Kumagai, S.; Taylor, A.W.; Naito, H.; Goto, S. Effects of exercise on brain function: role of free radicals. Appl. Physiol. Nutr. Metab. 2007, 32, 942–946. [Google Scholar] [CrossRef]
- Goto, S.; Naito, H.; Kaneko, T.; Chung, H.Y.; Radák, Z. Hormetic effects of regular exercise in aging: correlation with oxidative stress. Appl. Physiol. Nutr. Metab. 2007, 32, 948–953. [Google Scholar] [CrossRef]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Mitochondrial decay in aging. Biochim. Biophys. Acta 1995, 1271, 165–170. [Google Scholar]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; Hoffer, B.; Cullheim, S.; Mohammed, A.H.; Olson, L.; Larsson, N.G. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar]
- Terzioglu, M.; Larsson, N.G. Mitochondrial dysfunction in mammalian ageing. Novartis Found. Symp. 2007, 287, 197–208. [Google Scholar] [CrossRef]
- Boveris, A.; Alvarez, S.; Navarro, A. The role of mitochondrial nitric oxide synthase in inflammation and septic shock. Free Radic. Biol. Med. 2002, 33, 1186–1193. [Google Scholar] [CrossRef]
- Escames, G.; Acuña-Castroviejo, D.; López, L.C.; Tan, D.X.; Maldonado, M.D.; Sánchez-Hidalgo, M.; León, J.; Reiter, R.J. Pharmacological utility of melatonin in the treatment of septic shock: experimental and clinical evidence. J. Pharm. Pharmacol. 2006, 58, 1153–1165. [Google Scholar] [CrossRef]
- Alvarez, S.; Evelson, P.A. Nitric oxide and oxygen metabolism in inflammatory conditions: Sepsis and exposition to polluted ambients. Front. Biosci. 2007, 12, 964–974. [Google Scholar] [CrossRef]
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355. [Google Scholar] [CrossRef]
- Dahm, C.C.; Moore, K.; Murphy, M.P. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: Implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006, 281, 10056–10065. [Google Scholar] [CrossRef]
- Ducrocq, C.; Blanchard, B.; Pignatelli, B.; Ohshima, H. Peroxynitrite: An endogenous oxidizing and nitrating agent. Cell. Mol. Life Sci. 1999, 55, 1068–1077. [Google Scholar] [CrossRef]
- Blanchard, B.; Pompon, D.; Ducrocq, C. Nitrosation of melatonin by nitric oxide and peroxynitrite. J. Pineal Res. 2000, 29, 184–192. [Google Scholar]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef]
- Guenther, A.L.; Schmidt, S.I.; Laatsch, H.; Fotso, S.; Ness, H.; Ressmeyer, A.R.; Poeggeler, B.; Hardeland, R. Reactions of the melatonin metabolite AMK (N1-acetyl-5-methoxykynuramine) with reactive nitrogen species: Formation of novel compounds, 3-acetamidomethyl-6-methoxycinnolinone and 3-nitro-AMK. J. Pineal Res. 2005, 39, 251–260. [Google Scholar] [CrossRef]
- Trujillo, M.; Folkes, L.; Bartesaghi, S.; Kalyanaraman, B.; Wardman, P.; Radi, R. Peroxynitrite-derived carbonate and nitrogen dioxide radicals readily react with lipoic and dihydrolipoic acid. Free Radic. Biol. Med. 2005, 39, 279–288. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Celano, L.; Gil, M.; Carballal, S.; Durán, R.; Denicola, A.; Banerjee, R.; Alvarez, B. Inactivation of cystathionine β-synthase with peroxynitrite. Arch. Biochem. Biophys. 2009. [Google Scholar] [CrossRef]
- Klingen, A.R.; Palsdottir, H.; Hunte, C.; Ullmann, G.M. Redox-linked protonation state changes in cytochrome bc1 identified by Poisson-Boltzmann electrostatics calculations. Biochim. Biophys. Acta 2007, 1767, 204–221. [Google Scholar]
- Lesnefsky, E.J.; Hoppel, C.L. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim. Biophys. Acta 2008, 1777, 1020–1027. [Google Scholar]
- Lesnefsky, E.J.; Minkler, P.; Hoppel, C.L. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009, 46, 1008–1015. [Google Scholar] [CrossRef]
- Wenz, T.; Hielscher, R.; Hellwig, P.; Schägger, H.; Richers, S.; Hunte, C. Role of phospholipids in respiratory cytochrome bc1 complex catalysis and supercomplex formation. Biochim. Biophys. Acta 2009, 1787, 609–616. [Google Scholar]
- Petrosillo, G.; Matera, M.; Moro, N.; Ruggiero, F.M.; Paradies, G. Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free Radic. Biol. Med. 2009, 46, 88–94. [Google Scholar] [CrossRef]
- Basova, L.V.; Kurnikov, I.V.; Wang, L.; Ritov, V.B.; Belikova, N.A.; Vlasova, I.I.; Pacheco, A.A.; Winnica, D.E.; Peterson, J.; Bayir, H.; Waldeck, D.H.; Kagan, V.E. Cardiolipin switch in mitochondria: Shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 2007, 46, 3423–3434. [Google Scholar]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Bayir, H.; Tyurin, V.A.; Tyurina, Y.Y.; Viner, R.; Ritov, V.; Amoscato, A.A.; Zhao, Q.; Zhang, X.J.; Janesko-Feldman, K.L.; Alexander, H.; Basova, L.V.; Clark, R.S.; Kochanek, P.M.; Kagan, V.E. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann. Neurol. 2007, 62, 154–169. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, A.; Bayir, H.; Stoyanovsky, D.; Borisenko, G.G.; Tyurina, Y.Y.; Wipf, P.; Atkinson, J.; Greenberger, J.S.; Chapkin, R.S.; Belikova, N.A. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol. Nutr. Food Res. 2009, 53, 104–114. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; Greenberger, J.S.; Pitt, B.; Shvedova, A.A.; Borisenko, G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef]
- Zoccarato, F.; Cavallini, L.; Alexandre, A. Succinate is the controller of O2–/H2O2 release at mitochondrial complex I: negative modulation by malate, positive by cyanide. J. Bioenerg. Biomembr. 2009, 41, 387–393. [Google Scholar] [CrossRef]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; Grishin, N.V.; White, M.; Yang, X.J.; Zhao, Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Schlicker, C.; Gertz, M.; Papatheodorou, P.; Kachholz, B.; Becker, C.F.; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. [Google Scholar] [CrossRef]
- Liu, Y.; He, J.; Ji, S.; Wang, Q.; Pu, H.; Jiang, T.; Meng, L.; Yang, X.; Ji, J. Comparative studies of early liver dysfunction in senescence-accelerated mouse using mitochondrial proteomics approaches. Mol. Cell. Proteomics 2008, 7, 1737–1747. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J.; Miyahara, Y. Acutely administered melatonin restores hepatic mitochondrial physiology in old mice. Int. J. Biochem. Cell Biol. 2003, 35, 367–375. [Google Scholar] [CrossRef]
- Nishikawa, T.; Takahashi, J.A.; Fujibayashi, Y.; Fujisawa, H.; Zhu, B.; Nishimura, Y.; Ohnishi, K.; Higuchi, K.; Hashimoto, N.; Hosokawa, M. An early stage mechanism of the age-associated mitochondrial dysfunction in the brain of SAMP8 mice; an age-associated neurodegeneration animal model. Neurosci. Lett. 1998, 254, 69–72. [Google Scholar] [CrossRef]
