Influence of Different Polymer Types on the Overall Release Mechanism in Hydrophilic Matrix Tablets

Abstract
:1. Introduction
2. Results and Discussion
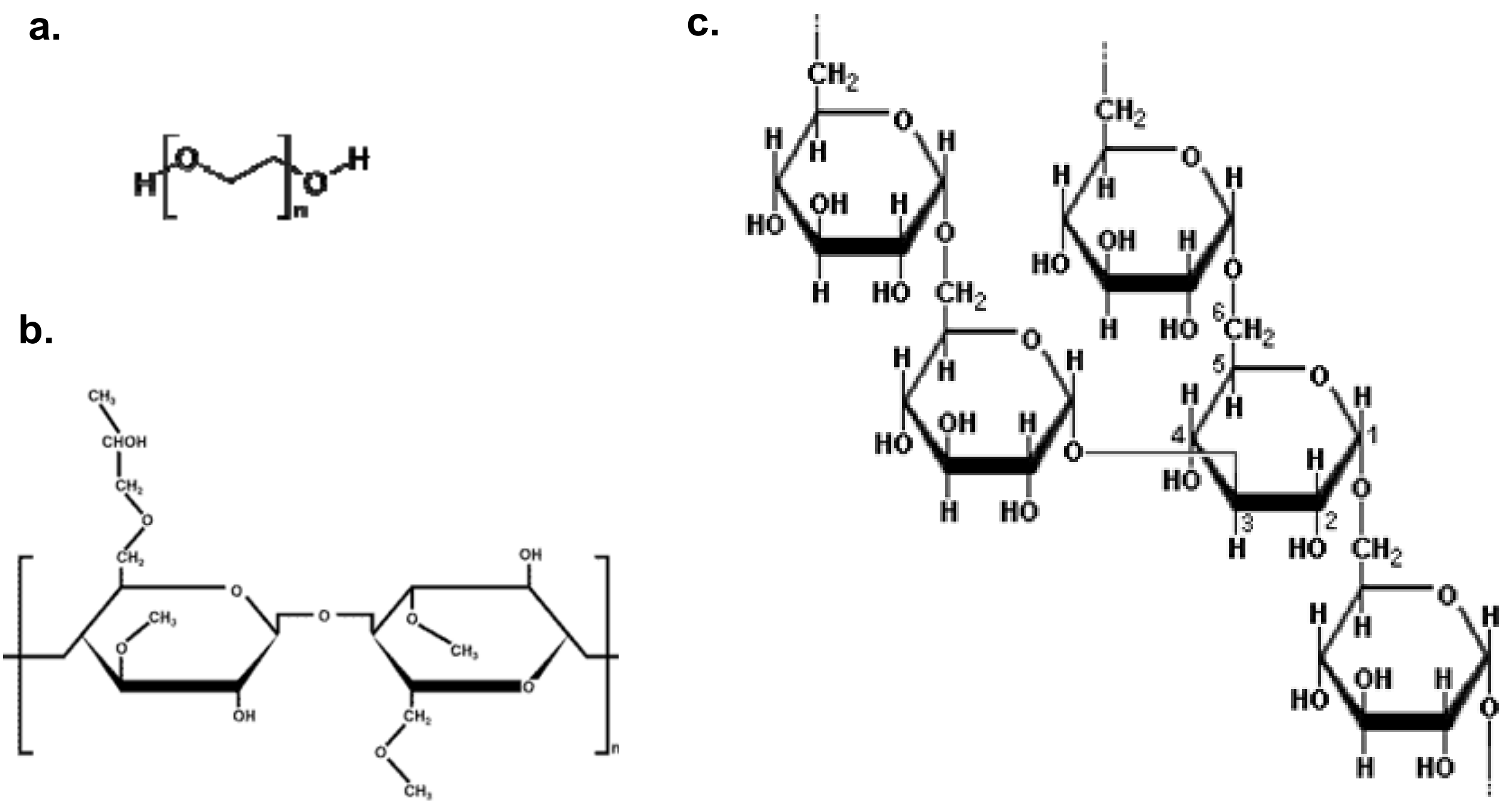
