General
All purifications by flash chromatography were performed on silica gel 230-400 mesh (Merck). Thin layer chromatography (TLC) was performed on silica gel IB-F (J.T. Baker). 1H-NMR spectra were recorded on either Varian 300 MHz or Bruker 400 MHz spectrometers. Resonances are reported as (solvent); δ = shift in ppm from tetramethyl silane (TMS) at 0 ppm and the multiplicity of signals as s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; br, broad; J values are in Hz. The numbering system used is shown in the schemes. IR spectra were measured on Nicolet FT-IR Impact 400D as KBr discs. UV-Visible spectra were obtained on a Hitachi U2000 spectrometer. FAB mass spectra were carried out using m-nitrobenzyl alcohol as matrix. Melting points were measured on a Laboratory Devices (Cambridge, MA) Mel-Temp apparatus and are uncorrected. High dilution reactions of acid chlorides with amines were carried out using infusion pump model 975 from Harvard Apparatus (MA, USA).
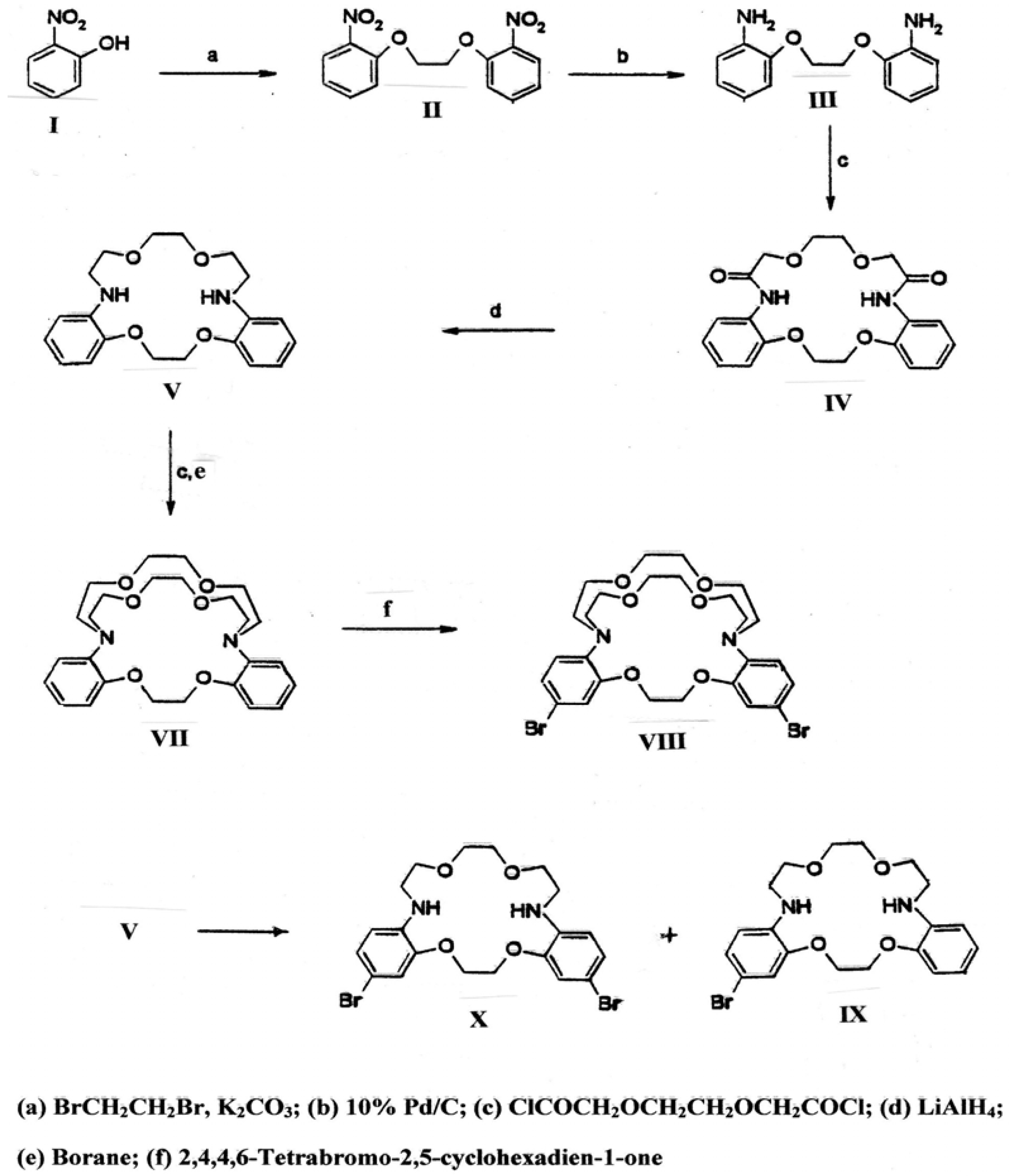
1,2-Bis (2-nitrophenoxy)ethane (II): A mixture of 2-nitrophenol (13.3 g, 96 mmol), 1,2-dibromoethane (10 g, 53 mmol) and K2CO3 (15.2 g, 110 mmol) in dimethylformamide (50 mL) was refluxed overnight. Additional 1,2-dibromoethane (10 g) was added and then refluxed for 2 hrs. The reaction was cooled to room temperature and water (50 mL) was added and the mixture stirred for 30 minutes. The resulting light brown precipitate was collected, washed with water, and recrystallized from acetone to afford yellow crystals (10.4 g, 71%); m.p.163-165 °C; IR (KBr): υmax 1,606, 1,525, 1,490, 1,445, 1,364, 1,254, 1,163, 937, 858, 777, 752 cm-1; 1H-NMR (DMSO-d6): δ 4.52 (s, 4H, OCH2CH2O), 7.13 (t, J = 7.4, 2H, 4-H), 7.42 (d, J = 8.4, 2H, 6-H), 7.64 (dt, J = 1.6 and 8.0 Hz, 2H, 5-H), 7.84 (dd, J = 1.8 and 8.4 Hz, 2H, 3-H).
