Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents


Abstract
:Introduction

- (a) The use of the selenide function due to the facile scission of Se from the organic moiety in these types of compounds. In addition, this proposed preliminary hypothesis concerning the action mechanism of these derivatives, related to the possible scission of Se from the organic moiety, allow us to determine the bond order. According to the Molecular Orbital Theory, the bond order (b.o.) is equivalent to the number of electrons in the antibonding molecular orbital minus the number of electrons in the bonding molecular orbital divided by two. This parameter can be taken as a quantitative descriptor for the bond strength and can be related to the aforementioned Se scission.
- (b) The appropriate choice of the substituent on the phenyl ring was made according to a synthetic accessibility and with the aim of assessing the following aspects:
- (b.1)
- Influence of the substituents on the Se charge and bond stability, as well as on the acidic character of the derivatives, expressed as pKa.
- (b.2)
- Modulation of the electronic distribution over the aromatic moiety with a variety of electron-donating and electron-withdrawing substituents, placed at different position with respect to the lateral chain.
- (b.3)
- Molecular volume, conformational behaviour and hydrophobic character (expressed as AlogP), considering the presence of the keto moiety, the aromatic/heteroaromatic ring, the methylene bridge and the carboxylic moiety as the structural basic pattern.
Results and Discussion
Chemistry

Biological Evaluation
Cytotoxic activity in PC-3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Ref. | R | Yield (%) | M. p. (ºC) | Recryst. Solvent | CHN | Anal (%) Calcd/Found |
| 1 | phenyl | 75 | 83a | Toluene | C9H8O3Se | C, 44.44/44.47; H, 3.29/3.24; N, 0.00/0.00. |
| 2 | 4-cyanophenyl | 78 | 147a | Toluene | C10H7NO3Se | C, 44.77/44.85; H, 2.61/2.67; N, 5.22/4.98. |
| 3 | 4-(trifluoromethyl)phenyl | 62 | 108a | Toluene | C10H7F3O3Se | C, 38.58/38.73; H, 2.25/2.15; N, 0.00/0.00. |
| 4 | 4-chlorophenyl | 19 | 135-136b,c | Toluene | C9H7ClO3Se | C, 38.95/38.71; H, 2.54/2.43; N, 0.00/0.00. |
| 5 | 4-methylphenyl | 53 | 92a | Toluene | C10H1003Se | C, 44.69/44.95; H, 3.89/3.89; N, 0.00/0.00. |
| 6 | 4-( tert-butyl)phenyl | 19 | 99-103b,c | Chloroform | C13H16O3Se | C, 52.18/52.04; H, 5.39/5.31; N, 0.00/0.05. |
| 7 | 4-methoxyphenyl | 16 | 104-107b | Chloroform/Carbon tetrachloride | C10H10O4Se | C, 43.97/43.49; H, 3.69/3.52; N, 0.00/0.00. |
| 8 | 2-chlorophenyl | 44 | 123-125b | Carbon tetrachloride | C9H7ClO3Se | C, 38.95/38.97; H, 2.54/2.52; N, 0.00/0.06. |
| 9 | 2-bromophenyl | 32 | 124-128b | Carbon tetrachloride | C9H7BrO3Se | C, 33.57/33.14; H, 2.19/2.02; N, 0.00/0.20. |
| 10 | 2-iodophenyl | 3 | 105-108b | Carbon tetrachloride | C9H7IO3Se | C, 29.29/29.14; H, 1.91/1.92; N, 0.00/0.05. |
| 11 | benzyl | 48 | 74a | Toluene | C9H10O3Se | C, 46.69/46.87; H, 3.89/3.89; N, 0.00/0.00. |
| 12 | 2-phenylethyl | 11 | 65-69b | Hexane | C11H12O3S ¼ H2O | C, 47.93/48.06; H, 4.57/4.47; N, 0.00/0.18. |
| 13 | 3,5-dichlorophenyl | 17 | 108-109b | Carbon tetrachloride | C9H6Cl2O3Se·½ H2O | C, 33.67/33.56; H, 2.20/1.84; N, 0.00/0.06. |
| 14 | 3,5-dimethoxyphenyl | 69 | 117a | Toluene | C11H12O5Se | C, 43.56/43.70; H, 3.96/3.88; N, 0.00/0.00. |