- Omata, N.; Murata, T.; Fujibayashi, Y.; Waki, A.; Sadato, N.; Yoshimoto, M.; Wada, Y.; Yonekura, Y. Age-related changes in energy production in fresh senescence-accelerated mouse brain slices as revealed by positron autoradiography. Dement. Geriatr. Cogn. Disord. 2001, 12, 78–84. [Google Scholar] [CrossRef]
- Tomobe, K.; Nomura, Y. Neurochemistry, neuropathology, and heredity in SAMP8: A mouse model of senescence. Neurochem. Res. 2009, 34, 660–669. [Google Scholar] [CrossRef]
- Carretero, M.; Escames, G.; López, L.C.; Venegas, C.; Dayoub, J.C.; García, L.; Acuña-Castroviejo, D. Long-term melatonin administration protects brain mitochondria from aging. J. Pineal Res. 2009, 47, 192–200. [Google Scholar] [CrossRef]
- Boveris, A.; Navarro, A. Brain mitochondrial dysfunction in aging. IUBMB Life 2008, 60, 308–314. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef]
- Naudí, A.; Caro, P.; Jové, M.; Gómez, J.; Boada, J.; Ayala, V.; Portero-Otín, M.; Barja, G.; Pamplona, R. Methionine restriction decreases endogenous oxidative molecular damage and increases mitochondrial biogenesis and uncoupling protein 4 in rat brain. Rejuvenation Res. 2007, 10, 473–484. [Google Scholar] [CrossRef]
- Fontán-Lozano, A.; López-Lluch, G.; Delgado-García, J.M.; Navas, P.; Carrión, A.M. Molecular bases of caloric restriction regulation of neuronal synaptic plasticity. Mol. Neurobiol. 2008, 38, 167–177. [Google Scholar] [CrossRef]
- Guarente, L. Mitochondria — a nexus for aging, calorie restriction, and sirtuins? Cell 2008, 132, 171–176. [Google Scholar]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta 2009. [Google Scholar] [CrossRef]
- Sen, S.; Phillis, J.W. α-Phenyl-tert-butyl-nitrone (PBN) attenuates hydroxyl radical production during ischemia-reperfusion injury of rat brain: an EPR study. Free Radic. Res. Commun. 1993, 19, 255–265. [Google Scholar] [CrossRef]
- Floyd, R.A.; Hensley, K. Nitrone inhibition of age-associated oxidative damage. Ann. N.Y. Acad. Sci. 2000, 899, 222–237. [Google Scholar]
- Floyd, R.A. Serendipitous findings while researching oxygen free radicals. Free Radic. Biol. Med. 2009, 46, 1004–1013. [Google Scholar] [CrossRef]
- Polovyanenko, D.N.; Plyusnin, V.F.; Reznikov, V.A.; Khramtsov, V.V.; Bagryanskaya, E.G. Mechanistic studies of the reactions of nitrone spin trap PBN with glutathiyl radical. J. Phys. Chem. B 2008, 112, 4841–4847. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Howard, B.J.; Yatin, S.; Allen, K.L.; Carney, J.M. Free radical oxidation of brain proteins in accelerated senescence and its modulation by N-tert-butyl-α-phenylnitrone. Proc. Natl. Acad. Sci. 1997, 94, 674–678. [Google Scholar] [CrossRef]
- Floyd, R.A.; Hensley, K.; Forster, M.J.; Kelleher-Andersson, J.A.; Wood, P.L. Nitrones, their value as therapeutics and probes to understand aging. Mech. Ageing Dev. 2002, 30, 1021–1031. [Google Scholar]
- Floyd, R.A.; Hensley, K.; Forster, M.J.; Kelleher-Andersson, J.A.; Wood, P.L. Nitrones as neuroprotectants and antiaging drugs. Ann N.Y. Acad. Sci. 2002, 959, 321–329. [Google Scholar]
- Kelicen, P.; Cantuti-Castelvetri, I.; Pekiner, C.; Paulson, K.E. The spin trapping agent PBN stimulates H2O2-induced Erk and Src kinase activity in human neuroblastoma cells. Neuroreport 2002, 13, 1057–1061. [Google Scholar] [CrossRef]
- Li, P.A.; He, Q.P.; Nakamura, L.; Csiszar, K. Free radical spin trap α-phenyl-N-tert-butyl-nitron inhibits caspase-3 activation and reduces brain damage following a severe forebrain ischemic injury. Free Radic. Biol. Med. 2001, 31, 1191–1197. [Google Scholar] [CrossRef]
- Kotake, Y.; Sang, H.; Miyajima, T.; Wallis, G.L. Inhibition of NF-κB, iNOS mRNA, COX2 mRNA, and COX catalytic activity by phenyl-N-tert-butylnitrone (PBN). Biochim. Biophys. Acta 1998, 1448, 77–84. [Google Scholar]
- Floyd, R.A.; Hensley, K.; Jaffery, F.; Maidt, L.; Robinson, K.; Pye, Q.; Stewart, C. Increased oxidative stress brought on by pro-inflammatory cytokines in neurodegenerative processes and the protective role of nitrone-based free radical traps. Life Sci. 1999, 65, 1893–1899. [Google Scholar] [CrossRef]
- Sang, H.; Wallis, G.L.; Stewart, C.A.; Kotake, Y. Expression of cytokines and activation of transcription factors in lipopolysaccharide-administered rats and their inhibition by phenyl N-tert-butylnitrone (PBN). Arch. Biochem. Biophys. 1999, 363, 341–348. [Google Scholar] [CrossRef]
- Pieper, G.M.; Nilakantan, V.; Zhou, X.; Khanna, A.K.; Johnson, C.P.; Roza, A.M.; Adams, M.B.; Hilton, G.; Felix, C.C. Treatment with α-phenyl-N-tert-butylnitrone, a free radical-trapping agent, abrogates inflammatory cytokine gene expression during alloimmune activation in rat cardiac allografts. J. Pharmacol. Exp. Ther. 2005, 312, 774–779. [Google Scholar]
- Hensley, K.; Pye, Q.N.; Maidt, M.L.; Stewart, C.A.; Robinson, K.A.; Jaffrey, F.; Floyd, R.A. Interaction of α-phenyl-N-tert-butyl nitrone and alternate electron acceptors with complex I indicates a substrate reduction site upstream form the rotenone binding site. J. Neurochem. 1998, 71, 2549–2557. [Google Scholar]
- Hagen, T.M.; Wehr, C.M.; Ames, B.N. Mitochondrial decay in aging. Reversal through supplementation of acetyl-L-carnitine and N-tert-butyl-α-phenyl-nitrone. Ann. N.Y. Acad. Sci. 1998, 854, 214–223. [Google Scholar]
- Floyd, R.A.; Kotake, Y.; Hensley, K.; Nakae, D.; Konishi, Y. Reactive oxygen species in choline deficiency induced carcinogenesis and nitrone inhibition. Mol. Cell. Biochem. 2002, 234–235, 195–203. [Google Scholar]
- Xu, K.; Puchowicz, M.A.; Sun, X.; LaManna, J.C. Mitochondrial dysfunction in aging rat brain following transient global ischemia. Adv. Exp. Med. Biol. 2008, 614, 379–386. [Google Scholar] [CrossRef]
- Atamna, H.; Paler-Martínez, A.; Ames, B.N. N-t-butyl hydroxylamine, a hydrolysis product of α-phenyl-N-t-butyl nitrone, is more potent in delaying senescence in human lung fibroblasts. J. Biol. Chem. 2000, 275, 6741–6748. [Google Scholar]
- Liu, J.; Atamna, H.; Kuratsune, H.; Ames, B.N. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann. N.Y. Acad. Sci. 2002, 959, 133–166. [Google Scholar]
- Saito, K.; Yoshioka, H. Protective effect of spin trap agent, N-tert-butyl-α-phenylnitrone on hyperoxia-induced oxidative stress and its potential as a nitric oxide donor. Free Radic. Res. 2002, 36, 143–149. [Google Scholar] [CrossRef]