1,2-Bis (2-aminophenoxy)ethane (III): A suspension of II (14.7 g, 48 mmol), 10% palladium on carbon (0.7 g) in methanol (400 mL) and ethyl acetate (50 mL) was hydrogenated overnight at atmospheric pressure and room temperature. The reaction mixture was filtered and the solvent removed in vacuo leaving a white solid, which was recrystallized from ethyl acetate to yield white crystals (9.5 g, 81%), m.p. 122-125 °C; IR (KBr): υmax 3,446 and 3,361 (NH), 1,610, 1,504, 1,461, 1,275, 1,213, 1,082, 947, 746, 737 cm-1; 1H-NMR (DMSO-d6): δ 4.26 (s, 4H, OCH2CH2O), 4.68 (s, 4H, NH2), 6.5 (dt, J = 2.0 and 8.0 Hz, 2H, 5-H), 6.65 (dt, J = 1.6 and 8.5 Hz, 2H, 4-H), 6.70 (dd, J = 1.2 and 7.4 Hz, 2H, 3-H), 6.85 (dd, J = 1.0 and 7.6 Hz, 2H, 6-H).
2,5,10,13-Tetraoxa-7,16-diaza-1(1,2),6(1,2)-benzacyclohexadecaphane-8,15-dione (IV): To a two liter three necked flask was added a solution of triethylamine (3.5 g, 35 mmol) in anhydrous toluene (400 mL) maintained at 0 °C with stirring. The bisamine (III) (3.06 g, 12.5 mmol) in THF (150 mL) and the diacid chloride, 2,6-dioxaoctanedioic acid chloride (2.69 g, 12.5 mmol) in toluene (150 mL) were added dropwise simultaneously through Teflon syringe needles (16 gauge, 24 inch length with KEL-Fluerhub) attached to 50 mL syringes which were controlled by an infusion pump (Harvard Apparatus, model 975). The addition proceeded at a rate of 0.3 mL/min. After the addition was complete, stirring was continued for a further two days at room temperature and the reaction mixture was filtered to remove the salts. Removal of the solvent in vacuo left a white solid which was recrystallized from ethyl acetate to afford white crystals of the diamide IV (3.1 g, 64%), m.p. 183-187 °C; IR (KBr): υmax 3,372, 3,356, 1,648, 1,607, 1,596, 1,460, 1,250, 1,121, 748 cm-1; 1H-NMR: (DMSO-d6) δ 3.80 (s, 4H, 11-CH2, 12-CH2), 4.11 (s, 4H, 9-CH2, 14-CH2), 4.47 (s, 4H, 3-CH2, 4-CH2), 6.93 (t, J = 6.5 Hz, 2H, 14-H, 64-H), 7.05 (t, J = 7.6 Hz, 2H, 15-H, 65-H), 7.90 (d, J = 8.0 Hz, 2H, 16’-H, 66-H), 8.4 (d, J = 7.4 Hz, 2H, 13-H, 63-H), 9.5 (s, 2H, -NH).