| 15 | 3,4,5-trimethoxyphenyl | 14 | 107-110b | Ethanol | C12H14O6Se | C, 43.26/43.32; H, 4.24/3.97; N, 0.00/0.11. |
| 16 | 3,4-methylendioxyphenyl | 47 | 106-113b | Carbon tetrachloride | C10H8O5Se | C, 41.83/41.56; H, 2.81/2.67; N, 0.00/0.02. |
| 17 | naphthyl | 37 | 130a | Toluene | C13H10O3Se | C, 53.24/52.99; H, 3.41/3.25; N, 0.00/0.00. |
| 18 | diphenylmethyl | 16 | 127-130b | Carbon tetrachloride | C16H14O3Se | C, 56.90/56.88; H, 4.33/4.11; N, 0.00/0.14. |
| 19 | 4-pyridyl | 11 | 119-121b,c | Ether / hexane | C8H7NO3Se | C, 39.36/39.39; H, 2.89/2.78; N, 5.74/5.67. |
| 20 | 3-pyridyl | 15 | 147-150b,c | Methanol | C8H7NO3Se | C, 39.36/39.57; H, 2.89/2.76; N,5.74/5.63. |
| 21 | 3-(2-chloropyridyl) | 157a | Toluene | C8H6ClNO3Se | C, 34.47/34.68; H, 2.15/2.17; N, 5.01/5.26. | |
| 22 | 3-(2-propylthiopyridyl) | 36 | 109-111b | Carbon tetrachloride | C11H13NO3SSe | C, 41.51/41.28; H, 4.12/3.90; N, 4.40/4.24. |
| 23 | 2-thienyl | 33 | 82-84b | Carbon tetrachloride | C7H6O3SSe | C, 33.75/33.45; H, 2.43/2.54; N, 0.00/0.07. |
| 24 | pyrazinyl | 10 | 138-140b | Isopropanol | C7H6N2O3Se | C, 34.30/34.18; H, 2.47/2.49; N, 1.43/1.37. |
| 25 | 2-quinolyl | 6 | 131-132b,c | Toluene | C12H9NO3Se | C, 49.00/49.33; H, 3.08/3.13; N, 4.76/4.75. |
| 26 | 3-quinolyl | 9 | 187-189b,c | Chloroform | C12H9NO3Se | C, 49.00/48.95; H, 3.08/3.23; N, 4.76/4.66. |
 | ||
|---|---|---|
| Compound | R | PC-3 cell line |
| IC50 (μM) | ||
| 1 | phenyl | 6.8 |
| 2 | 4-cyanophenyl | 10.0 |
| 3 | 4-(trifluoromethyl)phenyl | NEa |
| 4 | 4-chlorophenyl | NE |
| 5 | 4-methylphenyl | NE |
| 6 | 4-tert-butylphenyl | NE |
| 7 | 4-methoxyphenyl | NE |
| 8 | 2-chlorophenyl | NE |
| 9 | 2-bromophenyl | NE |
| 10 | 2-iodophenyl | NE |
| 11 | benzyl | 2.9 |
| 12 | 2-phenylethyl | NE |
| 13 | 3,5-dichlorophenyl | NE |
| 14 | 3,5-dimethoxyphenyl | 4.0 |
| 15 | 3,4,5-trimethoxyphenyl | NE |
| 16 | 3,4-methylenedioxyphenyl | NE |
| 17 | naphthyl | NE |
| 18 | diphenylmethyl | NE |
| 19 | 4-pyridyl | NE |
| 20 | 3-pyridyl | NE |
| 21 | 3-(2-chloropyridyl) | 10.0 |
| 22 | 3-(2-propylthiopyridyl) | NE |
| 23 | 2-thienyl | NE |
| 24 | pyrazinyl | NE |
| 25 | 2-quinolyl | NE |
| 26 | 3-quinolyl | NE |
| MSAb | 8.38 [33] | |
| Etoposide | 13.6 ± 2.2 [34] | |
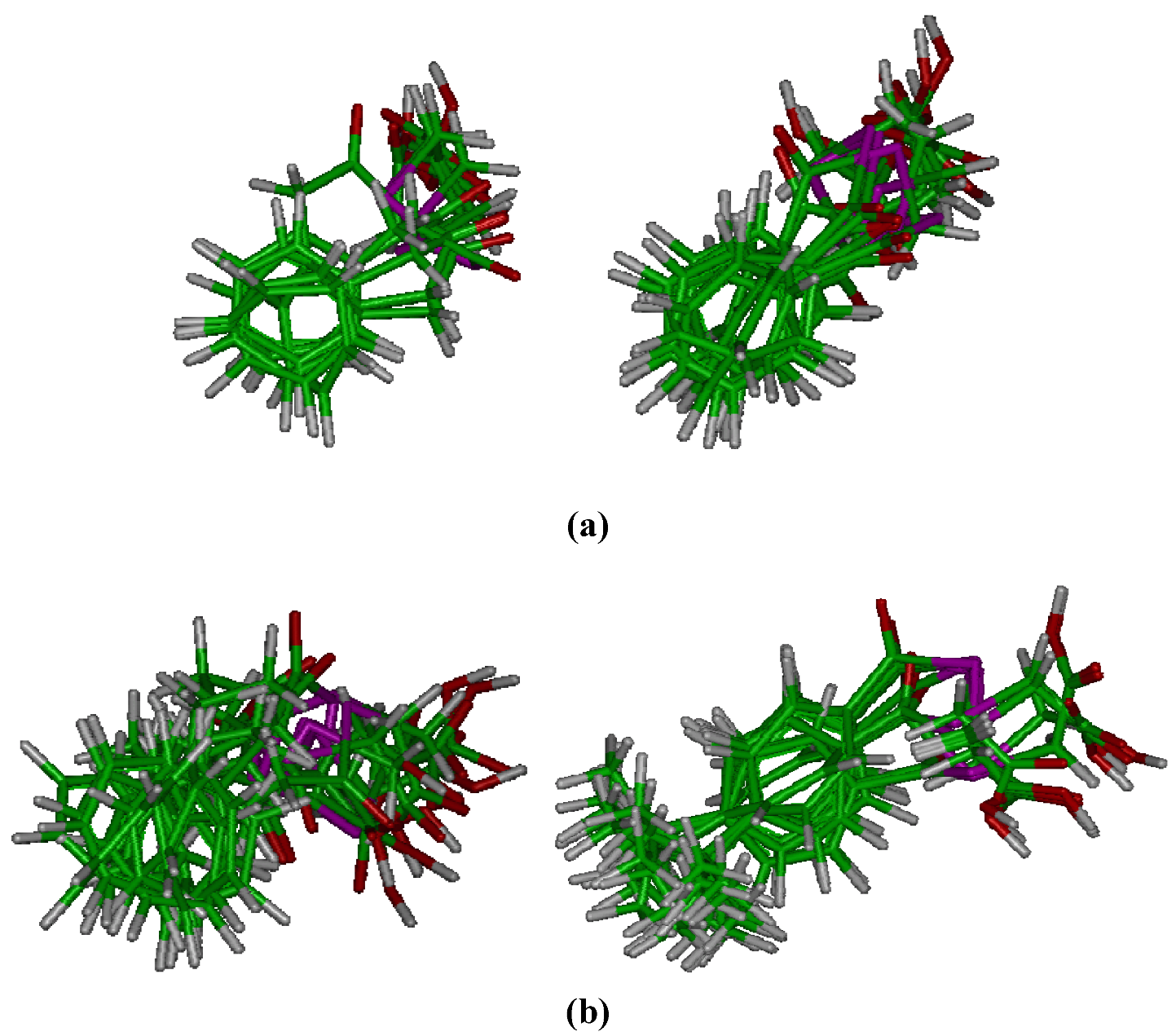
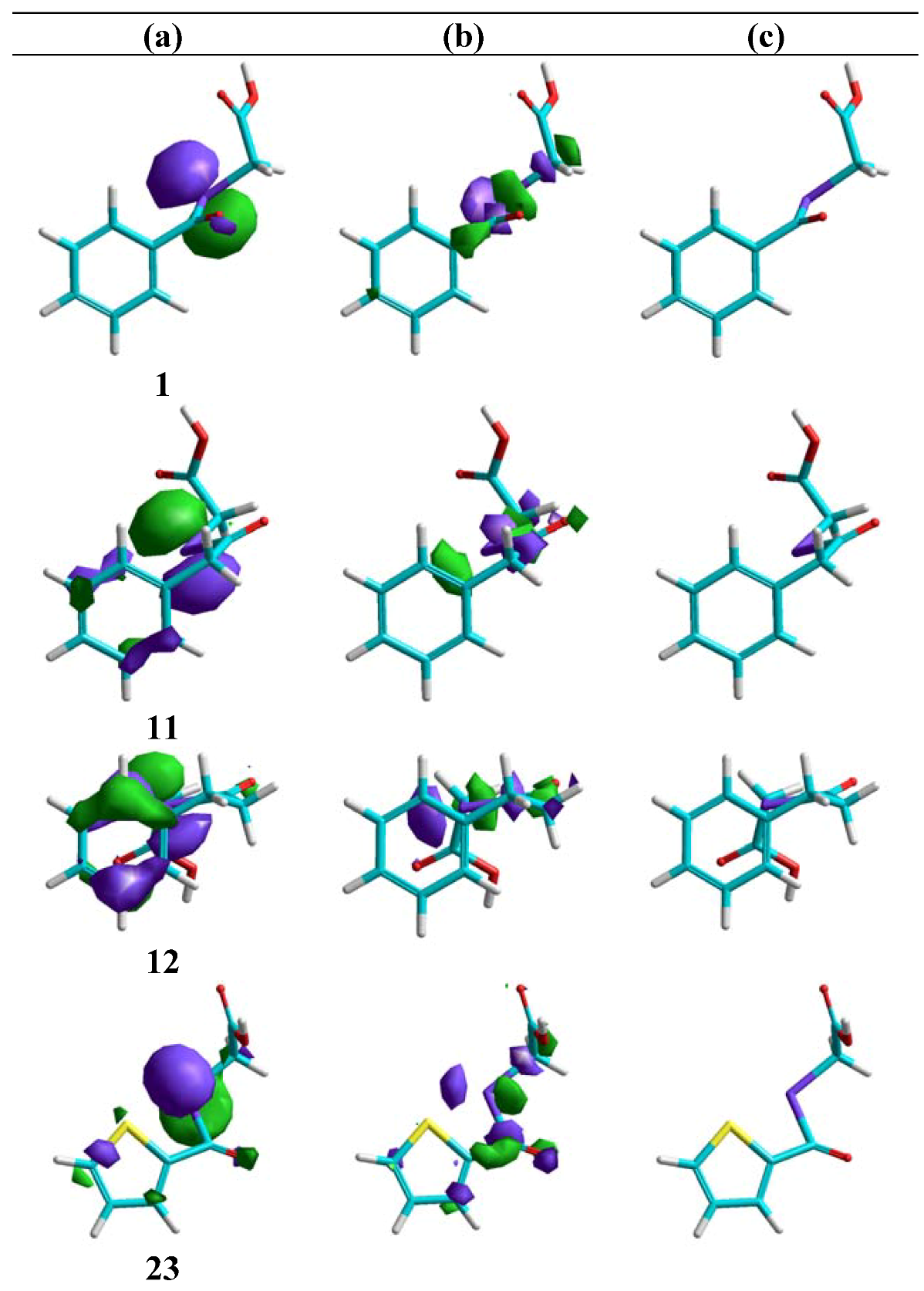
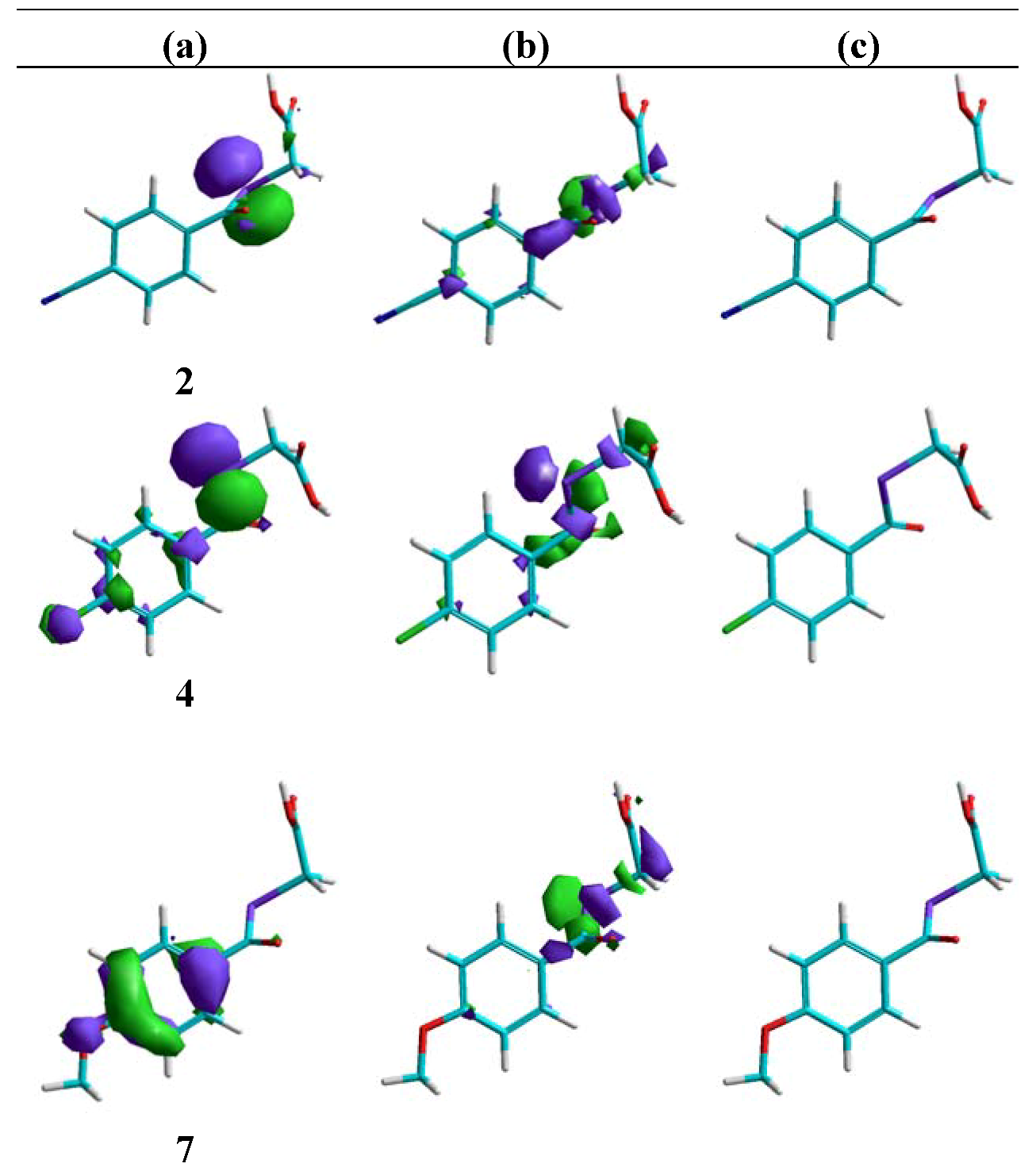
Molecular modelling

 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | X | Y | Z | n | R2 | R3 | R4 | R5 | R6 | AlogP | Volb | μ (D)c | IC50(μM)e |
| 1 | C | C | C | 0 | H | H | H | H | H | 1.795 | 68.58 | 3.465 | 6.8 |
| 2 | C | C | C | 0 | H | H | CN | H | H | 1.673 | 82.16 | 2.865 | 10.0 |
| 3 | C | C | C | 0 | H | H | CF3 | H | H | 2.737 | 92.64 | 1.137 | NE |
| 4 | C | C | C | 0 | H | H | Cl | H | H | 2.459 | 83.19 | 2.628 | NE |
| 5 | C | C | C | 0 | H | H | CH3 | H | H | 2.281 | 82.76 | 4.430 | NE |
| 6 | C | C | C | 0 | H | H | C(CH3)3 | H | H | 3.195 | 125.55 | 4.584 | NE |
| 7 | C | C | C | 0 | H | H | OCH3 | H | H | 1.778 | 89.51 | 5.337 | NE |
| 8 | C | C | C | 0 | Cl | H | H | H | H | 2.459 | 82.97 | 2.874 | NE |
| 9 | C | C | C | 0 | Br | H | H | H | H | 2.543 | 91.02 | 4.032 | NE |
| 10 | C | C | C | 0 | I | H | H | H | H | 2.373 | 100.49 | 3.259 | NE |
| 11 | C | C | C | 1 | H | H | H | H | H | 1.829 | 82.73 | 3.418 | 2.9 |
| Ref. | X | Y | Z | n | R2 | R3 | R4 | R5 | R6 | AlogP | Volb | μ ( D)c | IC50( μ M)d |
| 12 | C | C | C | 2 | H | H | H | H | H | 2.286 | 93.29 | 4.243 | NE |