- Saito, K.; Yoshioka, H. ESR characterization of a novel spin-trapping agent, 15N-labeled N-tert-butyl-α-phenylnitrone, as a nitric oxide donor. Biosci. Biotechnol. Biochem. 2002, 66, 2189–2193. [Google Scholar] [CrossRef]
- Novakov, CP.; Stoyanovsky, D.A. Comparative metabolism of N-tert-butyl-N-[1-(1-oxy-pyridin-4-yl)-ethyl]- and N-tert-butyl-N-(1-phenyl-ethyl)-nitroxide by the cytochrome P450 monooxygenase system. Chem. Res. Toxicol. 2002, 15, 749–753. [Google Scholar] [CrossRef]
- Spooren, A.A.; Evelo, C.T. In vitro haematotoxic effects of three methylated hydroxylamines. Arch. Toxicol. 1997, 71, 299–305. [Google Scholar] [CrossRef]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron accumulation during cellular senescence. Ann. N.Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar]
- Saito, K.; Ikeda, M.; Yoshioka, H.; Tomita, T. Nitric oxide and effect of a radical scavenger N-tert-butyl-α-phenylnitrone on stroke in a rat model. Pharmacology 2005, 73, 76–80. [Google Scholar] [CrossRef]
- Banerjee, R.; Saravanan, K.S.; Thomas, B.; Sindhu, K.M.; Mohanakumar, K.P. Evidence for hydroxyl radical scavenging action of nitric oxide donors in the protection against 1-methyl-4-phenylpyridinium-induced neurotoxicity in rats. Neurochem. Res. 2008, 33, 985–995. [Google Scholar] [CrossRef]
- Ha, K.S.; Kim, K.M.; Kwon, Y.G.; Bai, S.K.; Nam, W.D.; Yoo, Y.M.; Kim, P.K.; Chung, H.T.; Billiar, T.R.; Kim, Y.M. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB J. 2003, 17, 1036–1047. [Google Scholar] [CrossRef]
- Farinelli, S.E.; Park, D.S.; Greene, L.A. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J. Neurosci. 1996, 16, 2325–2334. [Google Scholar]
- Estévez, A.G.; Spear, N.; Thompson, J.A.; Cornwell, T.L.; Radi, R.; Barbeito, L.; Beckman, J.S. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. J. Neurosci. 1998, 18, 3708–3714. [Google Scholar]
- Robb, S.J.; Connor, J.R. Nitric oxide protects astrocytes from oxidative stress. Ann. N.Y. Acad. Sci. 2002, 962, 93–102. [Google Scholar] [CrossRef]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1α. FASEB J. 2006, 20, 1889–1891. [Google Scholar] [CrossRef]
- Paxinou, E.; Weisse, M.; Chen, Q.; Souza, J.M.; Hertkorn, C.; Selak, M.; Daikhin, E.; Yudkoff, M.; Sowa, G.; Sessa, W.C.; Ischiropoulos, H. Dynamic regulation of metabolism and respiration by endogenously produced nitric oxide protects against oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 11575–11580. [Google Scholar]
- Dubay, A.; Forster, M.J.; Sohal, R.S. Effect of the spin-trapping compound N-tert-butyl-α-phenylnitrone on protein oxidation and life span. Arch. Biochem. Biophys. 1995, 324, 249–254. [Google Scholar] [CrossRef]
- Poeggeler, B.; Durand, G.; Polidori, A.; Pappolla, M.A.; Vega-Naredo, I.; Coto-Montes, A.; Böker, J.; Hardeland, R.; Pucci, B. Mitochondrial medicine: neuroprotection and life extension by the new amphiphilic nitrone LPBNAH acting as a highly potent advanced antioxidant agent. J. Neurochem. 2005, 95, 962–973. [Google Scholar] [CrossRef]
- Durand, G.; Polidori, A.; Salles, J.P.; Pucci, B. Synthesis of a new family of glycolipidic nitrones as potential antioxidant drugs. Bioorg. Med. Chem. Lett. 2003, 13, 859–862. [Google Scholar] [CrossRef]
- Durand, G.; Polidori, A.; Ouari, O.; Tordo, P.; Gerome, V.; Rustin, P.; Pucci, B. Synthesis and preliminary biological evaluation of ionic and nonionic amphiphilic PBN. J. Med. Chem. 2003, 46, 5530–5535. [Google Scholar]
- Ortial, S.; Durand, G.; Poeggeler, B.; Polidori, A.; Pappolla, M.A.; Böker, J.; Hardeland, R.; Pucci, B. Fluorinated amphiphilic amino acid derivatives as antioxidant carriers: A new class of protective agents. J. Med. Chem. 2006, 49, 2812–2820. [Google Scholar] [CrossRef]
- Durand, G.; Poeggeler, B.; Böker, J.; Raynal, S. Fine-tuning the amphiphilicity: A crucial parameter in the design of potent α-phenyl-N-tert-butylnitrone analogues. J. Med. Chem. 2007, 50, 3976–3979. [Google Scholar] [CrossRef]
- Asanuma, T.; Yasui, H.; Inanami, O.; Waki, K.; Takahashi, M.; Iizuka, D.; Uemura, T.; Durand, G.; Polidori, A.; Kon, Y.; Pucci, B.; Kuwabara, M. A new amphiphilic derivative, N-{[4-(lactobionamido)methyl]benzylidene}-1,1-dimethyl-2-(octylsulfanyl)ethylamine N-oxide, has a protective effect against copper-induced fulminant hepatitis in Long-Evans Cinnamon rats at an extremely low concentration compared with its original form α-phenyl-N-(tert-butyl) nitrone. Chem. Biodivers. 2007, 4, 2253–2267. [Google Scholar] [CrossRef]
- Robertson, L.; Hartley, R.C. Synthesis of N-arylpyridinium salts bearing a nitrone spin trap as potential mitochondria-targeted antioxidants. Tetrahedron 2009, 65, 5284–5292. [Google Scholar] [CrossRef]
- El Fangour, S.; Marini, M.; Good, J.; McQuaker, S.J.; Shiels, P.G.; Hartley, R.C. Nitrones for understanding and ameliorating the oxidative stress associated with aging. Age (Dordr.) 2009. [Google Scholar] [CrossRef]
- Durand, G.; Poeggeler, B.; Ortial, S.; Polidori, A.; Böker, J.; Hardeland, R.; Pucci, B. Amphiphilic nitrone amines: A new class of protective agents acting as mitochondrial metabolism modifiers. Free Radic. Biol. Med. 2007, 43, S91–S92. [Google Scholar]
- Mastronardi, C.A.; Yu, W.H.; McCann, S.M. Resting and circadian release of nitric oxide is controlled by leptin in male rats. Proc. Natl. Acad. Sci. USA 2002, 99, 5721–5726. [Google Scholar] [CrossRef]
- Poeggeler, B.; Schulz, C.; Pappolla, M.A.; Bodó, E.; Tiede, S.; Lehnert, H.; Paus, R. Leptin and the skin: A new frontier. Exp. Dermatol. 2009. [Google Scholar] [CrossRef]
- Wang, C.H.; Lee, W.J.; Ghanta, V.K.; Wang, W.T.; Cheng, S.Y.; Hsueh, C.M. Molecules involve in the self-protection of neurons against glucose-oxygen-serum deprivation (GOSD)-induced cell damage. Brain Res. Bull. 2009, 79, 169–176. [Google Scholar] [CrossRef]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef]
- Shioda, S.; Funahashi, H.; Nakajo, S.; Yada, T.; Maruta, O.; Nakai, Y. Immunohistochemical localization of leptin receptor in the rat brain. Neurosci. Lett. 1998, 243, 41–44. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar]
- Mitchell, S.E.; Nogueiras, R.; Morris, A.; Tovar, S.; Grant, C.; Cruickshank, M.; Rayner, D.V.; Dieguez, C.; Williams, L.M. Leptin receptor gene expression and number in the brain are regulated by leptin level and nutritional status. J. Physiol. 2009, 587, 3573–3585. [Google Scholar] [CrossRef]