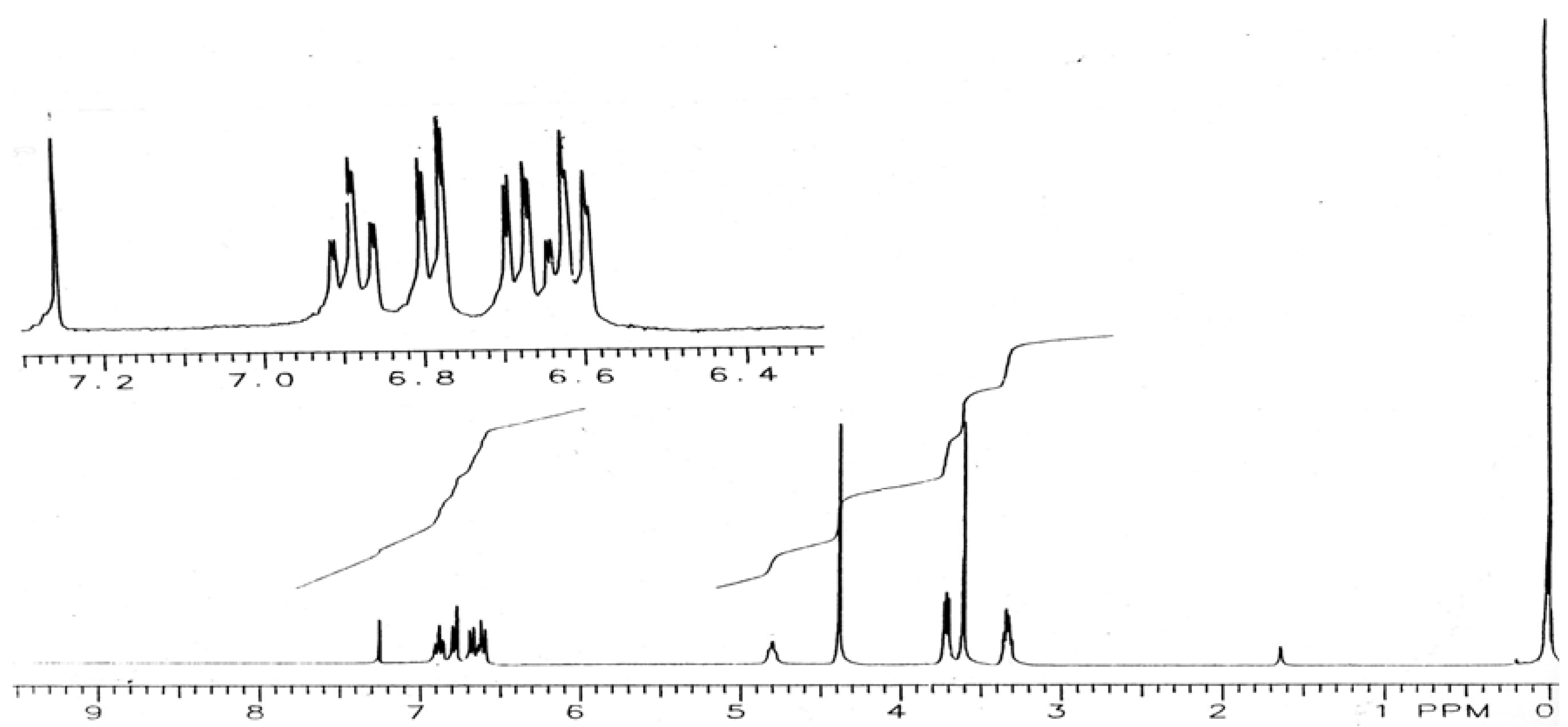
2,5,10,13-Tetraoxa-7,16-diaza-1(1,2),6(1,2)-benzacyclohexadecaphane (V): To the diamide IV (0.4 g, 1.0 mmol) was added LiAlH4 in THF (1.0 M, 10 mL) dropwise with stirring at room temperature. The reaction mixture was heated under reflux overnight. The excess of LiAlH4 was destroyed by the successive dropwise addition of water (0.4 mL), NaOH (15%, 0.4 mL) and water (1.2 mL). The reaction mixture was filtered and the filtrate was evaporated to dryness to leave a solid which was recrystallized from ethyl acetate/hexane to afford white crystals (0.24 g, 67%), m.p. 165-167 °C; IR (KBr): υmax 3,422, 1,597, 1,523, 1,448, 1,253, 1,222, 1,120, 941, 733, 715 cm-1; 1H-NMR (CDCl3): δ 3.33 (m, 4H, 8-CH2, 15-CH2), 3.60 (s, 4H, 11-CH2, 12-CH2), 3.71 (t, J = 4.7 Hz, 4H, 9-CH2, 14-CH2), 4.38 (s, 4H, 3-CH2, 4-CH2), 4.80 (br s, 2H, NH), 6.62 (dd, J = 1.43 and 7.80 Hz, 2H, 13-H, 63-H), 6.68 (dt, J = 1.53 and 7.70 Hz, 2H, 15-H, 65-H), 6.78 (dd, J = 1.4 and 7.95 Hz, 2H, 16-H, 66-H), 6.88 (dt, J = 1.40 and 7.62 Hz, 2H, 14-H, 64-H).
