Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents

Abstract
: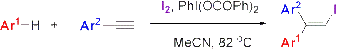
1. Introduction
2. Results and Discussion

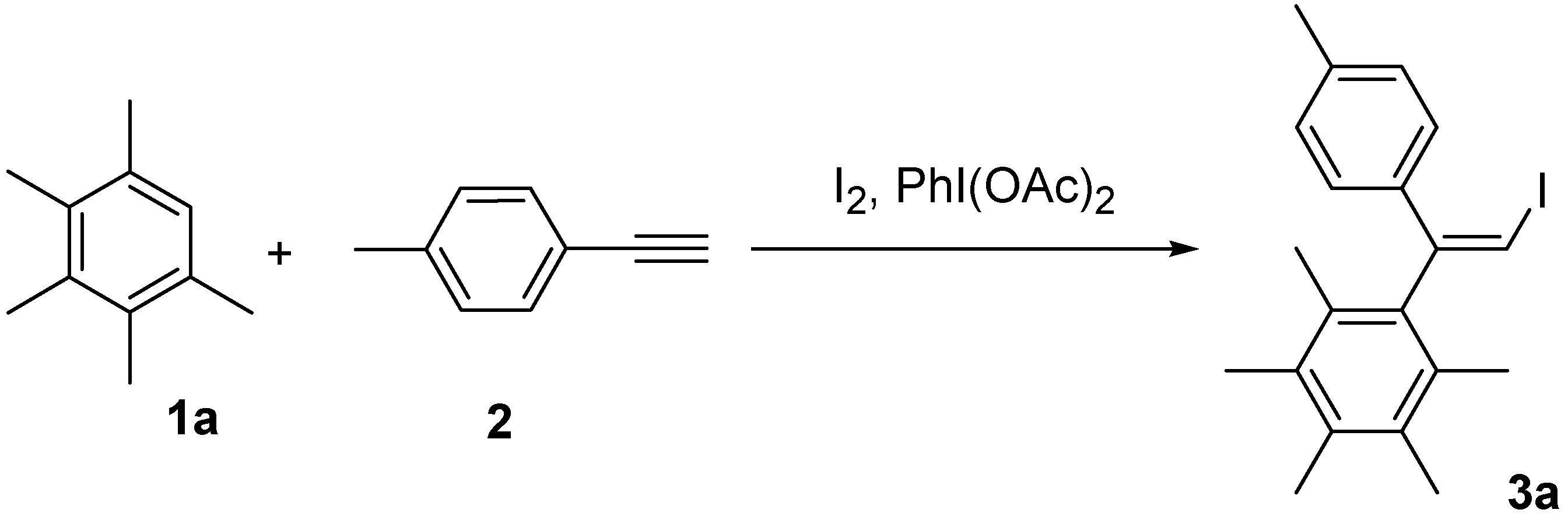
2.1. Optimization of Reaction Conditions

{kind=link}
{kind=link}
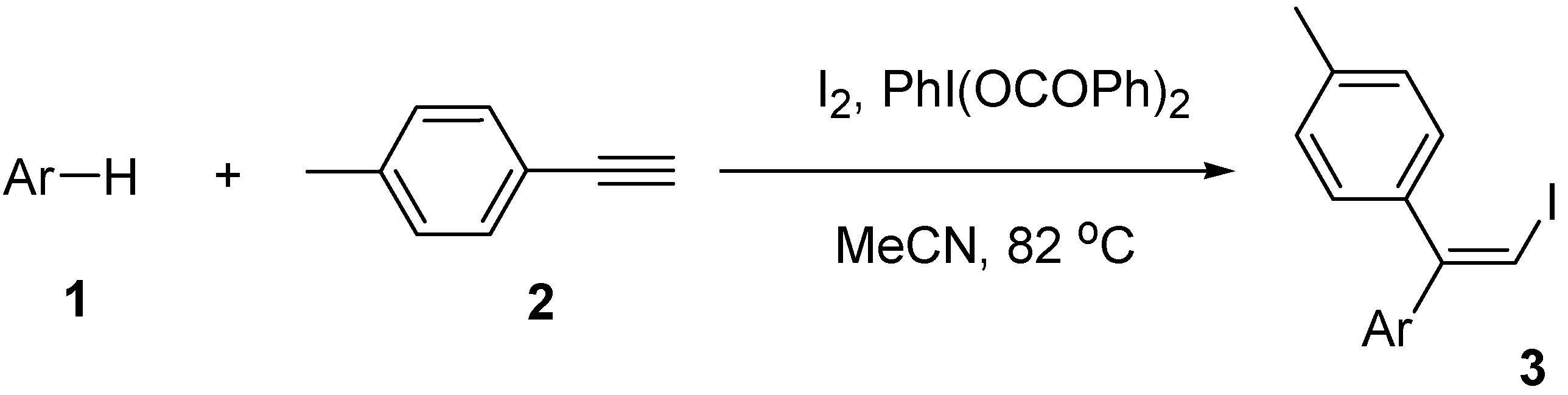{kind=link}
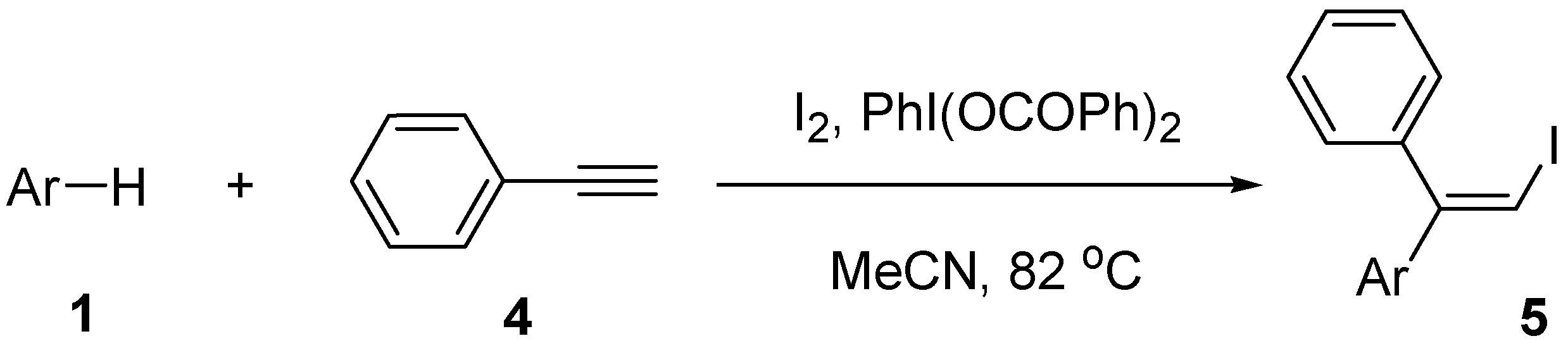{kind=link}
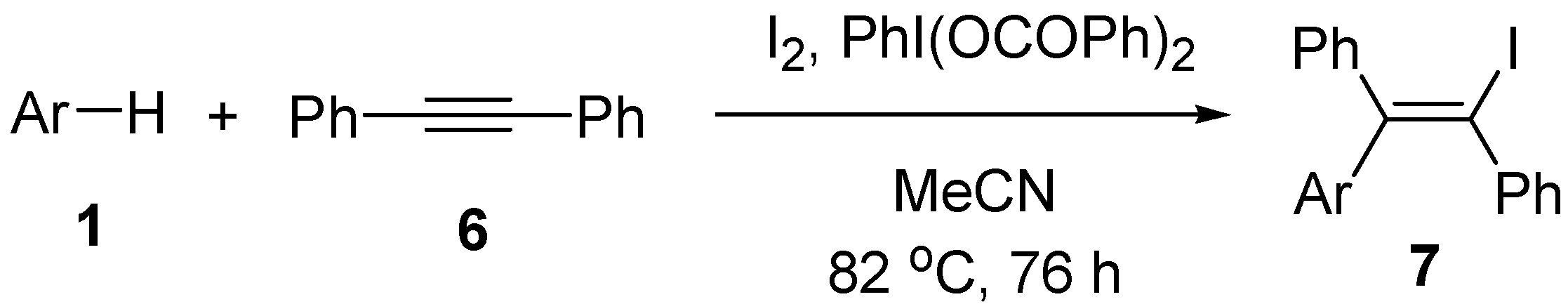{kind=link}
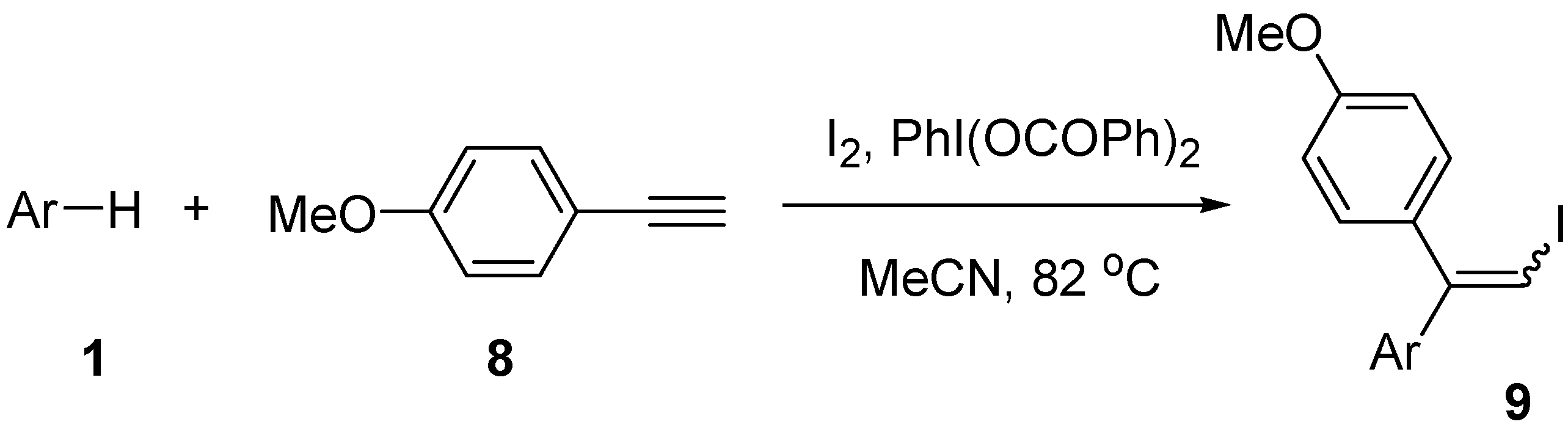{kind=link}
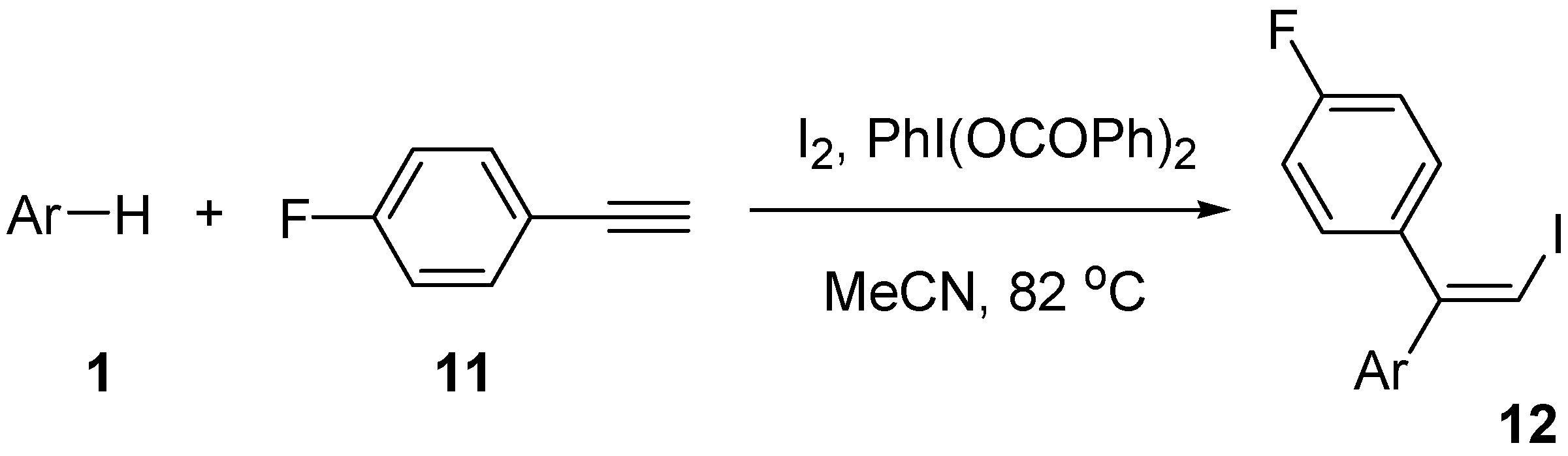{kind=link}
{kind=link}
| Entry | 1a (mmol) | PhI(OAc)2 (mmol) | Solvent (Amt.) | Temp. (oC) | Time (h) | Yield of 3a (%)* |