| 13 | C | C | C | 0 | H | Cl | H | Cl | H | 3.123 | 98.15 | 2.441 | NE |
| 14 | C | C | C | 0 | H | OCH3 | H | OCH3 | H | 1.762 | 111.05 | 4.591 | 4.0 |
| 15 | C | C | C | 0 | H | OCH3 | OCH3 | OCH3 | H | 1.745 | 131.49 | 5.090 | NE |
| 16 | C | C | C | 0 | H | -O-CH2-O- | H | H | 1.563 | 88.78 | 4.352 | NE | |
| 19 | C | C | N | 0 | H | H | - | H | H | 0.644 | 64.45 | 1.081 | NE |
| 20 | N | C | C | 0 | H | H | H | H | H | 0.644 | 64.64 | 3.543 | NE |
| 21 | N | C | C | 0 | Cl | H | H | H | H | 1.518 | 78.98 | 4.094 | 10.0 |
| 22 | N | C | C | 0 | S(CH2)2CH3 | H | H | H | H | 2.597 | 126.66 | 4.964 | NE |
| 23 | 2-thienyl | 1.520 | 64.02 | 3.315 | NE | ||||||||
| 24 | N | N | C | 0 | H | H | H | H | - | -0.078 | 60.35 | 3.096 | NE |
| MSA | - | - | - | - | - | - | - | - | - | - | - | - | 8.38 |
| Etoposide | - | - | - | - | - | - | - | - | - | - | - | - | 13.6 ± 2.2 |
 | |||||
|---|---|---|---|---|---|
| Ref. | R | AlogP | Volb | μ (D)c | IC50(μM)d |
| 17 | naphthyl | 2.703 | 106.65 | 3.759 | NE |
| 18 | diphenylmethyl | 3.324 | 144.34 | 3.632 | NE |
| 25 | 2-quinolyl | 2.409 | 102.41 | 4.451 | NE |
| 26 | 3-quinolyl | 1.981 | 102.22 | 2.685 | NE |
| MSA | - | - | - | - | 8.38 |
| Etoposide | - | - | - | - | 13.6 ± 2.2 |



 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | X | Y | Z | n | R2 | R3 | R4 | R5 | R6 | pKab | Bond order | HOMOc | LUMOd | EL-M | Q_See | IC50 | ||
| a | b | c | (M)f | |||||||||||||||
| 1 | C | C | C | 0 | H | H | H | H | H | 2,119 | 1,8315 | 0,9068 | 0,9618 | -9,210 | -1,552 | 7,658 | 0.0577 | 6.8 |
| 2 | C | C | C | 0 | H | H | CN | H | H | 2,057 | 1,8478 | 0,9050 | 0,9608 | -9,422 | -2,022 | 7,400 | 0.0778 | 10.0 |
| 3 | C | C | C | 0 | H | H | CF3 | H | H | 2,148 | 1,8492 | 0,9048 | 0,9608 | -9,399 | -1,894 | 7,505 | -0.0303 | NE |
| 4 | C | C | C | 0 | H | H | Cl | H | H | 2,188 | 1,7914 | 0,9659 | 0,9624 | -9,382 | -1,666 | 7,716 | -0.0394 | NE |
| 5 | C | C | C | 0 | H | H | CH3 | H | H | 2,184 | 1,8230 | 0,9037 | 0,9613 | -9,002 | -1,487 | 7,515 | 0.0491 | NE |
| 6 | C | C | C | 0 | H | H | C(CH3)3 | H | H | 2,182 | 1,8256 | 0,9027 | 0,9611 | -9,108 | -1,403 | 7,705 | 0.0485 | NE |
| 7 | C | C | C | 0 | H | H | OCH3 | H | H | 2,196 | 1,8159 | 0,8940 | 0,9592 | -9,039 | -1,208 | 7,831 | 0.0433 | NE |
| 8 | C | C | C | 0 | Cl | H | H | H | H | 2,360 | 1,8508 | 0,9320 | 0,9673 | -9,258 | -1,384 | 7,874 | 0.0292 | NE |
| 9 | C | C | C | 0 | Br | H | H | H | H | 2,346 | 1,8611 | 0,9172 | 0,9524 | -9,295 | -1,244 | 8,051 | 0.0134 | NE |
| 10 | C | C | C | 0 | I | H | H | H | H | 2,388 | 1,8721 | 0,9069 | 0,9626 | -8,864 | -1,582 | 7,282 | -0.0182 | NE |
| 11 | C | C | C | 1 | H | H | H | H | H | 2,370 | 1,8419 | 0,9742 | 0,9841 | -9,439 | -0,936 | 8,503 | 0.0681 | 2.9 |
| 12 | C | C | C | 2 | H | H | H | H | H | 2,378 | 1,8360 | 0,9799 | 0,9622 | -9,485 | -1,138 | 8,347 | 0.0580 | NE |
| 13 | C | C | C | 0 | H | Cl | H | Cl | H | 1,858 | 1,8482 | 0,9123 | 0,9626 | -9,410 | -1,961 | 7,449 | -0.0331 | NE |
| 14 | C | C | C | 0 | H | OCH3 | H | OCH3 | H | 2,791 | 1,9205 | 0,9126 | 0,9673 | -9,275 | -1,063 | 8,212 | 0.0693 | 4.0 |
| 15 | C | C | C | 0 | H | OCH3 | OCH3 | OCH3 | H | 2,431 | 1,8021 | 0,9528 | 0,9633 | -8,770 | -1,329 | 7,441 | 0.0652 | NE |
| 16 | C | C | C | 0 | H | -OCH2O- | H | H | 2,083 | 1,8413 | 0,9068 | 0,9620 | -9,223 | -1,630 | 7,593 | 0.0720 | NE | |