4,7,13,16,20,23-Hexaoxa-1,10-diaza-19(1,2)24(1,2)-dibenzabicyclo[8.8.6]tetracosaphane-2,9-dione (VI): To a stirred solution of triethylamine (2.61 g, 25.8 mmol) in toluene (500 mL) maintained at 0 °C was added the bisamine V (3.0 g, 8.4 mmol) in THF (200 mL) and the diacid chloride, 3,6-dioxaoctanedioic acid chloride (2.1 g, 9.8 mmol) in toluene (200 mL), simultaneously using 50 mL syringes. The rate of addition (0.3 mL/min) was controlled by an infusion pump. After addition was completed, the mixture was stirred at room temperature overnight. Removal of the solvent in vacuo left a semisolid which was crystallized from ethyl acetate to give white crystals (1.6 g, 40%), m.p. 186-189 °C; IR (KBr): υmax 1,672, 1,499, 1,452, 1,275, 1,132, 933, 755 cm-1; 1H-NMR (CDCl3): δ 3.05-4.75 (m, 24H), 6.96-7.16 (m, 6H), 7.25-7.40 (m, 12H).
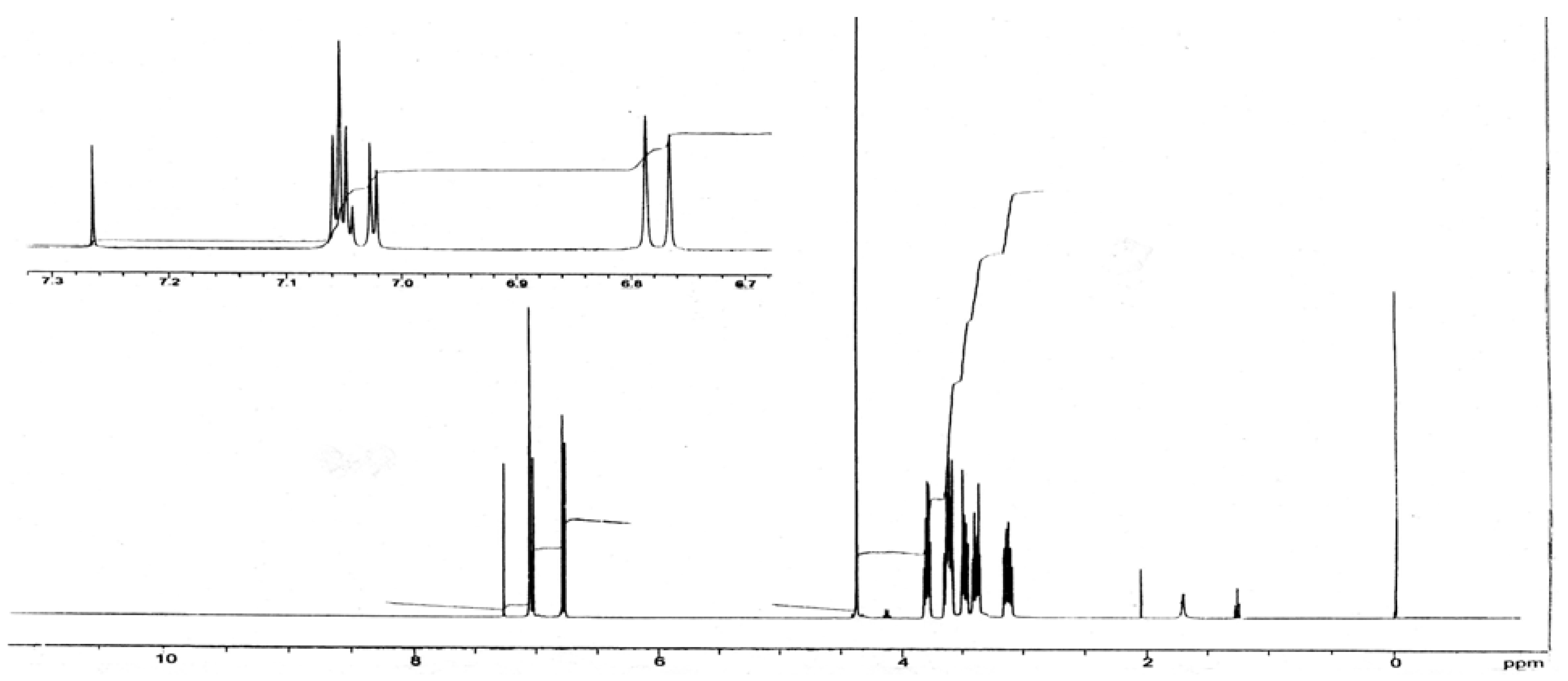
4,7,13,16,20,23-Hexaoxa-1,10-diaza-19(1,2)24(1,2)-dibenzabicyclo[8.8.6]tetracosaphane (VII): To a solution of diamide VI (0.2 g, 0.4 mmol) in THF (2 mL) was added borane in THF (1 M, 2 mL, 2 mmol), and the mixture was heated under reflux for 2 hrs, cooled and water (0.5 mL) carefully added followed by aqueous HCl (6 M, 4 mL). The solution was concentrated in vacuo and the resulting yellow solution was brought to pH 8 with aqueous LiOH. The aqueous layer was extracted with chloroform and dried over sodium sulfate. Removal of the solvent yielded a yellow oil which was triturated with ethyl acetate to give a white powder, m.p.141-143 °C; IR (KBr): υmax 1,593, 1,505, 1,449, 1,341, 1,250, 1,230, 1,135, 1,115, 1,056, 975, 940, 732 cm-1 ; 1H-NMR (CDCl3): δ 3.12 (m, 4H), 3.54 (m, 16H), 3.81 (m, 4H), 4.37 (s, 4H, 2CH21, 2CH22), 6.92 (m, 8H, aromatic).