|---|---|---|---|---|---|---|
| 1 | 1 | 1.25 | DCE (2 mL) | 45 | 28 | 12 |
| 2 | 5 | 3 | AcOH (2 mL) | 60 | 48 | 23 |
| 3 | 1.5 | 3 | MeCN (2 mL) | 60 | 48 | 31 |
| 4 | 5 | 3 | MeCN (2 mL) | 60 | 48 | 52 |
| 5 | 5 | 3 | MeCN (4 mL) | 78 | 56 | 67 |
| 6 | 10 | 3 | MeCN (4 mL) | 82 | 65 | 78 |
| 7 | 10 | 3 | EtOAc (4 mL) | 78 | 65 | 13 |
| Entry | Hypervalent iodine reagent | Yield of 3a (%) |
|---|---|---|
| 1 | PhI(OCOPh)2 | 86 |
| 2 | [Bis( m-chlorobenzoyloxy)iodo]benzene | 78 |
| 3 | [Bis( p-chlorobenzoyloxy)iodo]benzene | 65 |
| 4 | [Bis( p-nitrobenzoyloxy)iodo]benzene | 80 |
| 5 | [Bis( p-methylbenzoyloxy)iodo]benzene | 74 |
| 6 | [Bis( p-methoxybenzoyloxy)iodo]benzene | 70 |
| 7 | PhI(OH)OTs | 33a,b |
| 8 | AgOCOPh | 12c |
2.2. Scope of the iodoarylation reaction using I2 and PhI(OCOPh)2

| Entry | Arene | Time (h) | Product | Isolated yield (%) |
|---|---|---|---|---|
| 1 | Mesitylene (1b) | 65 | 3b | 75 |
| 2 | Durene (1c) | 67 | 3c | 56 |
| 3 | Bromomesitylene (1d) | 72 | 3d | 42a |
| 4 | p-Xylene (1e) | 72 | 3e | 33a |

| Entry | Arene | Time (h) | Product | Isolated yield (%) |
|---|---|---|---|---|
| 1 | 1a | 65 | 5a | 71 |
| 2 | 1b | 70 | 5b | 61 |
| 3 | 1c | 73 | 5c | 59 |
| 4 | 1d | 76 | 5d | 32 |
| 5 | 1e | 72 | 5e | 24 |


| Entry | Arene | Time (h) | Producta | Isolated yield (%) |
|---|---|---|---|---|
| 1 | 1a | 72 | 9a | 75 |
| 2 | 1b | 72 | 9b | 63 |
| 3 | 1c | 73 | 9c | 62 |
| 4 | 1d | 76 | 9d | 32 |
| 5 | 1e | 76 | 9e | 16 |

| Entry | ArH | Time (h) | Product | Isolated yield (%) |
|---|---|---|---|---|
| 1 | 1a | 72 | 12a | 69 |
| 2 | 1b | 72 | 12b | 67 |
| 3 | 1c | 73 | 12c | 56 |
| 4 | 1d | 76 | 12d | 27 |
| 5 | 1e | 76 | 12e | 23 |
2.3. Mechanistic consideration

3. Conclusions
4. Experimental
4.1. General
4.2. General procedure for the iodoarylation of alkynes
- Sample Availability: Samples of the compounds 3a-e, 5a-e, 9a-e and 12a-e are available from the authors.