| 19 | C | C | N | 0 | H | H | - | H | H | 1,985 | 1,7891 | 0,9918 | 0,9766 | -9,546 | -1,717 | 7,829 | 0.0825 | NE |
| 20 | N | C | C | 0 | H | H | H | H | H | 2,230 | 1,7775 | 0,9840 | 0,9767 | -9,483 | -1,663 | 7,820 | 0.0712 | NE |
| 21 | N | C | C | 0 | Cl | H | H | H | H | 2,145 | 1,8279 | 0,9540 | 0,9686 | -9,368 | -1,712 | 7,656 | 0.0129 | 10.0 |
| 22 | N | C | C | 0 | S(CH2)2CH3 | H | H | H | H | 2,486 | 1,8722 | 0,8969 | 0,9628 | -8,871 | -1,585 | 7,286 | -0.0093 | NE |
| 23 | 2-thienyl | 2,181 | 1,7872 | 0,9417 | 0,9598 | -9,204 | -1,696 | 7,508 | -0.0438 | NE | ||||||||
| 24 | N | N | C | 0 | H | H | H | H | - | 1,813 | 1,7829 | 1,0108 | 0,9746 | -9,406 | -2,030 | 7,376 | 0.1126 | NE |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | R | pKab | Bond order | HOMOc | LUMOd | ΔEL-M | Q_See | IC50 | ||
| a | b | c | (μM)f | |||||||
| 17 | Naphtyl | 2,655 | 1,8432 | 0,8921 | 0,9578 | -9,076 | -1,647 | 7,429 | 0.0644 | NE |
| 18 | Diphenylmethyl | 2,592 | 1,8829 | 0,9492 | 0,9689 | -9,456 | -1,110 | 8,346 | 0.0598 | NE |
| 25 | 2-quinolyl | 2,306 | 1,7884 | 1,0012 | 0,9606 | -9,214 | -1,918 | 7,296 | 0.0918 | NE |
| 26 | 3-quinolyl | 2,375 | 1,8453 | 0,8972 | 0,9589 | -9,222 | -1,881 | 7,341 | 0.0619 | NE |
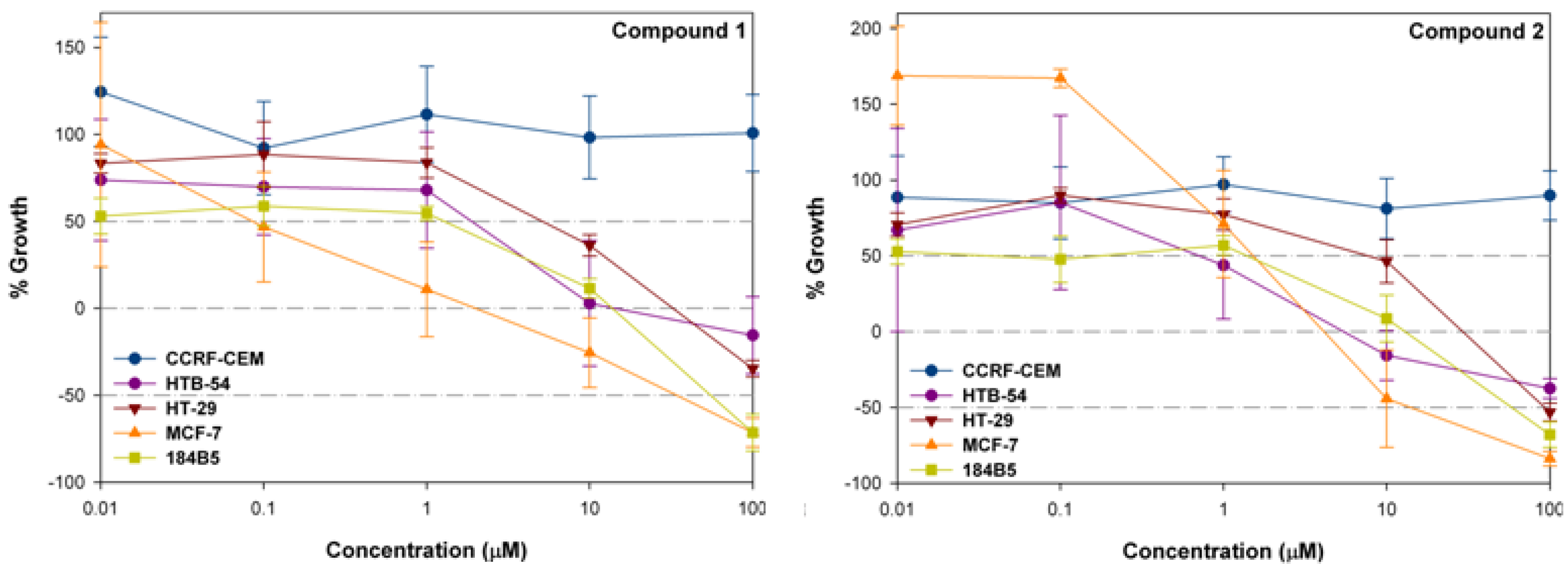
Cytotoxic activity in CCRF-CEM, HTB-54, HT-29, MCF-7 and 184B5
| Comp. | C.Pa (μM) | Cell lines | ||||
|---|---|---|---|---|---|---|
| CCRF-CEM | HTB-54 | HT-29 | MCF-7 | 184B5 | ||
| 1 | bGI50 | >100 | 0.58 | 7.39 | 0.09 | 1.97 |
| cTGI | >100 | 42.47 | 55.98 | 3.69 | 22.50 | |
| dLD50 | >100 | >100 | >100 | 58.01 | 76.73 | |
| 2 | GI50 | >100 | 1.90 | 8.95 | 2.64 | 2.29 |
| TGI | >100 | 9.91 | 51.95 | 6.54 | 20.14 | |
| LD50 | >100 | >100 | >100 | 23.12 | 78.91 | |
| 11 | GI50 | >100 | 11.14 | 16.19 | 0.006 | 1.05 |
| TGI | >100 | >100 | 72.19 | 4.18 | 9.64 | |
| LD50 | >100 | >100 | >100 | 53.49 | 72.34 | |
| 14 | GI50 | >100 | 16.82 | 5.99 | 1.57 | 0.0009 |
| TGI | >100 | >100 | 47.42 | 6.19 | 8.21 | |
| LD50 | >100 | >100 | >100 | 52.65 | 62.55 | |
| 21 | GI50 | >100 | 11.79 | 7.53 | 0.003 | 0.05 |
| TGI | >100 | >100 | 60.94 | 8.27 | 1.89 | |
| LD50 | >100 | >100 | >100 | 89.21 | 7.20 | |
| Doxorub. | GI50 | 0.033 | <0.01 | nde | 0.88 | nd |
| TGI | 0.071 | 1.25 | nd | >100 | nd | |
| LD50 | 0.29 | 3.45 | nd | >100 | nd | |

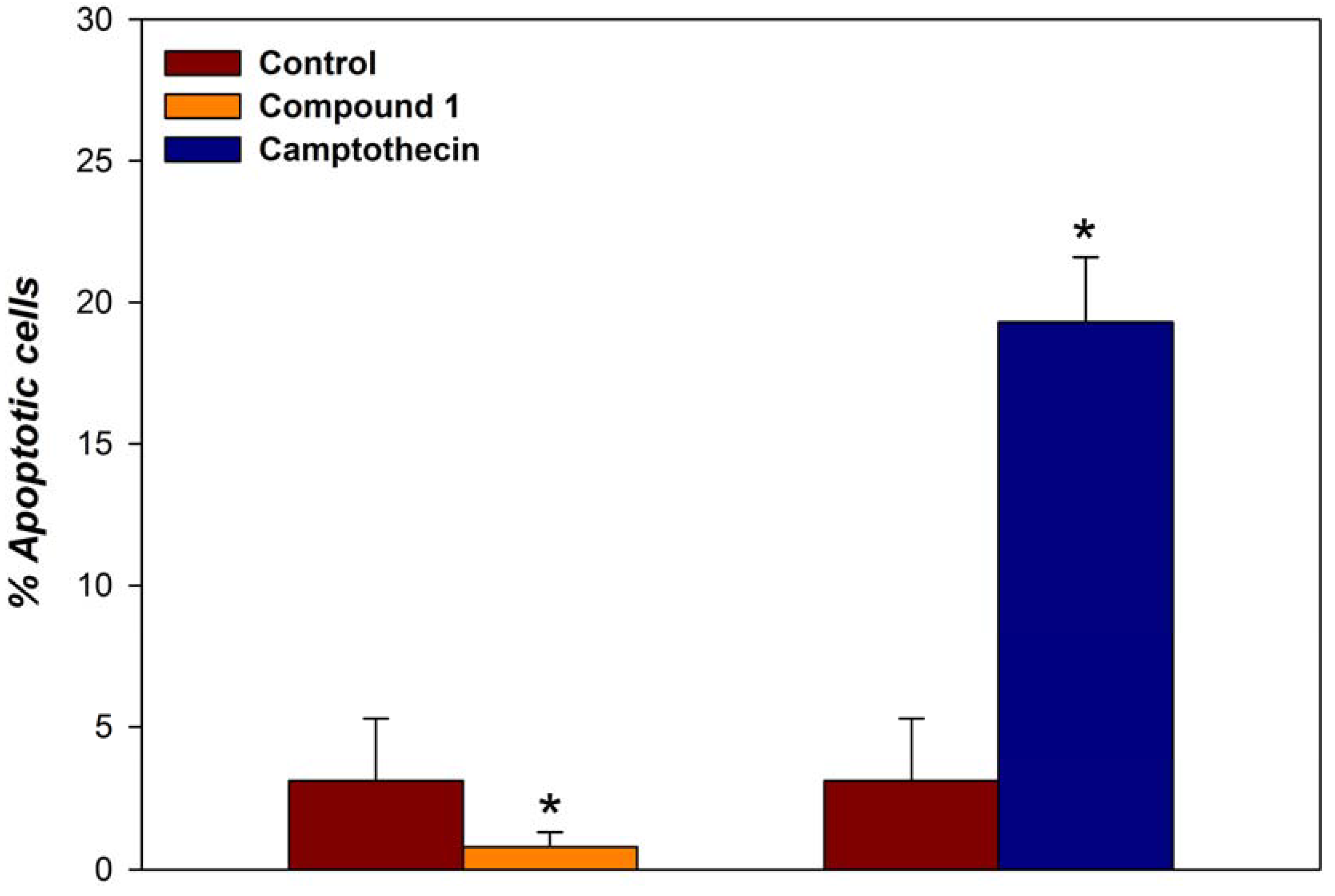
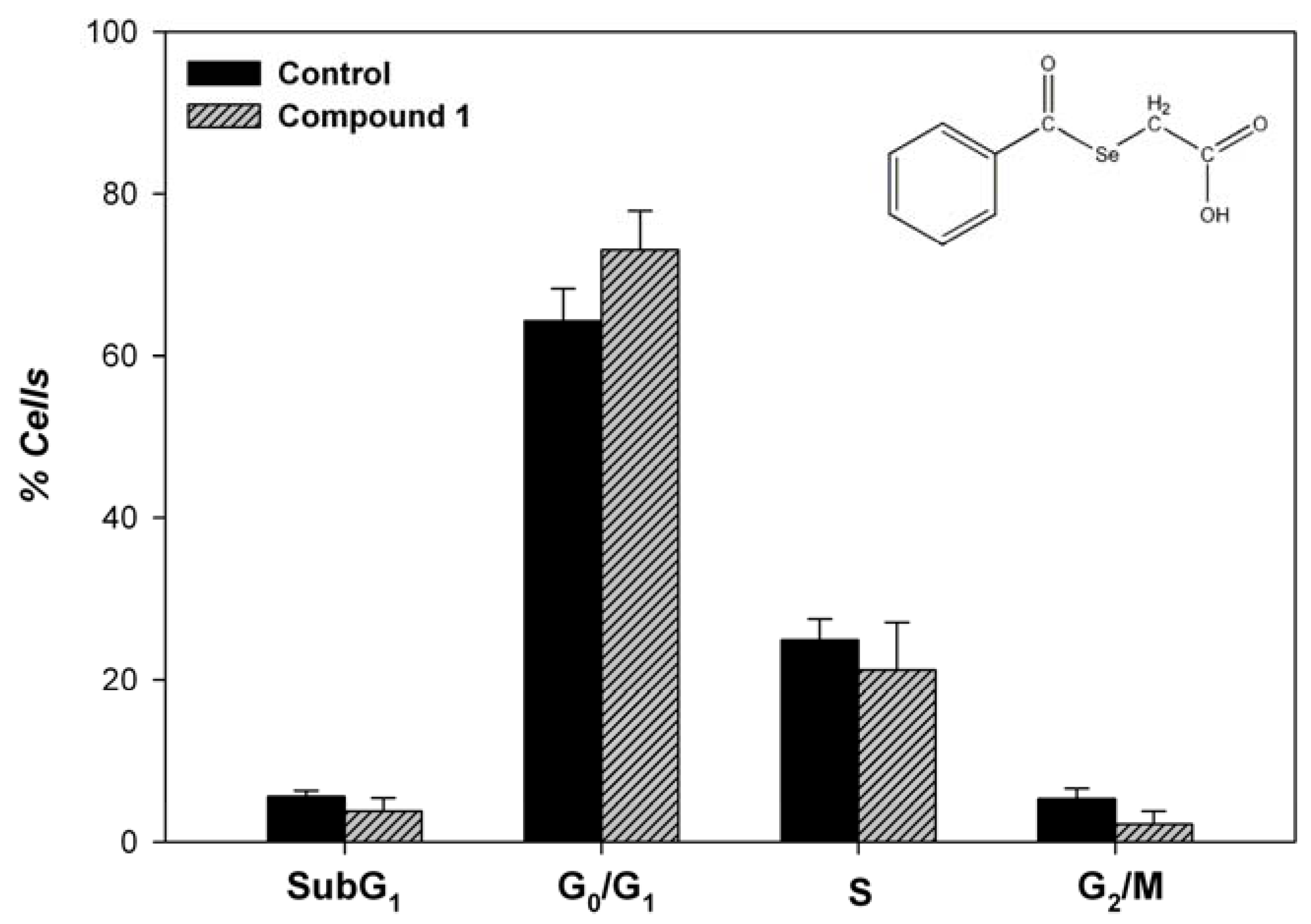
Apoptosis


Conclusions
Experimental
General
| Ref. | IR (KBr, υ in cm-1) | 1H NMR (400 MHz, δ/ppm, J in Hz) |
|---|---|---|
| 1 | 1702, 1678 | DMSO-d6, 3.81 (s, 2H, Se-CH2-COOH), 7.59 (m, 2H, H3 + H5), 7.75 (tt, 1H, H4, J4-5=J4-3=7.9, J4-6=J4-2=1.2), 7.90 (dd, 2H, H2 + H6, J6-5=J2-3=8.4), 12.80 (br s, 1H, COOH). |
| 2 | 2232, 1681 | CDCl3, 3.91 (s, 2H, Se-CH2-COOH), 7.81 (d, 2H, H3 + H5, J3-2=J5-6=8.7), 8.00 (d, 2H, H2 + H6). |
| 3 | 1694, 1712 | CDCl3 3.91 (s, 2H, Se-CH2-COOH), 7.78 (dd, 2H, H3+ H5, J3-2=J5-6=8.1, J3-CF3=J5-CF3=0.5), 8.02 (dd, 2H, H2 + H6, J2-CF3=J6-CF3=0.6). |
| 4 | 1697, 1686 | DMSO-d6, 3.81 (s, 2H, Se-CH2-COOH), 7.66 (d, 2H, H3+H5; J3-2= J5-6=8.5), 7.91 (d, 2H, H2+H6), 12.89 (br s, 1H, COOH). |
| 5 | 1691 | CDCl3, 2.43 (s, 3H, CH3), 3.85 (s, 2H, Se-CH2-COOH), 7.29 (d, 2H, H3+ H5, J3-2=J5-6= 8.1), 7.81 (d, 2H, H2 + H6). |
| 6 | 1715, 1666 | CDCl3, 1.36 (s, 9H, C-(CH3)3), 3.85 (s, 2H, Se-CH2-COOH), 7.51 (d, 2H, H3+H5, J3-2= J5-6=8.7), 7.86 (d, 2H, H2+H6). |
| 7 | 1707, 1692 | CDCl3, 3.83 (s, 2H, Se-CH2-COOH), 3.91 (s, 3H, CH3O), 6.97 (d, 2H, H3+H5, J3-2= 9.0), 7.90 (d, 2H, H2+H6). |