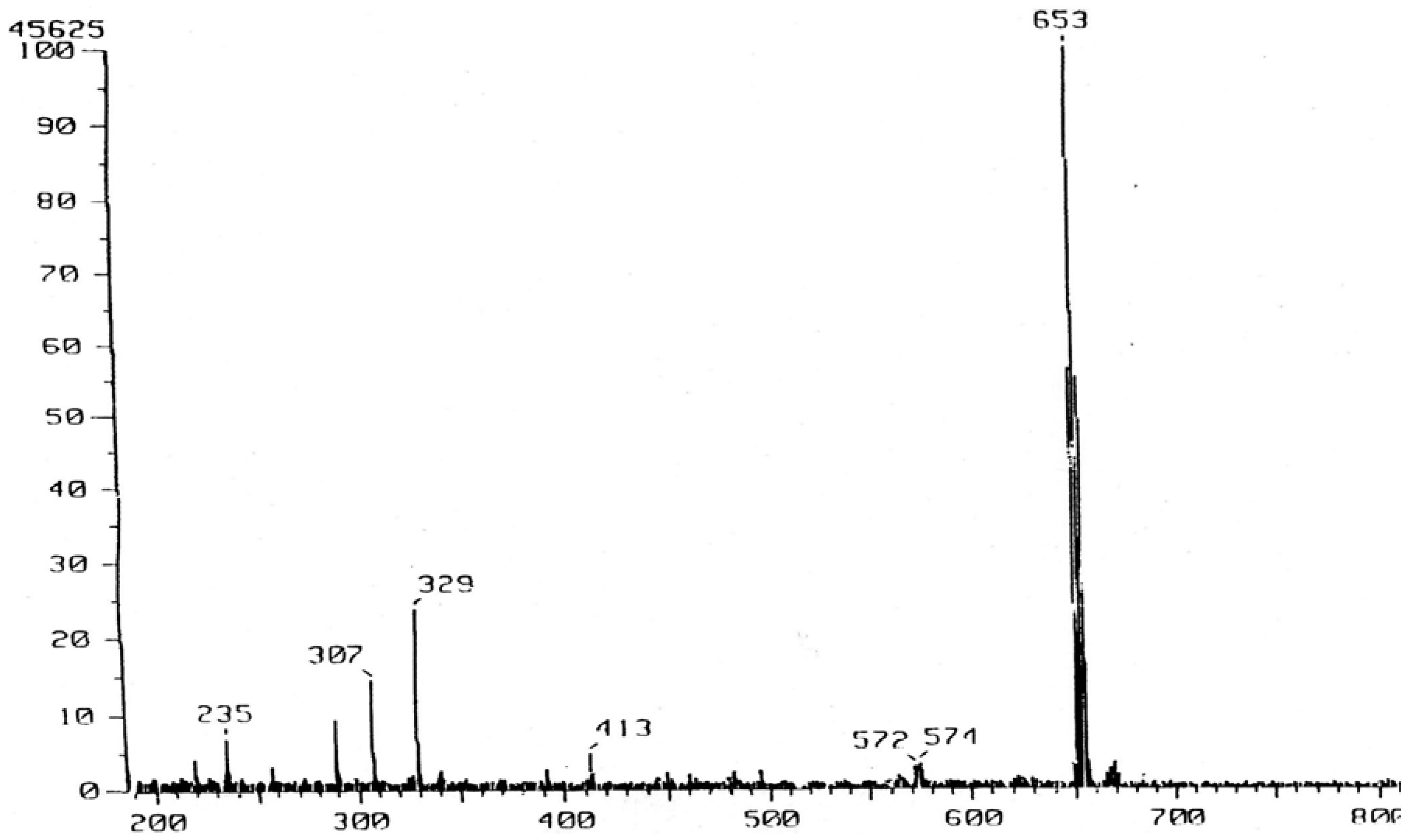
195,245-Dibromo-4,7,13,16,20,23-hexaoxa-1,10-diaza-19(1,2)24(1,2)-dibenzabicyclo[8.8.6]tetra-cosaphane (VIII): To a stirred solution of diamine VII (116 mg, 0.25 mmol) in CH2Cl2 (5 mL) kept at -78 °C was added portion-wise 2,4,4,6-tetrabromo-2,5-cyclohexadien-1-one (0.25 g, 0.61 mmol). The mixture was allowed to warm to room temperature and stirred overnight. The resulting yellow solution was extracted with aqueous NaOH (2 N, 10 mL), water and the organic layer was dried over sodium sulfate. Removal of the solvent afforded a yellow oil which was chromatographed using silica gel and methylene chloride and methanol (up to 5%) to give white solid which was recrystallized from ethyl acetate to yield white crystals (31 mg, 20 %), m.p. 204-206 °C; IR (KBr): υmax 1,580, 1,489, 1,232, 1,125, 959, 848, 817 cm-1; 1H-NMR (CDCl3): δ 3.09-3.16 (m, 4H), 3.36-3.42 (m, 4H), 3.46-3.52 (m. 4H), 3.58-3.66 (m, 8H), 3.76-3.83 (m, 4H), 4.48 (s, 4H, 2CH21, 2CH22), 6.78 (d, J = 8.0 Hz, 2H, 193-H, 243-H), 7.04 (dd, J = 8.0 and 2.4 Hz, 2H, 194-H, 244-H), 7.06 (d, J = 2.4 Hz, 2H, 196-H, 246-H); m/z (FAB) 651 (M+Na+).
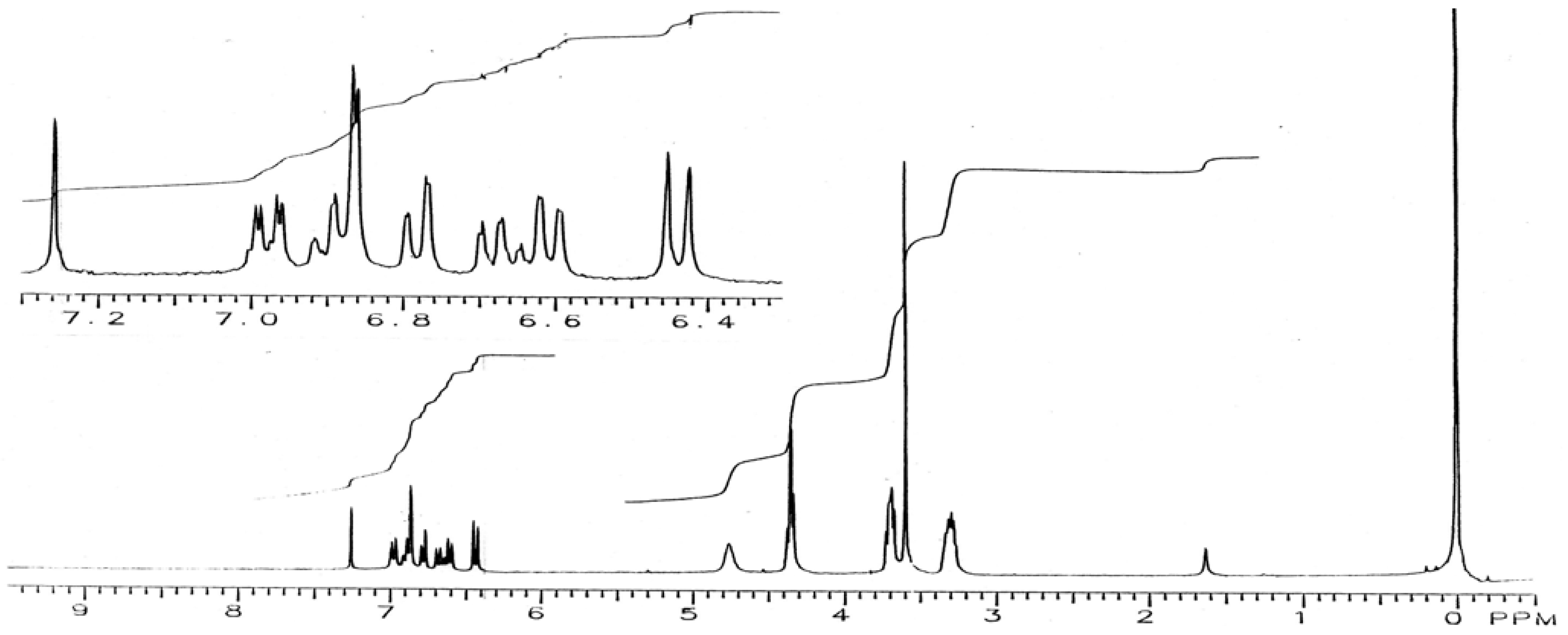