2.1. Characteristics of the polymer samples
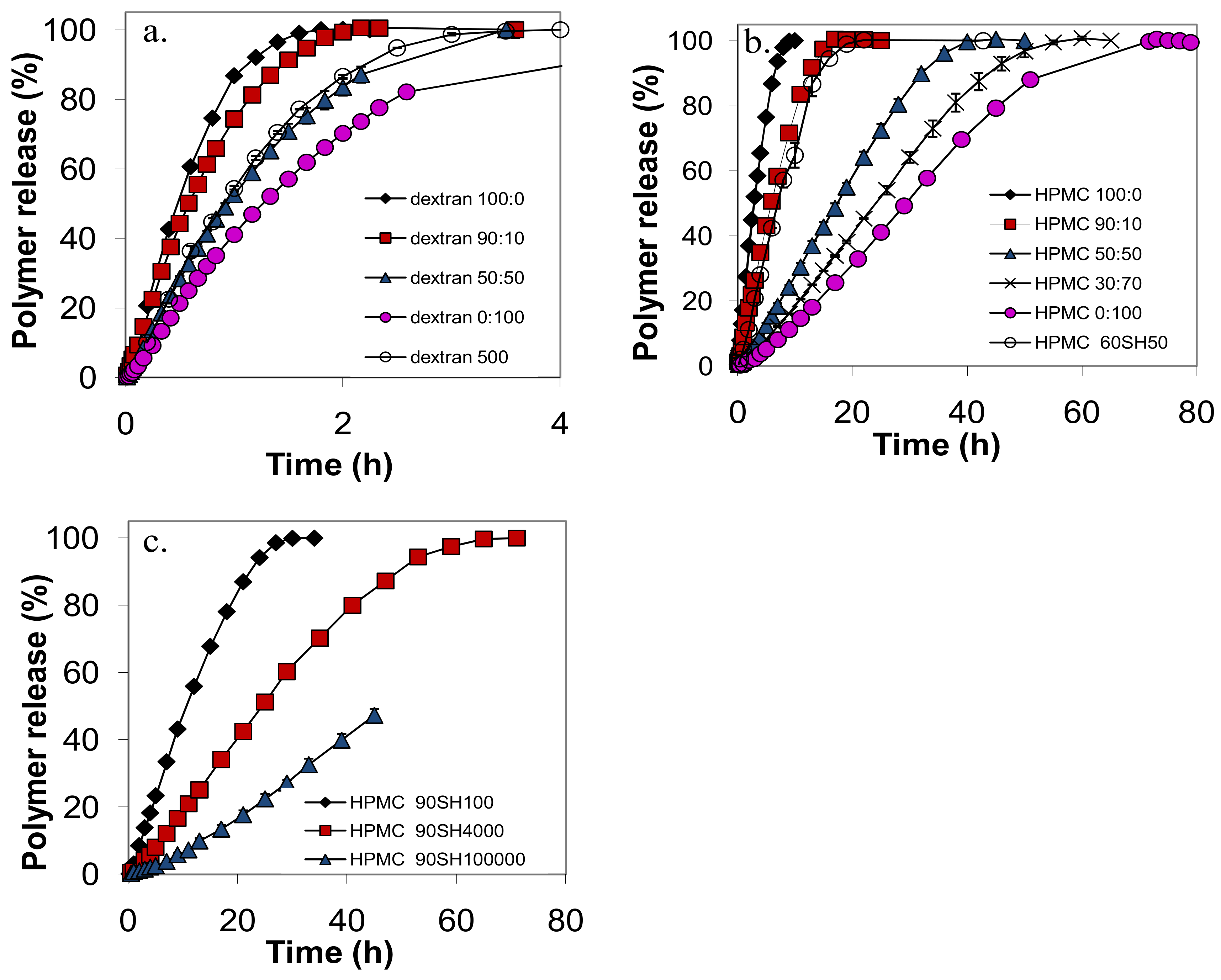
2.2. Release experiments
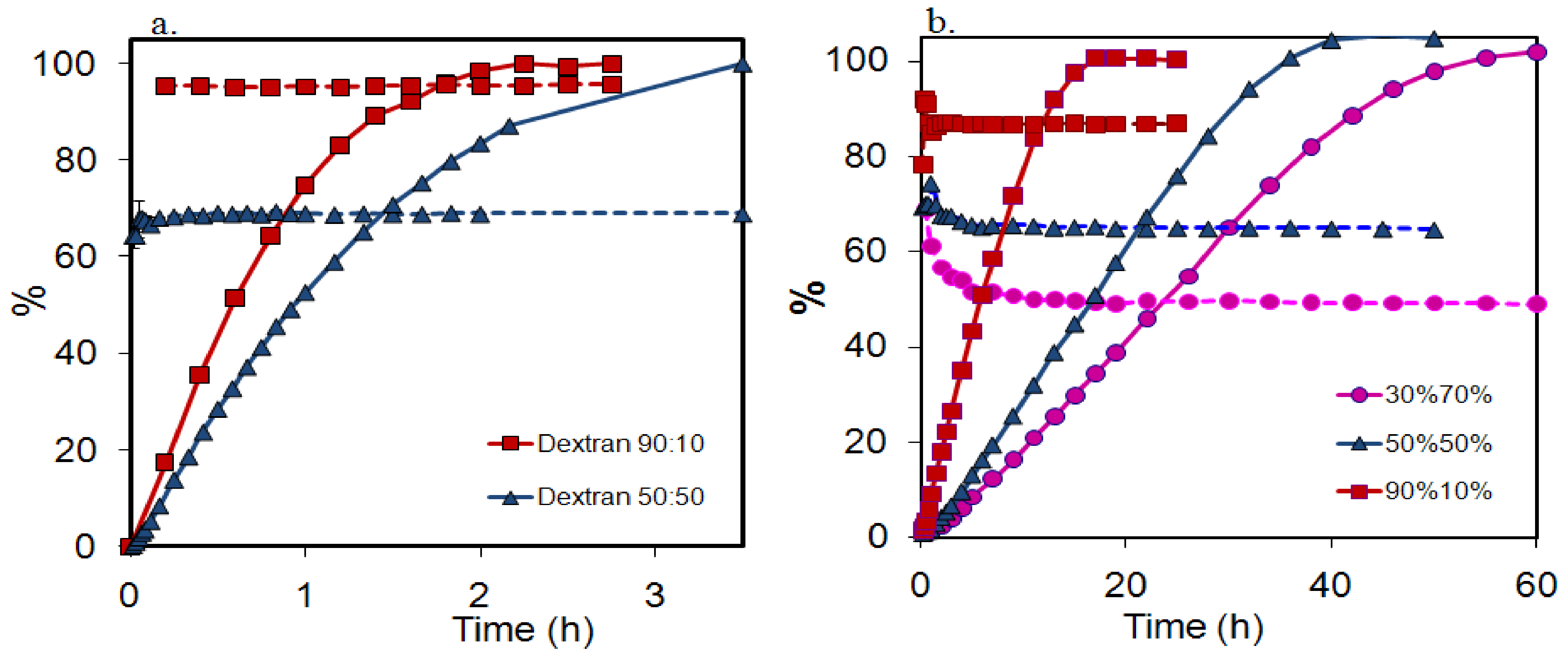