| 8 | 1707, 1688 | CDCl3, 3.89 (s, 2H, Se-CH2-COOH), 7.37-7.43 (m, 1H, H5), 7.49-7.50 (m, 2H, H4+H3), 7.76-7.79 (m, 1H, H6). |
| 9 | 1703, 1684 | CDCl3, 3.89 (s, 2H, Se-CH2-COOH), 7.40 (tt, 1H, H5, J5-6=1.8, J5-4=7.5 J5-3=8.9), 7.44 (tt, 1H, H4, J4-3=1.4 Hz, J4-6=7.6), 7.70 (dd, 1H, H3), 7.72 (dd, 1H, H6). |
| 10 | 1701, 1677 | CDCl3, 3.90 (s, 2H, Se-CH2-COOH), 7.23 (dt, 1H, H5, J5-3=1.5, J5-6=7.9, J5-4=7.7), 7.48 (dt, 1H, H4,J4-6=0.4, J4-3=7.7), 7.69 (dd, 1H, H3); 8.00 (dd, 1H, H6). |
| 11 | 1694, 1685 | CDCl3, 3.62 (s, 2H, Se-CH2-COOH), 3.90 (s, 2H, Ar-CH2-COSe), 7.32 (m, 2H, H3 + H5), 7.38 (m, 3H, H2 + H4 + H6). |
| 12 | 1708, 1686 | CDCl3, 3.03 (s, 4H, Ar-CH2-CH2-COSe + Ar-CH2-CH2-COSe), 3.69 (s, 2H, Se-CH2-COOH), 7.20-7.26 (m, 3H, H3+H4+H5), 7.30-7.34 (m, 2H, H2+H6). |
| 13 | 1699, 1667 | CDCl3, 3.84 (s, 2H, Se-CH2-COOH), 7.57 (s, 1H, H4), 7.70 (s, 2H, H2+H6), 8.44 (br s, 1H, COOH). |
| 14 | 1714, 1696 | CDCl3, 3.85 - 3.86 (s + s, 8H, 2 OCH3 + Se-CH2-COOH, JCH2-Se=72.2), 6.72 (dt, 1H, H4, J4-2=J4-6=0.8 Hz, J4-OCH3=2.3), 7.04 (dd, 2H, H2 + H6, J2-OCH3=J6-OCH3=2.2). |
| 15 | 1703, 1671 | CDCl3, 3.86 (s, 2H, Se-CH2-COOH), 3.94 (s, 6H, 3,5-diCH3O), 3.95 (s, 3H, 4-CH3O), 7.16 (s, 2H, H2). |
| 16 | 1701, 1675 | DMSO-d6, 3.76 (s, 2H, Se-CH2-COOH), 6.18 (s, 2H, O-CH2-O), 7.09 (dd, 1H, H5, J5-2= 0.9 Hz, J5-6=7.8), 7.34 (dd, 1H, H2, J2-6=2.9), 7.55 (dd, 1H, H6), 12.79 (br s, 1H, COOH). |
| 17 | 1719, 1671 | CDCl3, 3.92 (s, 2H, Se-CH2-COOH), 4.40 (br s, 1H, COOH), 7.63 (m, 2H, H4+ H5), 7.91 (m, 3H, H3 + H6 + H7), 8.00 (d, 1H, H8, J8-7=8.1), 8.48 (s, 1H, H2). |
| 18 | 1720, 1693 | CDCl3, 3.68 (s, 2H, Se-CH2-COOH), 5.23 (s, 1H, (Ph)2-CH-COSe), 7.31-7.40 (m, 10 H, 2H2, 2H6, 2H3, 2H5, 2 H4). |
| 19 | 1717, 1659 | DMSO–d6, 3,87 (s, 2H, Se-CH2-COOH), 7,78 (td, 2H, H3+H5), 8,85 (td, 2H, H2+H6). |
| 20 | 1713, 1673 | DMSO-d6, 3.87 (s, 2H, Se-CH2-COOH), 7.61-7.65 (m, 1H, H5), 8.26-8.30 (m, 1H, H4), 8.87-8.90 (m, 1H, H6), 9.02-9.03 (m, 1H, H2), 12.84 (br s, 1H, COOH). |
| 21 | 1724, 1690 | DMSO-d6, 3.87 (s, 2H, Se-CH2-COOH), 7.63 (ddd, 1H, H5, J5-4=7.7, J5-6=4.8, J5-Cl= 1.1), 8.24 (ddd, 1H, H4, J4-Cl=1.1), 8.63 (ddd, 1H, H6, J6-Cl=1.1), 12.90 (br s, 1H, COOH). |
| 22 | 1700, 1668 | DMSO-d6, 0.97 (t, 3H, CH3), 1.58-1.70 (m, 2H, S-CH2-CH2-CH3), 3.11 (dt, 2H, S-CH2-CH2-CH3), 3.81 (s, 2H, Se-CH2-COOH), 7.32 (dd, 1H, H4, J4-5=4.8, J4-6=7.8), 7.97 (dt, 1H, H5, J5-6=1.7), 8.29 (dd, 1H, H6), 12.89 (br s, 1H, COOH). |
| 23 | 1712, 1643 | DMSO-d6, 3.80 (s, 2H, Se-CH2-COOH), 7.30 (dd, 1H, H4, J4-5=3.9 Hz,J4-3=4.9 Hz), 8.02 (dd, 1H, H5, J5-3=1.1 Hz), 8.16 (dd, 1H, H3), 12.82 (bs, 1H, COOH). |
| 24 | 1703, 1674 | DMSO-d6, 3.74 (s, 2H, Se-CH2-COOH), 8.87 (dd, 1H, H6, J6-3=1.2 Hz, J6-5=2.4 Hz), 9.05 (dd, 1H, H5, J5-3=0.2 Hz), 9.06 (dd, 1H, H3), 12.73 (br s, 1H, COOH). |
| 25 | 1714, 1690 | DMSO-d6, 3.73 (s, 2H, Se-CH2-COOH), 7.82 (t, 1H, H7, J7-6=7.5, J7-8=8.0), 7.94 (t, 1H, H6, J6-5=8.3), 7.97 (d, 1H, H5), 8.15 (d, 1H, H8), 8.19 (d, 1H, H3, J3-4=8.4), 8.66 (d, 1H, H4), 12.68 (br s, 1H, COOH). |
| 26 | 1709, 1675 | DMSO-d6, 3,91 (s, 2H, Se-CH2-COOH), 7,77 (t, 1H, H6, J6-7=7,6 ), 7,98 (t, 1H, H7), 8,13 (d, 1H, H5, J5-8=8,3), 8,31 (d, 1H, H8), 9,07 (d, 1H, H4, J4-2=1,8), 9,23 (d, 1H, H2),12,75 (bs, 1H, COOH). |
General procedure for the preparation of selenylacetic acids 1–6, 8–26
Procedure for the preparation of (4-methoxyphenylselenyl)acetic acid (7)