References and Notes
- Diederich, F.; de Meijere, A. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed; Wiley-VCH: New York, NY, USA, 2004. [Google Scholar]
- Miyaura, N. Cross-Coupling Reactions: A Practical Guide; Springer: Berlin, Germany, 2002. [Google Scholar]
- Diederich, F.; Stang, P.J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Hossain, M.D.; Oyamada, J.; Kitamura, T. Direct Synthesis of Iodoarenes from Aromatic Substrates Using Molecular Iodine. Synthesis 2008, 690–692. [Google Scholar]
- Barluenga, J.; Rodriguez, M.A.; Gonzalez, J.M.; Campos, P.J. Iodo-carbofunctionalization of Alkynes with Aromatic Rings and Ipy2BF4. Tetrahedron Lett. 1990, 31, 4207–4210. [Google Scholar]
- Barluenga, J.; Rodriguez, M.A.; Campos, P.J. Electrophilic Addition of Positive Iodine to Alkynes through an Iodonium Mechanism. J. Org. Chem. 1990, 55, 3104–3105. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kitamura, T. Regio- and Stereoselective Iodoarylation of Arylacetylenes Using Molecular Iodine Promoted by Hypervalent Iodine. Tetrahedron Lett. 2009, 50, 4759–4761. [Google Scholar] [CrossRef]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Togo, H.; Iida, S. Synthetic Use of Molecular Iodine for Organic Synthesis. Synlett 2006, 2159–2175. [Google Scholar] [CrossRef]
- Stavber, S.; Jereb, M.; Zupan, M. Electrophilic Iodination of Organic Compounds Using Elemental Iodine or Iodides. Synlett 2008, 1487–1513. [Google Scholar]
- Finet, J.-P. Ligand Coupling Reactions with Heteroatomic Compounds; Pergamon: Oxford, UK, 1998; pp. 205–247. [Google Scholar]
- Zhdankin, V.V.; Stang, P.J. Alkynyliodonium Salts in Organic Synthesis. Tetrahedron 1998, 54, 10927–10966. [Google Scholar] [CrossRef]
- Wirth, T.; Hirt, U.H. Hypervalent Iodine Compounds: Recent Advances in Synthetic Applications. Synthesis 1999, 1271–1287. [Google Scholar] [CrossRef]
- Ochiai, M. Organic Synthesis Using Hypervalent Organoiodanes. In Chemistry of Hypervalent Compounds; Akiba, K.-Y., Ed.; Wiley-VCH: New York, NY, USA, 1999; pp. 359–388. [Google Scholar]
- Zhdankin, V.V.; Stang, P.J. Recent Developments in the Chemistry of Polyvalent Iodine Compounds. Chem. Rev. 2002, 102, 2523–2584. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin, Germany, 2003. [Google Scholar]
- Tohma, H.; Kita, Y. Hypervalent Iodine Reagents for the Oxidation of Alcohols and Their Application to Complex Molecule Synthesis. Adv. Synth. Catal. 2004, 346, 111–124. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodoarenes and Aryliodonium Salts. In Science of Synthesis; Thieme, Stuttgart: Germany, 2007; Volume 31a, pp. 161–234. [Google Scholar]
- Stang, P.J.; Boehshar, M.; Wingert, H.; Kitamura, T. Acetylenic Esters. Preparation and Characterization of Alkynyl Carboxylates via Polyvalent Iodonium Species. J. Am. Chem. Soc. 1988, 110, 3272–3278. [Google Scholar]
- Pretsh, E.; Clere, T.; Seibl, J.; Simon, W. Tables of Spectral Data for Structure Determination of Organic Compounds, 2nd ed; Springer-Verlag: New York, NY, USA, 1989; p. H215. [Google Scholar]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997; p. 42. [Google Scholar]
- Madsen, J.; Viuf, C.; Bols, M. A New Method for the Deprotection of Benzyl Ethers or the Selective Protection of Alcohols. Chem. Eur. J. 2000, 1140–1146. [Google Scholar]
- Bassi, P.; Tonellato, U. Reactivity of Vinyl Sulfonic Esters. XVI. Solvolytic Reactivity of β-Halovinyl Derivatives. J. Chem. Soc. Perkin Trans. 2 1974, 1283–1288. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rahman, M.A.; Kitamura, T. Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents. Molecules 2009, 14, 3132-3141. https://doi.org/10.3390/molecules14093132
Rahman MA, Kitamura T. Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents. Molecules. 2009; 14(9):3132-3141. https://doi.org/10.3390/molecules14093132
Chicago/Turabian StyleRahman, Md. Ataur, and Tsugio Kitamura. 2009. "Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents" Molecules 14, no. 9: 3132-3141. https://doi.org/10.3390/molecules14093132
APA StyleRahman, M. A., & Kitamura, T. (2009). Iodoarylation of Arylalkynes with Molecular Iodine in the Presence of Hypervalent Iodine Reagents. Molecules, 14(9), 3132-3141. https://doi.org/10.3390/molecules14093132