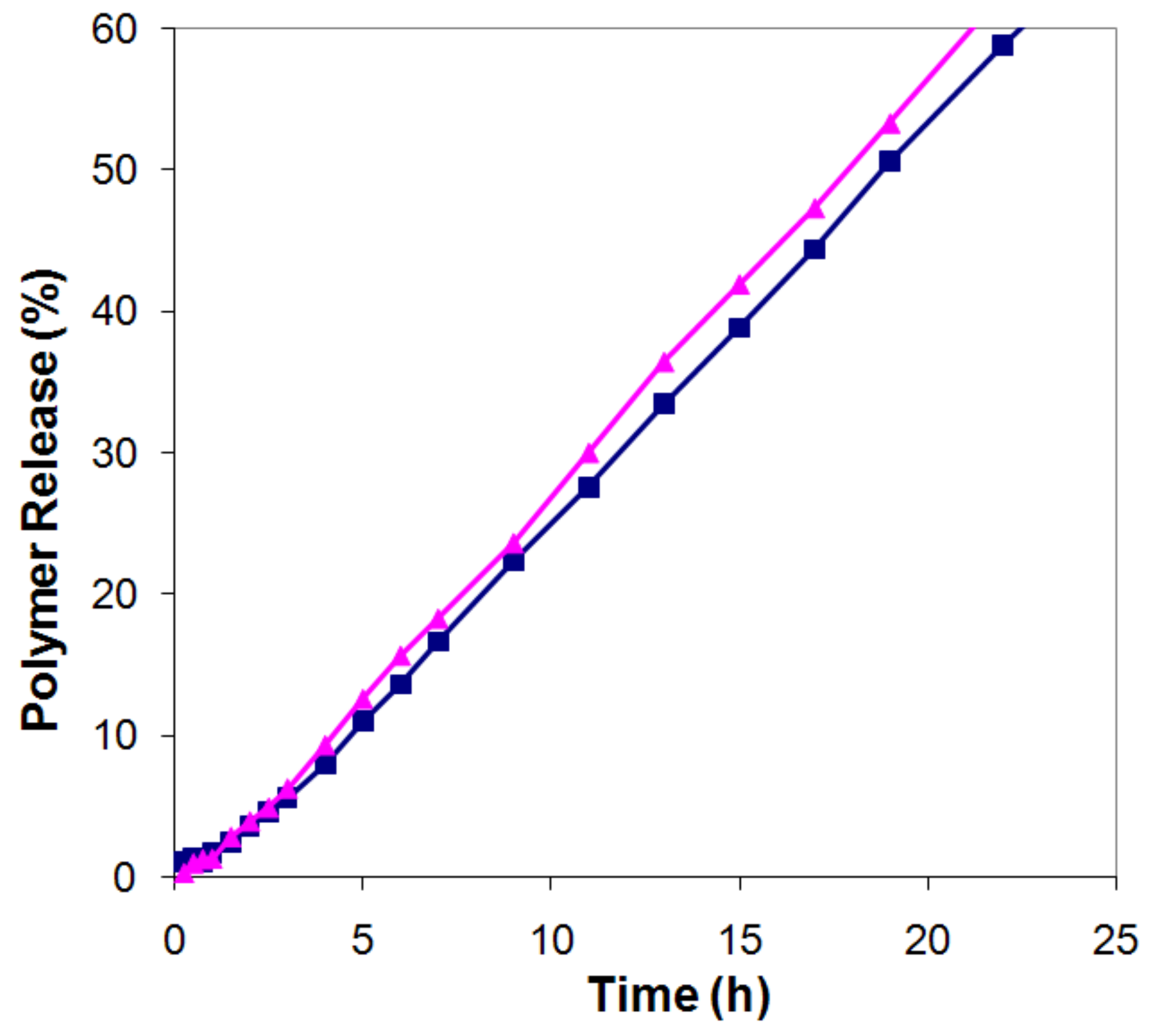
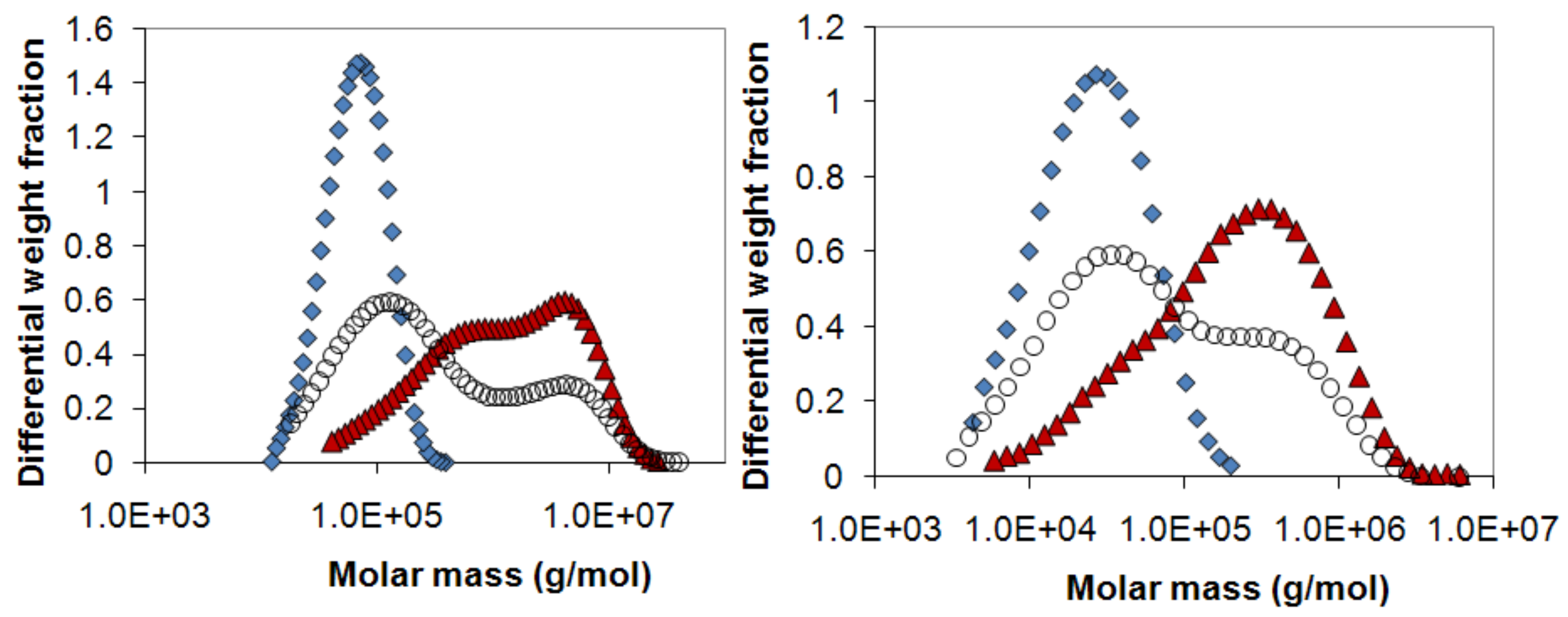
2.3. Absence of fractionation of polymer from polymer mixtures during release
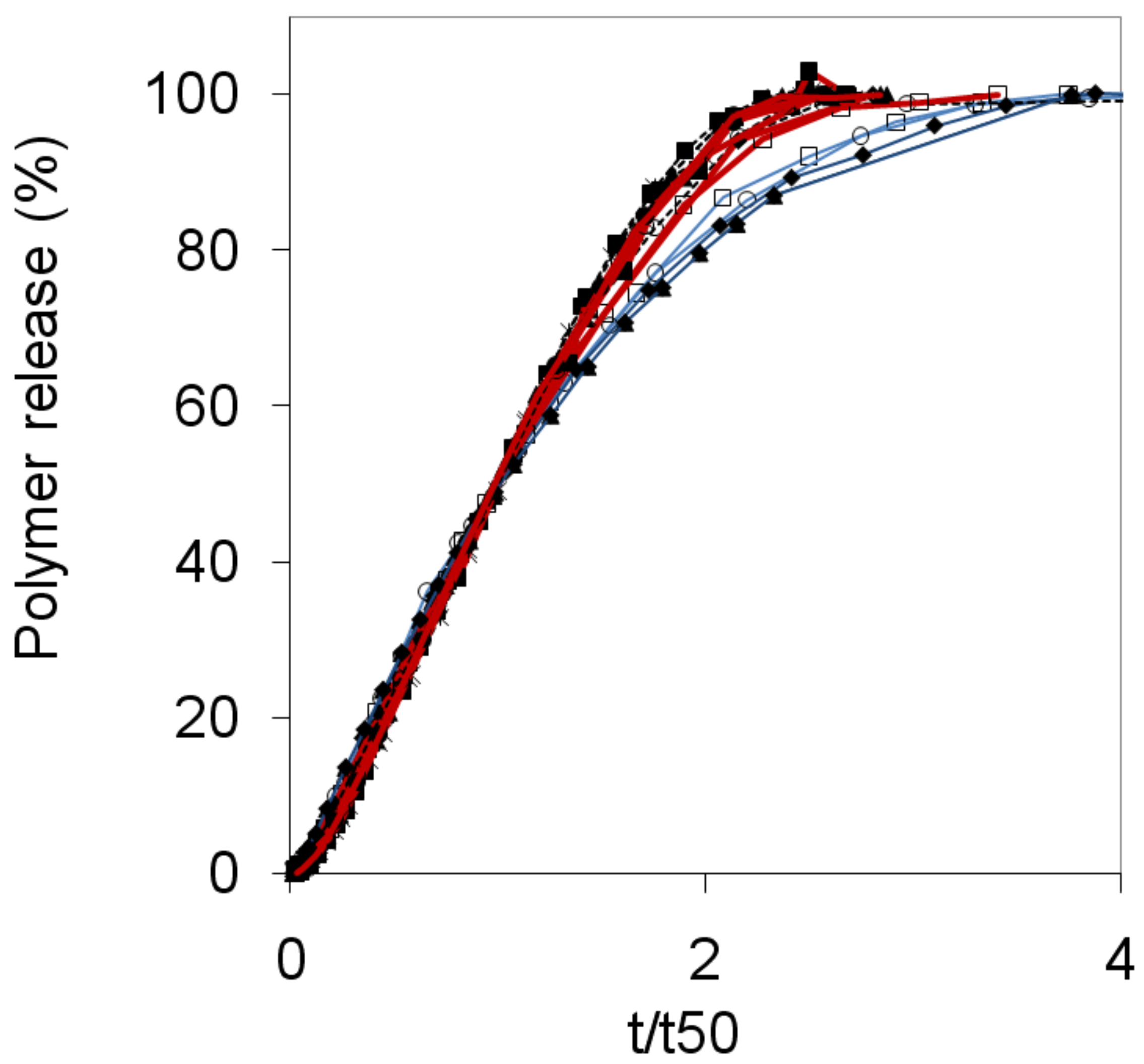