Cytotoxic activity in PC-3
Molecular modelling
Cytotoxic activity in CCRF-CEM, HTB-54, HT-29, MCF-7 and 184B5
Apoptosis and cell cycle
Acknowledgements
- Sample Availability: Samples of the compounds 1–26 are available from the authors.
References
- Varmus, H. The new era in cancer research. Science 2006, 312, 1162–1165. [Google Scholar] [CrossRef]
- Jariwalla, R.J.; Gangapurkar, B.; Nakamura, D. Differential sensitivity of various human tumour-derived cell types to apoptosis by organic derivatives of selenium. Br. J. Nutr. 2009, 101, 182–189. [Google Scholar] [CrossRef]
- Latreche, L.; Chavatte, L. Selenium incorporation into selenoproteins, implications in human health. In 10th International Symposium on Metal Ions in Biology and Medicine, Bastia, France; 2008; pp. 731–737. [Google Scholar]
- Clark, L.C.; Combs, G.F. Selenium-compounds and the prevention of cancer-research needs and public health implications. J. Nutr. 1986, 116, 170–173. [Google Scholar]
- Sonn, G.A.; Aronson, W.; Litwin, M.S. Impact of diet on prostate cancer: A review. Prostate Cancer Prostatic Dis. 2005, 8, 304–310. [Google Scholar] [CrossRef]
- El-Bayoumy, K.; Sinha, R.; Pinto, J.T.; Rivlin, R.S. Cancer chemoprevention by garlic and garlic-containing sulfur and selenium compounds. J. Nutr. 2006, 136, 864–869. [Google Scholar]
- Gonzalez, C.A.; Salas-Salvado, J. The potential of nuts in the prevention of cancer. Br. J. Nutr. 2006, 96, S87–S94. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sharma, A.; Desai, D.; Madhunapantula, S.V.; Huh, S.J.; Robertson, G.P.; Amin, S. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. J. Med. Chem. 2008, 51, 7820–7828. [Google Scholar] [CrossRef]
- Sanmartín, C.; Plano, D.; Palop, J.A. Selenium compounds and apoptotic modulation: A new perspective in cancer therapy. Mini Rev. Med. Chem. 2008, 8, 1020–1031. [Google Scholar] [CrossRef]
- Bardia, A.; Tleyjeh, I.M.; Cerhan, J.R.; Sood, A.K.; Limburg, P.J.; Erwin, P.J. Montori, V.M. Efficacy of antioxidant supplementation in reducing primary cancer incidence and mortality: Systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 23–34. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Systematic review: Primary and secondary prevention of gastrointestinal cancers with antioxidant supplements. Alimen. Pharmacol. Ther. 2008, 28, 689–703. [Google Scholar] [CrossRef]
- Connelly-Frost, A.; Poole, C.; Satia, J.A.; Kupper, L.L.; Millikan, R.C.; Sandler, R.S. Selenium, folate, and colon cancer. Nutr. Cancer 2009, 61, 165–178. [Google Scholar] [CrossRef]
- Smith, M.L.; Lancia, J.K.; Mercer, T.I.; Ip, C. Selenium compounds regulate p53 by common and distinctive mechanisms. Anticancer Res. 2004, 24, 1401–1408. [Google Scholar]
- Rikiishi, H. Apoptotic cellular events for selenium compounds involved in cancer prevention. J. Bioenerg. Biomembr. 2007, 39, 91–98. [Google Scholar] [CrossRef]
- Hurst, R.; Elliot, R.M.; Goldson, A.J.; Fainweather-Tait, S.J. Se-methylselenocysteine alters collagen gene and protein expression in human prostate cells. Cancer Lett. 2008, 269, 117–126. [Google Scholar] [CrossRef]
- Pinto, J.T.; Sinha, R.; Papp, K.; Facompre, N.D.; Desai, D.; El-Bayoumy, K. Differential effects of naturally occurring and synthetic organoselenium compounds on biomarkers in androgen responsive and androgen independent human prostate carcinoma cells. Int. J. Cancer 2007, 120, 1410–1417. [Google Scholar]