- Satoh, T.; Yoshino, S.; Katano, A.; Ishizuka, T.; Tomaru, T.; Shibusawa, N.; Hashimoto, K.; Yamada, M.; Mori, M. Isolation of a novel leptin receptor gene promoter preferentially functioning in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 389, 673–677. [Google Scholar] [CrossRef]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.L.; Kohno, D.; Uramura, K.; Sougawa, H.; Yada, T.; Wayner, M.J.; Sasaki, K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef]
- Fulco, M.; Sartorelli, V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 2008, 7, 3669–3679. [Google Scholar] [CrossRef]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial biogenesis and healthy aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef]
- Myers, M.G. Jr. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef]
- Procopio, C.; Andreozzi, F.; Laratta, E.; Cassese, A.; Beguinot, F.; Arturi, F.; Hribal, M.L.; Perticone, F.; Sesti, G. Leptin-stimulated endothelial nitric-oxide synthase via an adenosine 5'-monophosphate-activated protein kinase/Akt signaling pathway is attenuated by interaction with C-reactive protein. Endocrinology 2009, 150, 3584–3593. [Google Scholar] [CrossRef]
- Ho, P.W.L.; Liu, H.F.; Ho, J.W.M.; Zhang, W.Y.; Chu, A.C.Y.; Kwok, K.H.H.; Ge, X.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial uncoupling protein-2 (UCP2) mediates leptin protection against MPP+ toxicity in neuronal cells. Neurotox. Res. 2009. [Google Scholar] [CrossRef]
- Horvath, T.L.; Warden, C.H.; Hajos, M.; Lombardi, A.; Goglia, F.; Diano, S. Brain uncoupling protein 2: Uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J. Neurosci. 1999, 19, 10417–10427. [Google Scholar]
- Andrews, Z.B.; Horvath, T.L. Uncoupling protein-2 regulates lifespan in mice. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E621–E627. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Horvath, T.L. The role of mitochondrial uncoupling proteins in lifespan. Pflügers Arch. 2009. [Google Scholar] [CrossRef]
- Ceddia, R.B.; William, W.N. Jr.; Lima, F.B.; Flandin, P.; Curi, R.; Giacobino, J.P. Leptin stimulates uncoupling protein-2 mRNA expression and Krebs cycle activity and inhibits lipid synthesis in isolated rat white adipocytes. Eur. J. Biochem. 2000, 267, 5952–5958. [Google Scholar] [CrossRef]
- Tajima, D.; Masaki, T.; Hidaka, S.; Kakuma, T.; Sakata, T.; Yoshimatsu, H. Acute central infusion of leptin modulates fatty acid mobilization by affecting lipolysis and mRNA expression for uncoupling proteins. Exp. Biol. Med. (Maywood) 2005, 230, 200–206. [Google Scholar]
- Chu, A.C.; Ho, P.W.L.; Kwok, K.H.H.; Ho, J.W.M.; Chan, K.H.; Liu, H.F.; Kung, M.H.W.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP4 attenuates MPP+- and dopamine-induced oxidative stress, mitochondrial depolarization, and ATP deficiency in neurons and is interlinked with UCP2 expression. Free Radic. Biol. Med. 2009, 46, 810–820. [Google Scholar] [CrossRef]
- Deierborg, T.; Wieloch, T.; Diano, S.; Warden, C.H.; Horvath, T.L.; Mattiasson, G. Overexpression of UCP2 protects thalamic neurons following global ischemia in the mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1186–1195. [Google Scholar] [CrossRef]
- Kalra, S.P. Global life-long health benefits of repression of hypothalamic NPY system by central leptin gene therapy. Curr. Top. Med. Chem. 2007, 7, 1675–1681. [Google Scholar] [CrossRef]
- Hooft van Huijsduijnen, R.; Wälchli, S.; Ibberson, M.; Harrenga, A. Protein tyrosine phosphatases as drug targets: PTP1B and beyond. Expert Opin. Ther. Targets 2002, 6, 637–647. [Google Scholar] [CrossRef]
- McCarty, M.F.; Barroso-Aranda, J.; Contreras, F. The "rejuvenatory" impact of lipoic acid on mitochondrial function in aging rats may reflect induction and activation of PPAR-γ coactivator-1α. Med. Hypotheses 2009, 72, 29–33. [Google Scholar] [CrossRef]
- Chiarelli, F.; Di Marzio, D. Peroxisome proliferator-activated receptor-γ agonists and diabetes: Current evidence and future perspectives. Vasc. Health Risk Manag. 2008, 4, 297–304. [Google Scholar]
- Leahy, J.L. Thiazolidinediones in prediabetes and early type 2 diabetes: What can be learned about that disease's pathogenesis. Curr. Diab. Rep. 2009, 9, 215–220. [Google Scholar] [CrossRef]
- Festuccia, W.T.; Oztezcan, S.; Laplante, M.; Berthiaume, M.; Michel, C.; Dohgu, S.; Denis, R.G.; Brito, M.N.; Brito, N.A.; Miller, D.S.; Banks, W.A.; Bartness, T.J.; Richard, D.; Deshaies, Y. Peroxisome proliferator-activated receptor-γ-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology 2008, 149, 2121–2130. [Google Scholar] [CrossRef]
- Potula, R.; Ramirez, S.H.; Knipe, B.; Leibhart, J.; Schall, K.; Heilman, D.; Morsey, B.; Mercer, A.; Papugani, A.; Dou, H.; Persidsky, Y. Peroxisome proliferator-activated receptor-γ activation suppresses HIV-1 replication in an animal model of encephalitis. AIDS 2008, 22, 1539–1549. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Heilman, D.; Morsey, B.; Potula, R.; Haorah, J.; Persidsky, Y. Activation of peroxisome proliferator-activated receptor γ (PPARγ) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J. Immunol. 2008, 180, 1854–1865. [Google Scholar]
- Hyong, A.; Jadhav, V.; Lee, S.; Tong, W.; Rowe, J.; Zhang, J.H.; Tang, J. Rosiglitazone, a PPARγ agonist, attenuates inflammation after surgical brain injury in rodents. Brain Res. 2008, 1215, 218–224. [Google Scholar] [CrossRef]
- McTigue, D.M.; Tripathi, R.; Wie, P.; Lash, A.T. The PPARγ agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp. Neurol. 2007, 205, 396–406. [Google Scholar] [CrossRef]
- Roberts, T.J.; Chapman, A.C.; Cipolla, M.J. PPAR-γ agonist rosiglitazone reverses increased cerebral venous hydraulic conductivity during hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1347–H1353. [Google Scholar] [CrossRef]
- Shimazu, T.; Inoue, I.; Araki, N.; Asano, Y.; Sawada, M.; Furuya, D.; Nagoya, H.; Greenberg, J.H. A peroxisome proliferator-activated receptor-γ agonist reduces infarct size in transient but not in permanent ischemia. Stroke 2005, 36, 353–359. [Google Scholar] [CrossRef]
- Kovács, T. Therapy of Alzheimer disease [in Hungarian]. Neuropsychopharmacol. Hung. 2009, 11, 27–33. [Google Scholar]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARγ agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef]