2.4. Release and swelling
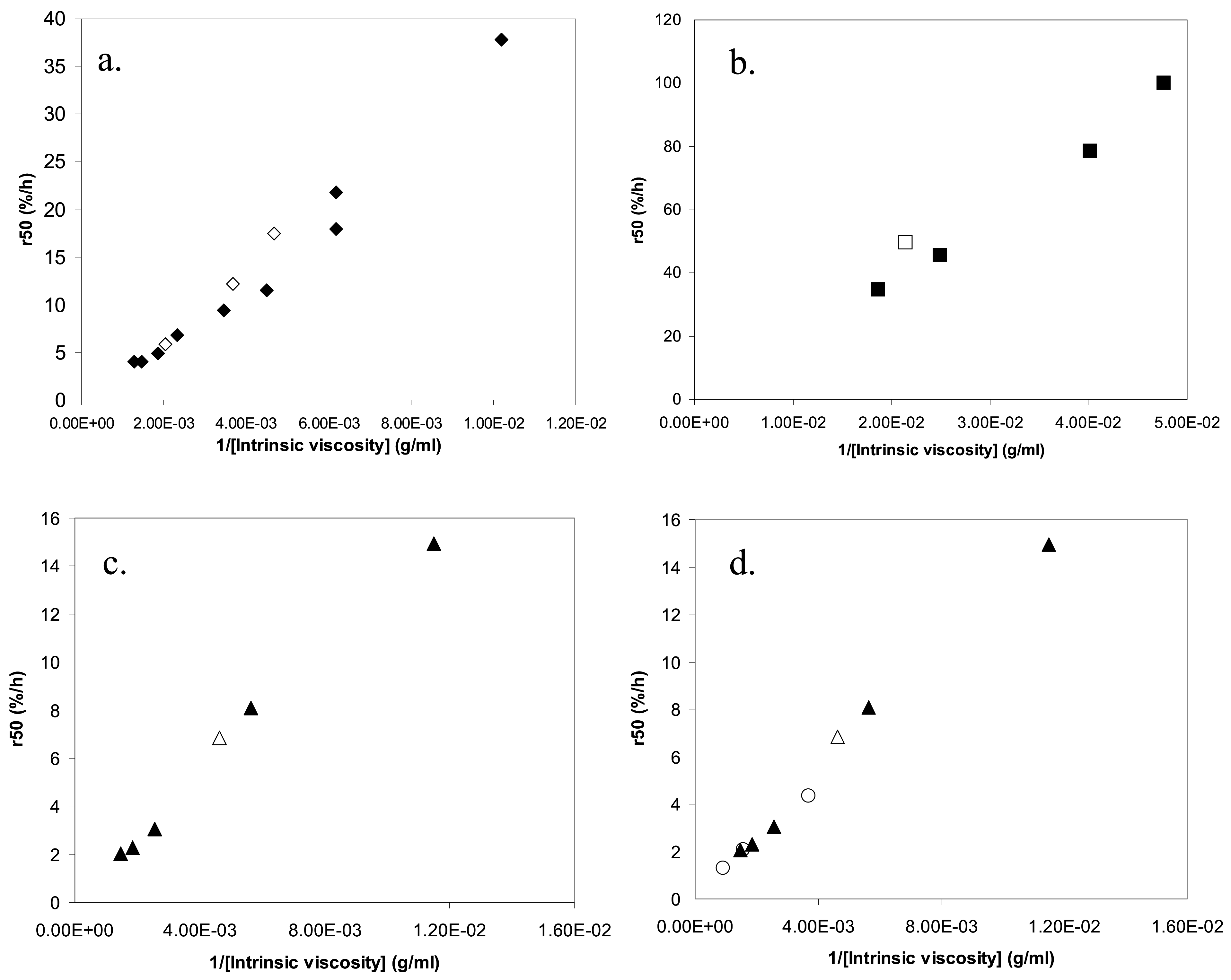
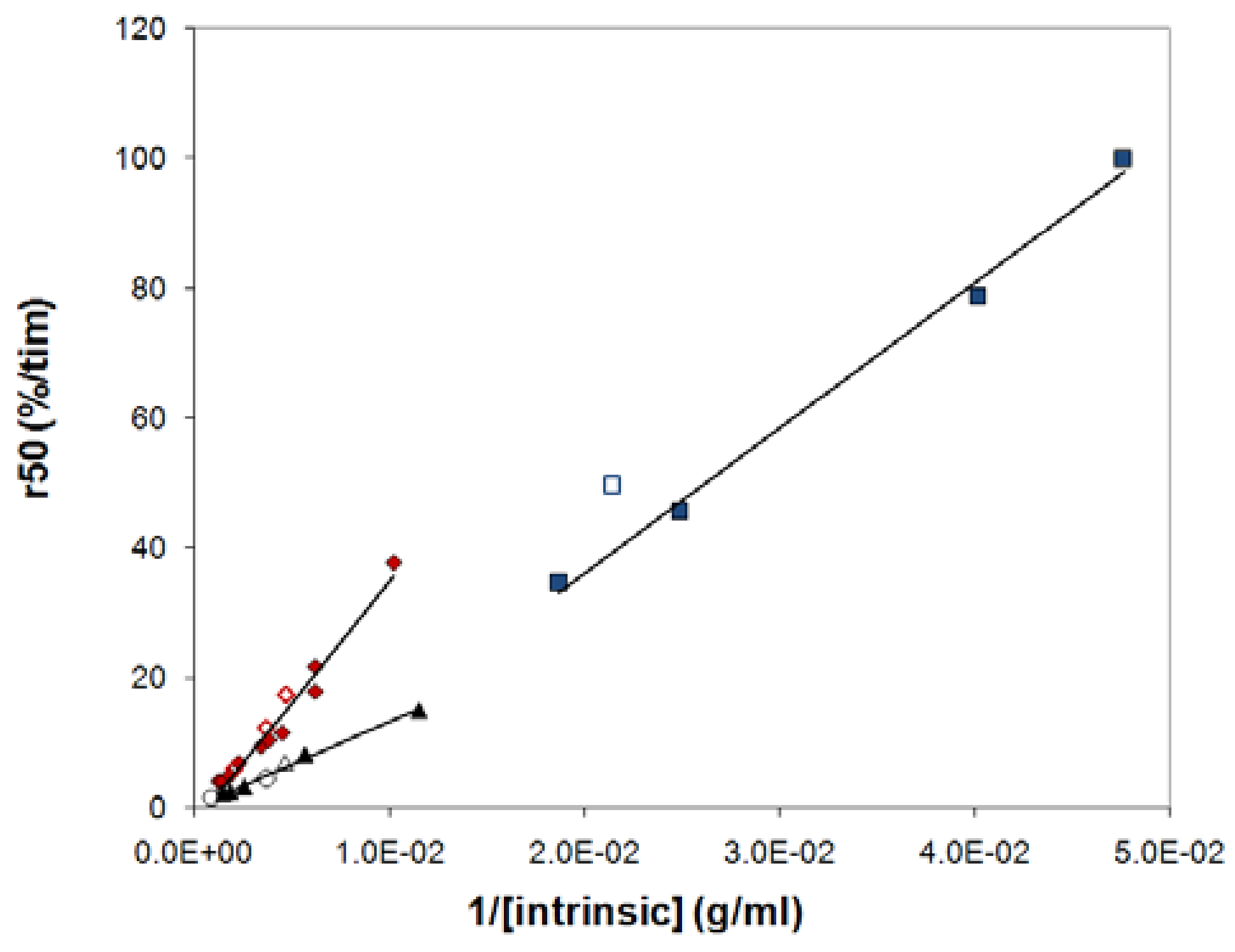
2.5. Relation between release and intrinsic viscosity