- Singh, U.; Null, K.; Sinha, R. In vitro growth inhibition of mouse mammary epithelial tumor cells by methylseleninic acid: Involvement of protein kinases. Mol. Nutr. Food Res. 2008, 52, 1281–1288. [Google Scholar] [CrossRef]
- Poerschke, R.L.; Franklin, M.R.; Moos, P.J. Modulation of redox status in human lung cell lines by organoselenocompounds: Selenazolidines, selenomethionine, and methylseleninic acid. Toxicol. Vitro 2008, 22, 1761–1767. [Google Scholar] [CrossRef]
- Talas, Z.S.; Ozdemir, I.; Yilmaz, I.; Gok, Y. Antioxidative effects of novel synthetic organoselenium compound in rat lung and kidney. Ecotoxicol. Environ. Saf. 2009, 72, 916–921. [Google Scholar] [CrossRef]
- Shiah, H.S.; Lee, W.S.; Juang, S.H.; Hong, P.C.; Lung, C.C.; Chang, C.J.; Chou, K.M.; Chang, F.Y. Mitochondria-mediated and p53-associated apoptosis induced in human cancer cells by a novel selenophene derivative, D-501036. Biochem. Pharmacol. 2007, 73, 610–619. [Google Scholar]
- Xing, F.; Li, S.L.; Ge, X.Y.; Wang, C.; Zeng, H.H.; Li, D.; Dong, L. The inhibitory effect of a novel organoselenium compound BBSKE on the tongue cancer Tca8113 in vitro and in vivo. Oral Oncol. 2008, 44, 963–969. [Google Scholar] [CrossRef]
- Barbosa, N.B.D.; Nogueira, C.W.; Guecheva, T.N.; Bellinaso, M.D.; Rocha, J.B.T. Diphenyl diselenide supplementation delays the development of N-nitroso-N-methylurea-induced mammary tumors. Arch. Toxicol. 2008, 82, 655–663. [Google Scholar] [CrossRef]
- Xiao, H.; Parkin, K.L. Induction of Phase II enzyme activity by various selenium compounds. Nutr. Cancer 2006, 55, 210–223. [Google Scholar] [CrossRef]
- Rosa, R.M.; Picada, J.D.; Saffi, J.; Henriques, J.A.P. Cytotoxic, genotoxic, and mutagenic effects of diphenyl diselenide in Chinese hamster lung fibroblast. Mut. Res. 2007, 628, 87–98. [Google Scholar] [CrossRef]
- Zhao, G.H.; Lin, H.K.; Zhu, S.R.; Sun, H.W.; Chen, Y.T. Dinuclear palladium(II) complexes containing two monofunctional [Pd(en)(pyridine)Cl]+ units bridged by Se or S. Synthesis, characterization, cytotoxicity and kinetic studies of DNA-binding. J. Inorg. Biochem. 1998, 70, 219–226. [Google Scholar] [CrossRef]
- Abele, E.; Popelis, J.; Shestakova, I.; Domracheva, I.; Arsenyan, P.; Lukevics, E. Synthesis and cytotoxicity of di(3-indolyl) selenide derivatives. Khimiya Geterotsiklicheskikh Soedinenii 2004, 6, 868–872. [Google Scholar]
- Zeng, H.W.; Briske-Anderson, M.; Idso, J.P.; Hunt, C.D. The selenium metabolite methylselenol inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2006, 136, 1528–1532. [Google Scholar]
- Ohta, Y.; Suzuki, K.T. Methylation and demethylation of intermediates selenide and methylselenol in the metabolism of selenium. Toxicol. Appl. Pharmacol. 2008, 226, 169–177. [Google Scholar] [CrossRef]
- Plano, D.; Sanmartín, C.; Moreno, E.; Prior, C.; Calvo, A.; Palop, J.A. Novel potent organoselenium compounds as cytotoxic agents in prostate cancer cells. Bioorg. Med. Chem. Lett. 2007, 17, 6853–6859. [Google Scholar] [CrossRef]
- Plano, D.; Moreno, E.; Font, M.; Encío, I.; Palop, J.A.; Sanmartín, C. Synthesis and biological evaluation of selenadiazole derivatives as anticancer agents. Bioorg. Med. Chem. 2009. (Submitted for publication).. [Google Scholar]