- Jiang, Q.; Heneka, M.; Landreth, G.E. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer's disease: Therapeutic implications. CNS Drugs 2008, 22, 1–14. [Google Scholar]
- Escribano, L.; Simón, A.M.; Pérez-Mediavilla, A.; Salazar-Colocho, P.; Del Río, J.; Frechilla, D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochem. Biophys. Res. Commun. 2009, 379, 406–410. [Google Scholar] [CrossRef]
- Mogi, M.; Li, J.M.; Tsukuda, K.; Iwanami, J.; Min, L.J.; Sakata, A.; Fujita, T.; Iwai, M.; Horiuchi, M. Telmisartan prevented cognitive decline partly due to PPAR-γ activation. Biochem. Biophys. Res. Commun. 2008, 375, 446–449. [Google Scholar] [CrossRef]
- Tsukuda, K.; Mogi, M.; Iwanami, J.; Min, L.J.; Sakata, A.; Jing, F.; Iwai, M.; Horiuchi, M. Cognitive deficit in amyloid-β-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-γ activation. Hypertension 2009, 54, 782–787. [Google Scholar] [CrossRef]
- Stolar, M.W.; Hoogwerf, B.J.; Gorshow, S.M.; Boyle, P.J.; Wales, D.O. Managing type 2 diabetes: Going beyond glycemic control. J. Manag. Care Pharm. 2008, 14 (Suppl. B), s2–s19. [Google Scholar]
- Madsen, K.G.; Grönberg, G.; Skonberg, C.; Jurva, U.; Hansen, S.H.; Olsen, J. Electrochemical oxidation of troglitazone: Identification and characterization of the major reactive metabolite in liver microsomes. Chem. Res. Toxicol. 2008, 21, 2035–2041. [Google Scholar] [CrossRef]
- Tahara, K.; Nishikawa, T.; Hattori, Y.; Iijima, S.; Kouno, Y.; Abe, Y. Production of a reactive metabolite of troglitazone by electrochemical oxidation performed in nonaqueous medium. J. Pharm. Biomed. Anal. 2009, 50, 1030–1036. [Google Scholar] [CrossRef]
- Sapko, M.T.; Guidetti, P.; Yu, P.; Tagle, D.A.; Pellicciari, R.; Schwarcz, R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington's disease. Exp. Neurol. 2006, 197, 31–40. [Google Scholar] [CrossRef]
- Ceresoli-Borroni, G.; Guidetti, P.; Schwarcz, R. Acute and chronic changes in kynurenate formation following an intrastriatal quinolinate injection in rats. J. Neural Transm. 1999, 106, 229–242. [Google Scholar] [CrossRef]
- Poeggeler, B.; Rassoulpour, A.; Wu, H.Q.; Guidetti, P.; Roberts, R.C.; Schwarcz, R. Dopamine receptor activation reveals a novel, kynurenate-sensitive component of striatal N-methyl-D-aspartate neurotoxicity. Neuroscience 2007, 148, 188–197. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar]
- Parsons, C.G.; Danysz, W.; Quack, G.; Hartmann, S.; Lorenz, B.; Wollenburg, C.; Baran, L.; Przegalinski, E.; Kostowski, W.; Krzascik, P.; Chizh, B.; Headley, P.M. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: Electrophysiological, biochemical and behavioral characterization. J. Pharmacol. Exp. Ther. 1997, 283, 1264–1275. [Google Scholar]
- Carpenedo, R.; Pittaluga, A.; Cozzi, A.; Attucci, S.; Galli, A.; Raiteri, M.; Moroni, F. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur. J. Neurosci. 2001, 13, 2141–2147. [Google Scholar] [CrossRef]
- Rassoulpour, A.; Wu, H.Q.; Ferré, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef]
- Zsizsik, B.K.; Hardeland, R. Formation of kynurenic and xanthurenic acids from kynurenine and 3-hydroxykynurenine in the dinoflagellate Lingulodinium polyedrum: Role of a novel, oxidative pathway. Comp. Biochem. Physiol. 2002, 133C, 383–392. [Google Scholar]
- Politi, V.; D'Alessio, S.; Di Stazio, G.; De Luca, G. Antioxidant properties of indole-3-pyruvic acid. Adv. Exp. Med. Biol. 1996, 398, 291–298. [Google Scholar] [CrossRef]
- Lapin, I.P.; Politi, V. Anxiolytic effect of indole-3-pyruvic acid (IPA) in mice. Pharmacol. Res. 1993, 28, 129–134. [Google Scholar] [CrossRef]
- Lapin, I.P.; Politi, V. Antiethanol effects of indol-3-ylpyruvic acid in mice. Alcohol Alcohol. 1994, 29, 265–268. [Google Scholar]
- Bartolini, B.; Corniello, C.; Sella, A.; Somma, F.; Politi, V. The enol tautomer of indole-3-pyruvic acid as a biological switch in stress responses. Adv. Exp. Med. Biol. 2003, 527, 601–608. [Google Scholar] [CrossRef]
- Politi, V.; De Luca, G.; Gallai, V.; Puca, F.; Comin, M. Clinical experiences with the use of indole-3-pyruvic acid. Adv. Exp. Med. Biol. 1999, 467, 227–232. [Google Scholar] [CrossRef]
- Gardoni, F.; Di Luca, M. New targets for pharmacological intervention in the glutamatergic synapse. Eur. J. Pharmacol. 2006, 545, 2–10. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Wenk, G.L. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003, 9, 275–308. [Google Scholar] [CrossRef]
- Areosa, S.A.; Sheriff, F.; McShane, R. Memantine for dementia. Cochrane Database Syst. Rev. 2005, 3, CD003154. [Google Scholar] [CrossRef]
- Kos, T.; Popik, P.; Pietraszek, M.; Schäfer, D.; Danysz, W.; Dravolina, O.; Blokhina, E.; Galankin, T.; Bespalov, A.Y. Effect of 5-HT3 receptor antagonist MDL 72222 on behaviors induced by ketamine in rats and mice. Eur. Neuropsychopharmacol. 2006, 16, 297–310. [Google Scholar] [CrossRef]
- Chipana, C.; Torres, I.; Camarasa, J.; Pubill, D.; Escubedo, E. Memantine protects against amphetamine derivatives-induced neurotoxic damage in rodents. Neuropharmacology 2008, 54, 1254–1263. [Google Scholar] [CrossRef]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef]
- Yokogoshi, H.; Kobayashi, M.; Mochizuki, M.; Terashima, T. Effect of theanine, γ-glutamylethylamide, on brain monoamines and striatal dopamine release in conscious rats. Neurochem. Res. 1998, 23, 667–673. [Google Scholar] [CrossRef]
- Nathan, P.J.; Lu, K.; Gray, M.; Oliver, C. The neuropharmacology of L-theanine (N-ethyl-L-glutamine): A possible neuroprotective and cognitive enhancing agent. J. Herb. Pharmacother. 2006, 6, 21–30. [Google Scholar]
- Yokogoshi, H.; Mochizuki, M.; Saitoh, K. Theanine-induced reduction of brain serotonin concentration in rats. Biosci. Biotechnol. Biochem. 1998, 62, 816–817. [Google Scholar] [CrossRef]
- Kakuda, T. Neuroprotective effects of the green tea components theanine and catechins. Biol. Pharm. Bull. 2002, 25, 1513–1518. [Google Scholar] [CrossRef]
- Egashira, N.; Hayakawa, K.; Mishima, K.; Kimura, H.; Iwasaki, K.; Fujiwara, M. Neuroprotective effect of γ-glutamylethylamide (theanine) on cerebral infarction in mice. Neurosci. Lett. 2004, 363, 58–61. [Google Scholar] [CrossRef]
- Cho, H.S.; Kim, S.; Lee, S.Y.; Park, J.A.; Kim, S.J.; Chun, H.S. Protective effect of the green tea component, L-theanine on environmental toxins-induced neuronal cell death. Neurotoxicology 2008, 29, 656–662. [Google Scholar] [CrossRef]