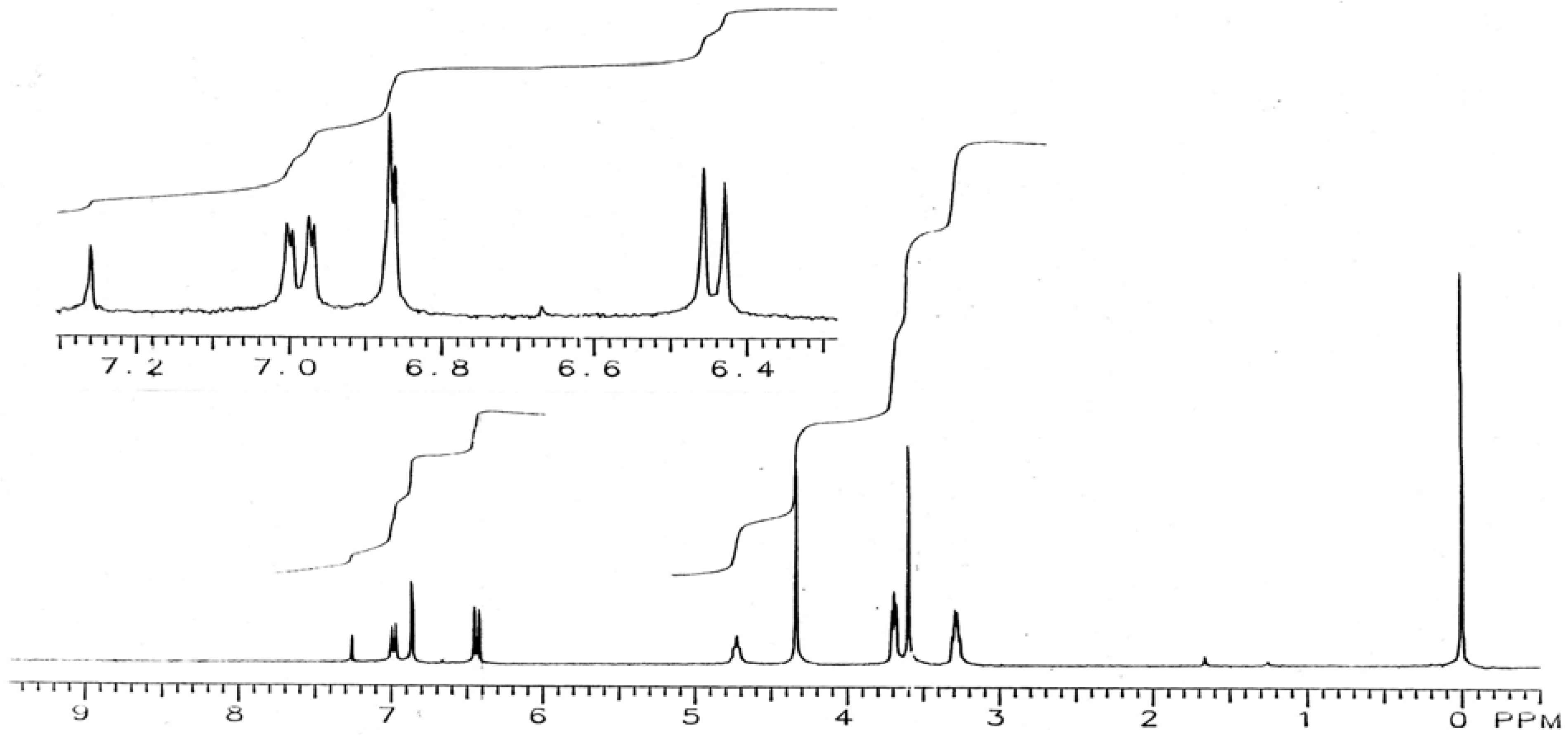
15-Bromo-2,5,10,13-tetraoxa-7,16-diaza-1(1,2),6(1,2)-benzenecyclohexadecaphane (IX): To a stirred solution of diamine V (150 mg, 0.4 mmol) and pyridine (98 mg, 1.2 mmol) in CH2Cl2 (10 mL) and kept at -78 °C was added slowly 0.78 M bromine solution in CH2Cl2 (1.3 mL). The mixture was allowed to warm to room temperature. The reaction mixture was washed with aqueous Na2CO3 and dried over sodium sulfate. Removal of the solvent yielded a residue which was chromatographed on a TLC plate (200 µm layer of silica gel IB-F, 1% CH3OH in CH2Cl2 as solvent) to give two major products: the monobromo-derivative IX (22 mg, 13%), m.p. 158-161 °C; IR (KBr): υmax 3,405, 1,603, 1,520, 1,453, 1,261, 1,202, 1,141, 1,096, 942, 838, 799, 724 cm-1; 1H-NMR (CDCl3): δ 3.31 (m, 4H, 8-CH2, 15-CH2), 3.60 (s, 4H, 11-CH2, 12-CH2), 3.70 (m, 4H, 9-CH2, 14-CH2), 4.36 (m, 4H, 3-CH2, 4-CH2), 4.76 (br s, 2H, NH), 6.44 (d, J = 8.8 Hz, 1H, 13-H), 6.61 (d, J = 7.5 Hz, 1H, 63-H), 6.68 (t, J = 7.5 Hz, 1H, 65-H), 6.78 (d, J = 7.5 Hz, 1H, 66-H), 6.86 (d, J = 2.0 Hz, 1H, 16-H), 6.90 (t, J = 7.5 Hz, 1H, 64-H), 6.98 (dd, J = 2 and 8.7 Hz, 1H, 14-H); and the dibromo derivative X (20 mg, 10%), m.p. 210-214 °C; IR (KBr): υmax 3,405, 1,598, 1,509, 1,463, 1,406, 1,346, 1,217, 1,203, 1,146, 1,097, 939, 843, 794 cm-1; 1H-NMR (CDCl3): δ 3.29 (q, J = 5 Hz, 4H, 8-CH2, 15-CH2), 3.60 (s, 4H, 11-CH2, 12-CH2), 3.69 (t, J = 5 Hz, 4H, 9-CH2, 14-CH2), 4.33 (s, 4H, 3-CH2, 4-CH2), 4.73 (br t, J = 5 Hz, 2H, NH), 6.44 (d, J = 8.7 Hz, 2H, 13-H, 63-H), 6.86 (d, J = 2 Hz, 2H, 16-H, 66-H), 6.99 (dd, J = 2 and 8.7 Hz, 2H, 14-H, 64-H).
15,65-Dibromo-2,5,10,13-tetraoxa-7,16-diaza-1(1,2),6(1,2)-benzenecyclohexadecaphane (X): To a stirred solution of of diamine V (1.4 g, 4 mmol) in CH2Cl2 (50 mL) maintained at -78 °C was added finely powdered 2,4,4,6-tetrabromo-2,5-cyclohexadien-1-one (3.4 g, 8 mmol). After addition was completed, the reaction mixture was allowed to warm to room temperature and stirred overnight. The resulting yellow solution was extracted with aqueous 2N sodium hydroxide. The organic layer was washed with water and dried over sodium sulfate. Removal of the solvent afforded a yellow solid which was recrystallized from ethyl acetate to yield white flakes (1.6 g, 78%), m.p. 212-215 °C, 1H-NMR spectral data were identical to those given above.