2.6. Quantitative aspects
3. Experimental
3.1. Materials
3.2. Tablet preparation
3.3. Moisture content
3.4. Release experiment
3.5. Analysis of release samples and determinations of the average molecular weight
3.6. Viscometry
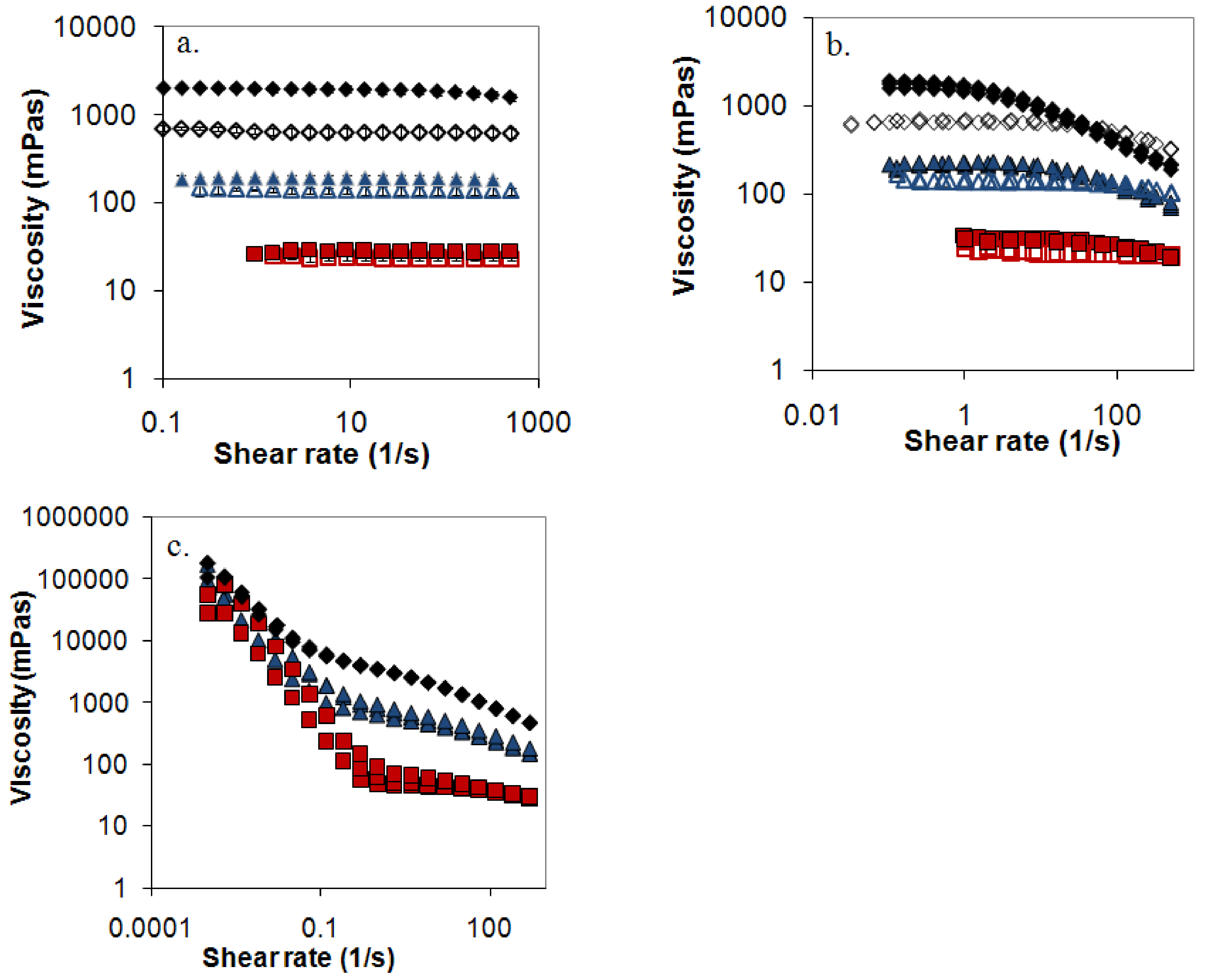
3.7. Rheometry
4. Conclusions
- they all developed a gel upon contact with the dissolution medium.