- de Mejia, E.G.; Ramirez-Mares, M.V.; Puangpraphant, S. Bioactive components of tea: Cancer, inflammation and behavior. Brain Behav. Immun. 2009, 23, 721–731. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J. Oxidative and excitatory mechanisms of developmental neurotoxicity: Transcriptional profiles for chlorpyrifos, diazinon, dieldrin, and divalent nickel in PC12 cells. Environ. Health Perspect. 2009, 117, 587–596. [Google Scholar]
- Hardeland, R.; Poeggeler, B. Actions of melatonin, its structural and functional analogs in the central nervous system and the significance of metabolism. Cent. Nerv. Syst. Agents Med. Chem. 2007, 7, 289–303. [Google Scholar] [CrossRef]
- Corton, J.C.; Brown-Borg, H.M. Peroxisome proliferator-activated receptor γ coactivator 1 in caloric restriction and other models of longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1494–1509. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Alaynick, W.A. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion 2008, 8, 329–337. [Google Scholar] [CrossRef]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; Yang, Y.; Chen, Y.; Hirschey, M.D.; Bronson, R.T.; Haigis, M.; Guarente, L.P.; Farese, R.V., Jr.; Weissman, S.; Verdin, E.; Schwer, B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar]
- Dilova, I.; Easlon, E.; Lin, S.J. Calorie restriction and the nutrient sensing signaling pathways. Cell. Mol. Life Sci. 2007, 64, 752–767. [Google Scholar] [CrossRef]
- Fulco, M.; Sartorelli, V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 2008, 7, 3669–3679. [Google Scholar] [CrossRef]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1α and PGC-1β regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef]
- Viña, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009. [Google Scholar] [CrossRef]
- Goldberg, D.M. Does wine work? Clin. Chem. 1995, 41, 14–16. [Google Scholar]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; Moon, R.C.; Pezzuto, J.M. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar]
- Wendler, J.; Hardeland, R. Phenolic food constituents as free-radical scavengers and luminophores: I. Resveratrol. In Studies on Antioxidants and their Metabolites; Hardeland, R., Ed.; Cuvillier: Göttingen, Germany, 2006; pp. 38–47. [Google Scholar]
- Burkhardt, S.; Reiter, R.J.; Tan, D.X.; Hardeland, R.; Cabrera, J.; Karbownik, M. DNA oxidatively damaged by chromium(III) and H2O2 is protected by melatonin, N1-acetyl-N2-formyl-5-methoxykynuramine, resveratrol and uric acid. Int. J. Biochem. Cell Biol. 2001, 33, 775–783. [Google Scholar] [CrossRef]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; Geny, B.; Laakso, M.; Puigserver, P.; Auwerx, J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Yousuf, S.; Atif, F.; Ahmad, M.; Hoda, N.; Ishrat, T.; Khan, B.; Islam, F. Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res. 2009, 1250, 242–253. [Google Scholar]
- Zhang, H.; Schools, G.P.; Lei, T.; Wang, W.; Kimelberg, H.K.; Zhou, M. Resveratrol attenuates early pyramidal neuron excitability impairment and death in acute rat hippocampal slices caused by oxygen-glucose deprivation. Exp. Neurol. 2008, 212, 44–52. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Enhanced tolerance against early and late apoptotic oxidative stress in mammalian neurons through nicotinamidase and sirtuin mediated pathways. Curr. Neurovasc. Res. 2008, 5, 159–170. [Google Scholar] [CrossRef]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef]
- Pallàs, M.; Casadesús, G.; Smith, M.A.; Coto-Montes, A.; Pelegri, C.; Vilaplana, J.; Camins, A. Resveratrol and neurodegenerative diseases: Activation of SIRT1 as the potential pathway towards neuroprotection. Curr. Neurovasc. Res. 2009, 6, 70–81. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Raval, A.P.; Lin, H.W.; Dave, K.R.; Defazio, R.A.; Della Morte, D.; Kim, E.J.; Perez-Pinzon, M.A. Resveratrol and ischemic preconditioning in the brain. Curr. Med. Chem. 2008, 15, 1545–1551. [Google Scholar] [CrossRef]
- Smith, J.J.; Kenney, R.D.; Gagne, D.J.; Frushour, B.P.; Ladd, W.; Galonek, H.L.; Israelian, K.; Song, J.; Razvadauskaite, G.; Lynch, A.V.; Carney, D.P.; Johnson, R.J.; Lavu, S.; Iffland, A.; Elliott, P.J.; Lambert, P.D.; Elliston, K.O.; Jirousek, M.R.; Milne, J.C.; Boss, O. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst. Biol. 2009, 3, article no. 31. [Google Scholar]
- Chaudhary, N.; Pfluger, P.T. Metabolic benefits from Sirt1 and Sirt1 activators. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 431–437. [Google Scholar] [CrossRef]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L.; Wang, M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 2009. [Google Scholar] [CrossRef]
- Xie, Z.; Dong, Y.; Zhang, M.; Cui, M.Z.; Cohen, R.A.; Riek, U.; Neumann, D.; Schlattner, U.; Zou, M.H. Activation of protein kinase Cζ by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J. Biol. Chem. 2006, 281, 6366–6375. [Google Scholar]
- Shin, S.M.; Cho, I.J.; Kim, S.G. Resveratrol protects mitochondria against oxidative stress through AMP-activated protein kinase-mediated glycogen synthase kinase-3beta inhibition downstream of poly(ADP-ribose)polymerase-LKB1 pathway. Mol. Pharmacol. 2009, 76, 884–895. [Google Scholar] [CrossRef]
- Albani, D.; Polito, L.; Forloni, G. Sirtuins as novel targets for Alzheimer's disease and other neurodegenerative disorders: Experimental and genetic evidence. J. Alzheimers Dis. 2009, ahead . [Google Scholar] [CrossRef]
- Pirola, L.; Fröjdö, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef]
- Allard, S.J.; Perez, E.; Zou, S.; de Cabo, R. Dietary activators of Sirt1. Mol. Cell. Endocrinol. 2009, 299, 58–63. [Google Scholar] [CrossRef]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; De Fabiani, E.; Altucci, L.; Kazantsev, A. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar]
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D'Mello, S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One 2008, 3, article no. e4090. [Google Scholar]
- Allison, S.J.; Milner, J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 2007, 6, 2669–2677. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef]
- Rathaus, M.; Lerrer, B.; Cohen, H.Y. DeubiKuitylation: A novel DUB enzymatic activity for the DNA repair protein, Ku70. Cell Cycle 2009, 8, 1843–1852. [Google Scholar] [CrossRef]