195,245-Dibromo-4,7,13,16,20,23-hexaoxa-1,10-diaza-19(1,2)24(1,2)-dibenzabicyclo[8.8.6]tetra-cosaphane-2,9-dione (XI): To a stirred solution of triethylamine (1.45 g, 14.3 mmol) in toluene (300 mL) maintained at 0 οC were added the bisamine X (1.4 g, 2.7 mmol) in THF (100 mL), and 3,6-dioxa-octanedioic acid chloride (0.6 g, 2.8 mmol) in toluene (100 mL), simultaneously at a rate of 0.3 mL/min with the aid of an infusion pump. After the addition was completed, the mixture was stirred at room temperature overnight. The mixture was filtered and removal of solvent in vacuo gave an oil which was chromatographed using silica and CH2Cl2 and methanol (10%) to yield a white solid (0.27 g,15%), m.p. 282-289 οC (decomposition); IR (KBr): υmax1,684, 1,586, 1,494, 1,403, 1,273, 1,134, 1,102, 952, 853, 739 cm-1; 1H-NMR (CDCl3): δ 3.25-4.75 (m, 24H), 6.94-7.03 (m, 2H), 7.10-7.20 (m, 4H).
Conversion of XI to VIII: To a solution of diamide XI (200 mg, 0.3 mmol) in THF (4 mL) was added borane in THF (1 M, 3 mL), and the mixture heated under reflux for 2 hrs. The reaction mixture was worked up as described above to yield white solid, which showed IR (KBr) and 1H-NMR data similar to those of VIII reported above.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