- the shapes of the release curves were similar for mixed and nonmixed samples, with a slower initial rate, an almost linear release rate during the main release period and a slower release rate at the end of the release experiment.
- the polymers showed virtually no fractionation during release.
- the release rate from a tablet of a given polymer could, to a good approximation, be predicted by the average intrinsic viscosity of the polymer sample.
Acknowledgements
References
- Lapidus, H.; Lordi, N.G. Some factors affecting the release of a water-soluble drug from a compressed hydrophilic matrix. J. Pharm. Sci. 1966, 55, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, H.; Lordi, N.G. Drug release from compressed hydrophilic matrixes. J. Pharm. Sci. 1968, 57, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.E.; Dale, L.B.; Christenson, G.L. Utilization of hydrophilic gums for the control of drug release from tablet formulations I. Disintegration and dissolution behavior. J. Pharm. Sci. 1966, 55, 974–976. [Google Scholar]
- Larsson, A.; Abrahmsén-Alami, S.; Juppo, A.M. Oral Extended Release Formulations. In Pharmaceutical Manufacturing Handbook: Production and Processes, 1st ed.; Gad, S.C., Ed.; Wiley: Hoboken, NJ, USA, 2008; Chapter 6.8. [Google Scholar]
- Fukuda, M.; Peppas, N.A.; McGinity, J. Properties of sustained release hot-melt extruded tablets containing chitosan and xanthan gum. Int. J. Pharm. 2006, 310, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Ju, R.T.C.; Nixon, P.R.; Patel, M.V. Drug Release from Hydrophilic Matrixes 1. New Scaling Laws for Predicting Polymer and Drug Release Based on the Polymer Disentanglement Concentration and the Diffusion Layer. J. Pharm. Sci. 1995, 84, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Körner, A.; Larsson, A.; Piculell, L.; Wittgren, B. Tuning the polymer release from hydrophilic matrix tablets by mixing short and long matrix polymers. J. Pharm. Sci. 2005, 94, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Apicella, A.; Cappello, B.; Del Nobile, M.A.; La Rotonda, M.I.; Mensitieri, G.; Nicolais, L. Poly(Ethylene oxide) (PEO) and different molecular weight PEO blends monolithic devices for drug release. Biomaterials 1993, 14, 83–90. [Google Scholar] [CrossRef]
- Gao, P.; Skoug, J.W.; Nixon, P.R.; Ju, T.R.; Stemm, N.L.; Sung, K.C. Swelling of hydroxypropyl methylcellulose matrix tablets. 2. Mechanistic study of the influence of formulation variables on matrix performance and drug release. J. Pharm. Sci. 1996, 85, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Neau, S.H.; Chow, M.Y.; Durrani, M.J. Fabrication and characterization of extruded and spheronized beads containing Carbopol 974P, NF resin. Int. J. Pharm. 1996, 131, 47–55. [Google Scholar] [CrossRef]
- Miller-Chou, B.A.; Koenig, J.L. A review of polymer dissolution. Prog. Polym. Sci. 2003, 28, 1223–1270. [Google Scholar] [CrossRef]
- Narasimhan, B.; Peppas, N.A. Advances in Polymer Science; Springer: Berlin; New York, 1997; Vol. 128, pp. 158–207. [Google Scholar]
- Ueberreiter, K. Diffusion in Polymers, 1st ed.; Crank, J., Park, C.S., Eds.; Academic Press: London and New York, 1968; pp. 220–257. [Google Scholar]
- Körner, A.; Larsson, A.; Piculell, L.; Wittgren, B. Molecular information on the dissolution of polydisperse polymers: Mixtures of long and short poly(ethylene oxide). J. Phys. Chem. 2005, 109, 11530–11537. [Google Scholar] [CrossRef] [PubMed]
- Borgquist, P.; Körner, A.; Piculell, L.; Larsson, A.; Axelsson, A. A model for the drug release from a polymer matrix tablet—effects of swelling and dissolution. J. Control. Release 2006, 113, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Körner, A.; Andersson, Å.; Larsson, A.; Piculell, L. Swelling and polymer erosion for poly(ethylene oxide) tablets of different molecular weights polydispersities. J. Pharm. Sci. 2009, in press. [Google Scholar] [CrossRef] [PubMed]
- Wittgren, B.; Wahlund, K.-G. Fast molar mass and size characterization of polysaccharides using asymmetrical flow field flow fractionation – multi angle light scattering. J. Chromatogr. A 1997, 760, 205–218. [Google Scholar] [CrossRef]