- De Athayde, P.F.; De Souza, A.G.; De Morais, S.A.; Botelho, J.R.; Barbosa, J.M.; Miller, J.; Lira, B.F. Synthesis and characterization of three new organo-selenium compounds. A convenient synthesis of aroylselenoglycolic acids. Arkivoc 2004, 6, 22–26. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Inmunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, H.; Hawthorn, L.; Ganther, H.E.; Ip, C. Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 2003, 63, 52–59. [Google Scholar]
- Van Brussel, J.P.; Oomen, M.A.; Vosselbeld, P.J.M.; Wiemer, E.A.C.; Sonneveld, P.; Mickisch, G.H.J. Identification of multidrug resistance-associated protein 1 and glutathione as multidrug resistance mechanisms in human prostate cancer cells: chemosensitization with leukotriene D4 antagonists and buthionine sulfoximine. BJU Int. 2004, 93, 1333–1338. [Google Scholar] [CrossRef]
- Font, M.; Ardaiz, E.; Cordeu, L.; Cubedo, E.; García-Foncillas, J.; Sanmartín, C.; Palop, J.A. Structural characteristics of novel symmetrical diaryl derivatives with nitrogenated functions. Requirements for cytotoxic activity. Bioorg. Med. Chem. 2006, 14, 1942–1948. [Google Scholar]
- Accerlys Software Inc. Discovery Studio Modeling Environment Release 1.7.; Accerlys Software Inc.: San Diego, USA, 2007.
- MOPAC MOPAC2009, Stewart, J.P. Stewart Computational Chemistry, Colorado Springs, CO, USA. Available online: http://OpenMOPAC.net,2008.
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Available online: http://dtp.nci.nih.gov/branches/btb/ivclsp.html accessed on 27 June 2009.
- Philchenkov, A.; Zavelevich, M.; Khranovskaya, N.; Surai, P. Comparative analysis of apoptosis induction by selenium compounds in human lymphoblastic leukemia MT-4 cells. Exp. Oncol. 2007, 29, 257–261. [Google Scholar]
- Naithani, R. Organoselenium compounds in cancer chemoprevention. Mini Rev. Med. Chem. 2008, 8, 657–668. [Google Scholar] [CrossRef]
- Fimognari, C.; Nüsse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. Dreiding: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure–activity relationships. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally-occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: An analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semi-empirical methods I-Method. J. Comp. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Li, Y.Z.; Pan, J.; Li, J.L.; Lee, J.H.; Tunkey, C.; Saraf, K.; Garbe, J.C.; Whitley, M.Z.; Jelinsky, S.A.; Stampfer, M.R.; Haney, S.A. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol. Cancer 2007, 6, Article 7. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sanmartín, C.; Plano, D.; Domínguez, E.; Font, M.; Calvo, A.; Prior, C.; Encío, I.; Palop, J.A. Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents. Molecules 2009, 14, 3313-3338. https://doi.org/10.3390/molecules14093313
Sanmartín C, Plano D, Domínguez E, Font M, Calvo A, Prior C, Encío I, Palop JA. Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents. Molecules. 2009; 14(9):3313-3338. https://doi.org/10.3390/molecules14093313
Chicago/Turabian StyleSanmartín, Carmen, Daniel Plano, Enrique Domínguez, María Font, Alfonso Calvo, Celia Prior, Ignacio Encío, and Juan Antonio Palop. 2009. "Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents" Molecules 14, no. 9: 3313-3338. https://doi.org/10.3390/molecules14093313
APA StyleSanmartín, C., Plano, D., Domínguez, E., Font, M., Calvo, A., Prior, C., Encío, I., & Palop, J. A. (2009). Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents. Molecules, 14(9), 3313-3338. https://doi.org/10.3390/molecules14093313