- Rose, G.; Dato, S.; Altomare, K.; Bellizzi, D.; Garasto, S.; Greco, V.; Passarino, G.; Feraco, E.; Mari, V.; Barbi, C.; BonaFe, M.; Franceschi, C.; Tan, Q.; Boiko, S.; Yashin, A.I.; De Benedictis, G. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 2003, 38, 1065–1070. [Google Scholar] [CrossRef]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; Franceschi, C.; Passarino, G.; De Benedictis, G. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar]
- Hardeland, R. Melatonin: Signaling mechanisms of a pleiotropic agent. BioFactors 2009, 35, 183–192. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin, hormone of darkness and more – occurrence, control mechanisms, actions and bioactive metabolites. Cell. Mol. Life Sci. 2008, 65, 2001–2018. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Hardeland, R.; Lopez-Burillo, S.; Mayo, J.C.; Sainz, R.M.; Reiter, R.J. Melatonin - a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J. Pineal Res. 2003, 34, 75–78. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.M.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin - Nature’s most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [Google Scholar] [CrossRef]
- Hardeland, R.; Pandi-Perumal, S.R. Melatonin, a potent agent in antioxidative defense: Actions as a natural food constituent, gastrointestinal factor, drug and prodrug. Nutr. Metab.(Lond.) 2005, 2, 22. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; Shoji, M.; Abe, K.; Leone, A.; Grundke-Ikbal, I.; Wilson, G.L.; Ghiso, J.; Williams, C.; Refolo, L.M.; Pappolla, M.A.; Chain, D.G.; Neria, E. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer's disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar]
- Tricoire, H.; Locatelli, A.; Chemineau, P.; Malpaux, B. Melatonin enters the cerebrospinal fluid through the pineal recess. Endocrinology 2002, 143, 84–90. [Google Scholar] [CrossRef]
- Messner, M.; Hardeland, R.; Rodenbeck, A.; Huether, G. Tissue retention and subcellular distribution of continuously infused melatonin in rats under near physiological conditions. J. Pineal Res. 1998, 25, 251–259. [Google Scholar] [CrossRef]
- López, A.; García, J.A.; Escames, G.; Venegas, C.; Ortiz, F.; López, L.C.; Acuña-Castroviejo, D. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J. Pineal Res. 2009, 46, 188–198. [Google Scholar] [CrossRef]
- Tan, D.X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: a potent, endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Reiter, R.J. Oxidative damage in the central nervous system: Protection by melatonin. Prog. Neurobiol. 1998, 56, 359–384. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress. A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef]
- Tan, D.X.; Reiter, R.J.; Manchester, L.C.; Yan, M.T.; El-Sawi, M.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Allegra, M.; Hardeland, R. Chemical and physical properties and potential mechanisms: Melatonin as a broad spectrum antioxidant and free radical scavenger. Curr. Top. Med. Chem. 2002, 2, 181–197. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Tamura, H. Melatonin defeats neurally-derived free radicals and reduces the associated neuromorphological and neurobehavioral damage. J. Physiol. Pharmacol. 2007, 58 (Suppl. 6), 5–22. [Google Scholar]
- Rosen, J.; Than, N.N.; Koch, D.; Poeggeler, B.; Laatsch, H.; Hardeland, R. Interactions of melatonin and its metabolites with the ABTS cation radical: extension of the radical scavenger cascade and formation of a novel class of oxidation products, C2-substituted 3-indolinones. J. Pineal Res. 2006, 41, 374–381. [Google Scholar] [CrossRef]
- Ressmeyer, A.R.; Mayo, J.C.; Zelosko, V.; Sáinz, R.M.; Tan, D.X.; Poeggeler, B.; Antolín, I.; Zsizsik, B.K.; Reiter, R.J.; Hardeland, R. Antioxidant properties of the melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK): scavenging of free radicals and prevention of protein destruction. Redox Rep. 2003, 8, 205–213. [Google Scholar] [CrossRef]
- Hardeland, R.; Poeggeler, B. Melatonin beyond its classical functions. Open Physiol. J. 2008, 1, 1–23. [Google Scholar] [CrossRef]
- Golombek, D.A.; Escolar, E.; Cardinali, D.P. Melatonin-induced depression of locomotor activity in hamsters: Time-dependency and inhibition by the central-type benzodiazepine antagonist Ro 15-1788. Physiol. Behav. 1991, 49, 1091–1097. [Google Scholar] [CrossRef]
- Golombek, D.A.; Escolar, E.; Burin, L.J.; De Brito Sánchez, M.G.; Fernández Duque, D.; Cardinali, D.P. Chronopharmacology of melatonin: Inhibition by benzodiazepine antagonism. Chronobiol. Int. 1992, 9, 124–131. [Google Scholar] [CrossRef]
- Golombek, D.A.; Fernández Duque, D.; De Brito Sánchez, M.G.; Burin, L.; Cardinali, D.P. Time-dependent anticonvulsant activity of melatonin in hamsters. Eur. J. Pharmacol. 1992, 210, 253–258. [Google Scholar] [CrossRef]
- Muñoz-Hoyos, A.; Sánchez-Forte, M.; Molina-Carballo, A.; Escames, G.; Martin-Medina, E.; Reiter, R.J.; Molina-Font, J.A.; Acuña-Castroviejo, D. Melatonin's role as an anticonvulsant and neuronal protector: Experimental and clinical evidence. J. Child Neurol. 1998, 13, 501–509. [Google Scholar] [CrossRef]
- Molina-Carballo, A.; Muñoz-Hoyos, A.; Sánchez-Forte, M.; Uberos-Fernández, J.; Moreno-Madrid, F.; Acuña-Castroviejo, D. Melatonin increases following convulsive seizures may be related to its anticonvulsant properties at physiological concentrations. Neuropediatrics 2007, 38, 122–125. [Google Scholar] [CrossRef]
- Fenoglio-Simeone, K.; Mazarati, A.; Sefidvash-Hockley, S.; Shin, D.; Wilke, J.; Milligan, H.; Sankar, R.; Rho, J.M.; Maganti, R. Anticonvulsant effects of the selective melatonin receptor agonist ramelteon. Epilepsy Behav. 2009, 16, 52–57. [Google Scholar] [CrossRef]
- Solmaz, I.; Gürkanlar, D.; Gökçil, Z.; Göksoy, C.; Ozkan, M.; Erdoğan, E. Antiepileptic activity of melatonin in guinea pigs with pentylenetetrazol-induced seizures. Neurol. Res. 2009. [Google Scholar] [CrossRef]
- Pozo, D.; Reiter, J.R.; Calvo, J.R.; Guerrero, J.M. Physiological concentrations of melatonin inhibit nitric oxide synthase in rat cerebellum. Life Sci. 1994, 55, PL455–PL460. [Google Scholar] [CrossRef]
- León, J.; Vives, F.; Crespo, E.; Camacho, E.; Espinosa, A.; Gallo, M.A.; Escames, G.; Acuña-Castroviejo, D. Modification of nitric oxide synthase activity and neuronal response in rat striatum by melatonin and kynurenine derivatives. J. Neuroendocrinol. 1998, 10, 297–302. [Google Scholar]