- Rajabi-Siahboomi, A.R.; Bowtell, R.W.; Mansfield, P.; Henderson, A.; Davies, M.C.; Melia, C.D. Structure and behaviour in hydrophilic matrix sustained release dosage forms: 2. NMR-imaging studies of dimensional changes in the gel layer and core of HPMC tablets undergoing hydration. J. Control. Release 1994, 31, 121–128. [Google Scholar] [CrossRef]
- Clasen, C.; Kulicke, W.-M. Determination of viscoelastic and rheo-optical material functions of water-soluble cellulose derivatives. Prog. Polym. Sci. 2001, 26, 1839–1919. [Google Scholar] [CrossRef]
- de Gennes, P.G. Scaling Concepts in Polymer Physics, 1st ed.; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Morris, E.R.; Cutler, A.N.; Ross-Murphy, S.B.; Rees, D.A. Concentration and shear rate dependence of viscosity in random coil polysaccharide solutions. Carbohydr. Polym. 1981, 1, 5–21. [Google Scholar] [CrossRef]
- Atkins, P.W. Physical Chemistry, 6th ed.; Oxford University Press: Oxford, England, 1998. [Google Scholar]
Sample Availability: Samples of the PEO and HPMC are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tablet Composition | Mn/105 (g/mol) | Mw/105 (g/mol) | PI | [η] (dl/g) | r50 (%/h) | t50 (h) |
|---|---|---|---|---|---|---|
| Dextran | ||||||
| 100:0 | 0.49±0.01 | 0.76±0.01 | 1.56±0.01 | 0.21 | 100.0 | 0.5 |
| 90:10 | 0.38±0.01 | 3.61±0.26 | 9.40±0.74 | 0.25 | 78.6 | 0.6 |
| 50:50 | 1.18±0.01 | 15.2±0.15 | 13.0±0.72 | 0.40 | 45.8 | 0.9 |
| 0:100 | 4.42±0.15 | 26.8±0.35 | 6.06±0.19 | 0.54 | 34.7 | 1.3 |
| T500 | 2.26±0.04 | 6.09±0.06 | 2.64±0.03 | 0.47 | 49.6 | 0.9 |
| HPMC | ||||||
| 60SH100:0 | 0.18±0.01 | 0.34±0.01 | 1.87±0.03 | 0.87 | 14.9 | 2.8 |
| 60SH90:10 | 0.17±0.01 | 0.69±0.01 | 3.04±1.72 | 1.77 | 8.1 | 6.0 |
| 60SH50:50 | 0.29±0.01 | 2.07±0.01 | 7.28±0.13 | 3.91 | 3.1 | 17.4 |
| 60SH30:70 | 0.42±0.01 | 2.85±0.04 | 6.75±0.05 | 5.41 | 2.3 | 24.2 |
| 60SH0:100 | 0.80±0.01 | 3.87±0.12 | 4.84±0.16 | 6.31 | 2.0 | 29.1 |
| 60SH50 | 0.36±0.01 | 0.870±0.00 | 2.38±0.02 | 2.16 | 6.8 | 7.4 |
| 90SH100 | 0.44±0.01 | 1.19±0.003 | 2.69±0.02 | 2.71 | 4.4 | 10.5 |
| 90SH4000 | 0.97±0.004 | 3.01±0.02 | 3.11±0.02 | 6.40 | 2.1 | 24.3 |
| 90SH100000 | 2.05±0.06 | 6.14±0.09 | 3.00±0.13 | 11.0 | 1.3 | 46.8 |
| PEO | ||||||
| PEO 100:0 | 0.25±0.01b | 1.22±0.05a | 4.78b | 1.0b | 37.8a | 1.3a |
| PEO 90:10 | 0.36±0.01b | 3.02±0.08a | 8.39b | 1.6b | 21.8a | 2.3a |
| PEO 88:12 | 0.40±0.01c | 3.15±0.03c | 7.74c | 1.6 | 17.9c | 2.9c |
| PEO 80:20 | 0.50±0.01c | 4.35±0.1c | 8.77c | 2.2 | 11.5c | 4.6c |
| PEO 70:30 | 0.52±0.01b | 6.95±0.06a | 13.4b | 2.9b | 9.4a | 5.8a |
| PEO 50:50 | 0.64±0.02b | 12.2±0.33a | 19.1b | 4.3b | 6.8a | 8.5a |
| PEO 30:70 | 0.87±0.01b | 14.5±0.13a | 16.7b | 5.4b | 4.9a | 11.2a |
| PEO 10:90 | 1.56±0.06b | 19.3±0.37a | 12.4b | 6.8b | 4.0a | 14.1a |
| PEO 0:100 | 2.34±0.01b | 21.9±0.07a | 9.37b | 7.8b | 4.0a | 14.0a |
| PEO 0.3 | 1.01±0.02b | 3.9±0.03b | 3.88b | 2.7b | 11.6b | 4.3b |
| PEO 0.9 | 1.56±0.09b | 9.7±0.11b | 6.28b | 4.9b | 5.9b | 9.4b |
| PEO 0.3M | 2.00±0.03c | 2.34±0.02c | 1.17c | 2.1 | 17.4c | 2.9c |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Körner, A.; Piculell, L.; Iselau, F.; Wittgren, B.; Larsson, A. Influence of Different Polymer Types on the Overall Release Mechanism in Hydrophilic Matrix Tablets. Molecules 2009, 14, 2699-2716. https://doi.org/10.3390/molecules14082699
Körner A, Piculell L, Iselau F, Wittgren B, Larsson A. Influence of Different Polymer Types on the Overall Release Mechanism in Hydrophilic Matrix Tablets. Molecules. 2009; 14(8):2699-2716. https://doi.org/10.3390/molecules14082699
Chicago/Turabian StyleKörner, Anna, Lennart Piculell, Frida Iselau, Bengt Wittgren, and Anette Larsson. 2009. "Influence of Different Polymer Types on the Overall Release Mechanism in Hydrophilic Matrix Tablets" Molecules 14, no. 8: 2699-2716. https://doi.org/10.3390/molecules14082699
APA StyleKörner, A., Piculell, L., Iselau, F., Wittgren, B., & Larsson, A. (2009). Influence of Different Polymer Types on the Overall Release Mechanism in Hydrophilic Matrix Tablets. Molecules, 14(8), 2699-2716. https://doi.org/10.3390/molecules14082699