- Chang, H.M.; Ling, E.A.; Chen, C.F.; Lue, J.H.; Wen, C.Y.; Shieh, J.Y. Melatonin attenuates the neuronal NADPH-d/NOS expression in the nodose ganglion of acute hypoxic rats. J. Pineal Res. 2002, 32, 65–73. [Google Scholar] [CrossRef]
- Chang, H.M.; Tseng, C.Y.; Wei, I.H.; Lue, J.H.; Wen, C.Y.; Shieh, J.Y. Melatonin restores the cytochrome oxidase reactivity in the nodose ganglia of acute hypoxic rats. J. Pineal Res. 2005, 39, 206–214. [Google Scholar] [CrossRef]
- Tjong, Y.W.; Li, M.F.; Hung, M.W.; Fung, M.L. Melatonin ameliorates hippocampal nitric oxide production and large conductance calcium-activated potassium channel activity in chronic intermittent hypoxia. J. Pineal Res. 2008, 44, 234–243. [Google Scholar] [CrossRef]
- Escames, G.; Acuña-Castroviejo, D.; López, L.C.; Tan, D.X.; Maldonado, M.D.; Sánchez-Hidalgo, M.; León, J.; Reiter, R.J. Pharmacological utility of melatonin in the treatment of septic shock: experimental and clinical evidence. J. Pharm. Pharmacol. 2006, 58, 1153–1165. [Google Scholar] [CrossRef]
- Escames, G.; López, L.C.; Ortiz, F.; López, A.; García, J.A.; Ros, E.; Acuña-Castroviejo, D. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. FEBS J. 2007, 274, 2135–2147. [Google Scholar] [CrossRef]
- Hardeland, R.; Backhaus, C.; Fadavi, A.; Hess, M. N1-acetyl-5-methoxykynuramine contrasts with other tryptophan metabolites by a peculiar type of NO scavenging: cyclization to a cinnolinone prevents formation of unstable nitrosamines. J. Pineal Res. 2007, 43, 104–105. [Google Scholar] [CrossRef]
- Hardeland, R.; Backhaus, C.; Fadavi, A. Reactions of the NO redox forms NO+, •NO and HNO (protonated NO–) with the melatonin metabolite N1-acetyl-5-methoxykynuramine. J. Pineal Res. 2007, 43, 382–388. [Google Scholar] [CrossRef]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef]
- León, J.; Escames, G.; Rodríguez, M.I.; López, L.C.; Tapias, V.; Entrena, A.; Camacho, E.; Carrión, M.D.; Gallo, M.A.; Espinosa, A.; Tan, D.X.; Reiter, R.J.; Acuña-Castroviejo, D. Inhibition of neuronal nitric oxide synthase activity by N1-acetyl-5-methoxykynuramine, a brain metabolite of melatonin. J. Neurochem. 2006, 98, 2023–2033. [Google Scholar] [CrossRef]
- Tapias, V.; Escames, G.; López, L.C.; López, A.; Camacho, E.; Carrión, M.D.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Acuña-Castroviejo, D. Melatonin and its brain metabolite N1-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in parkinsonian mice. J. Neurosci. Res. 2009, 87, 3002–3010. [Google Scholar] [CrossRef]
- Srinivasan, V.; Pandi-Perumal, S.R.; Maestroni, G.J.M.; Esquifino, A.I.; Hardeland, R.; Cardinali, D.P. Role of melatonin in neurodegenerative diseases. Neurotox. Res. 2005, 7, 293–318. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Jou, M.J.; Korkmaz, A.; Manchester, L.C.; Paredes, S.D. Biogenic amines in the reduction of oxidative stress: Melatonin and its metabolites. Neuroendocrinol. Lett. 2008, 29, 391–398. [Google Scholar]
- Sharman, E.H.; Bondy, S.C. Effects of age and dietary antioxidants on cerebral electron transport chain activity. Neurobiol. Aging 2001, 22, 629–634. [Google Scholar] [CrossRef]
- Bondy, S.C.; Sharman, E.H. Melatonin and the aging brain. Neurochem. Int. 2007, 50, 571–580. [Google Scholar] [CrossRef]
- Petrosillo, G.; Fattoretti, P.; Matera, M.; Ruggiero, F.M.; Bertoni-Freddari, C.; Paradies, G. Melatonin prevents age-related mitochondrial dysfunction in rat brain via cardiolipin protection. Rejuvenation Res. 2008, 11, 935–943. [Google Scholar] [CrossRef]
- Rodríguez, M.I.; Escames, G.; López, L.C.; López, A.; García, J.A.; Ortiz, F.; Sánchez, V.; Romeu, M.; Acuña-Castroviejo, D. Improved mitochondrial function and increased life span after chronic melatonin treatment in senescent prone mice. Exp. Gerontol. 2008, 43, 749–756. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar]
- Acuña-Castroviejo, D.; Escames, G.; León, J.; Carazo, A.; Khaldy, H. Mitochondrial regulation by melatonin and its metabolites. Adv. Exp. Med. Biol. 2003, 527, 549–557. [Google Scholar] [CrossRef]
- Gutierrez-Cuesta, J.; Tajes, M.; Jiménez, A.; Coto-Montes, A.; Camins, A.; Pallàs, M. Evaluation of potential pro-survival pathways regulated by melatonin in a murine senescence model. J. Pineal Res. 2008, 45, 497–505. [Google Scholar] [CrossRef]
- Chang, H.M.; Wu, U.I.; Lan, C.T. Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J. Pineal Res. 2009, 47, 211–220. [Google Scholar] [CrossRef]
- Tajes, M.; Gutierrez-Cuesta, J.; Ortuño-Sahagun, D.; Camins, A.; Pallàs, M. Anti-aging properties of melatonin in an in vitro murine senescence model: involvement of the sirtuin 1 pathway. J. Pineal Res. 2009, 47, 228–237. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: a connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef]
- Nowak, A.; Rahman, H.; Heer, C.; Schueth, A.; Laatsch, H.; Hardeland, R. Reactions of the melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK) with the tyrosine side-chain fragment, 4-ethylphenol. Redox Rep. 2008, 13, 102–108. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Huether, G.; Schneider, A.; Bach, A.; Sirén, A.L.; Hardeland, R.; Bähr, M.; Nave, K.A.; Ehrenreich, H. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–321. [Google Scholar] [CrossRef]
- Hardeland, R. New approaches in the management of insomnia: weighing the advantages of prolonged release melatonin and synthetic melatoninergic agonists. Neuropsychiatr. Dis. Treat. 2009, 5, 341–354. [Google Scholar] [CrossRef]
- Hardeland, R. Tasimelteon, a melatonin agonist for the treatment of insomnia and circadian rhythm sleep disorders. Curr. Opin. Investig. Drugs 2009, 10, 691–701. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hardeland, R. Neuroprotection by Radical Avoidance: Search for Suitable Agents. Molecules 2009, 14, 5054-5102. https://doi.org/10.3390/molecules14125054
Hardeland R. Neuroprotection by Radical Avoidance: Search for Suitable Agents. Molecules. 2009; 14(12):5054-5102. https://doi.org/10.3390/molecules14125054
Chicago/Turabian StyleHardeland, Rüdiger. 2009. "Neuroprotection by Radical Avoidance: Search for Suitable Agents" Molecules 14, no. 12: 5054-5102. https://doi.org/10.3390/molecules14125054
APA StyleHardeland, R. (2009). Neuroprotection by Radical Avoidance: Search for Suitable Agents. Molecules, 14(12), 5054-5102. https://doi.org/10.3390/molecules14125054