Protecting Groups in Carbohydrate Chemistry: Influence on Stereoselectivity of Glycosylations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
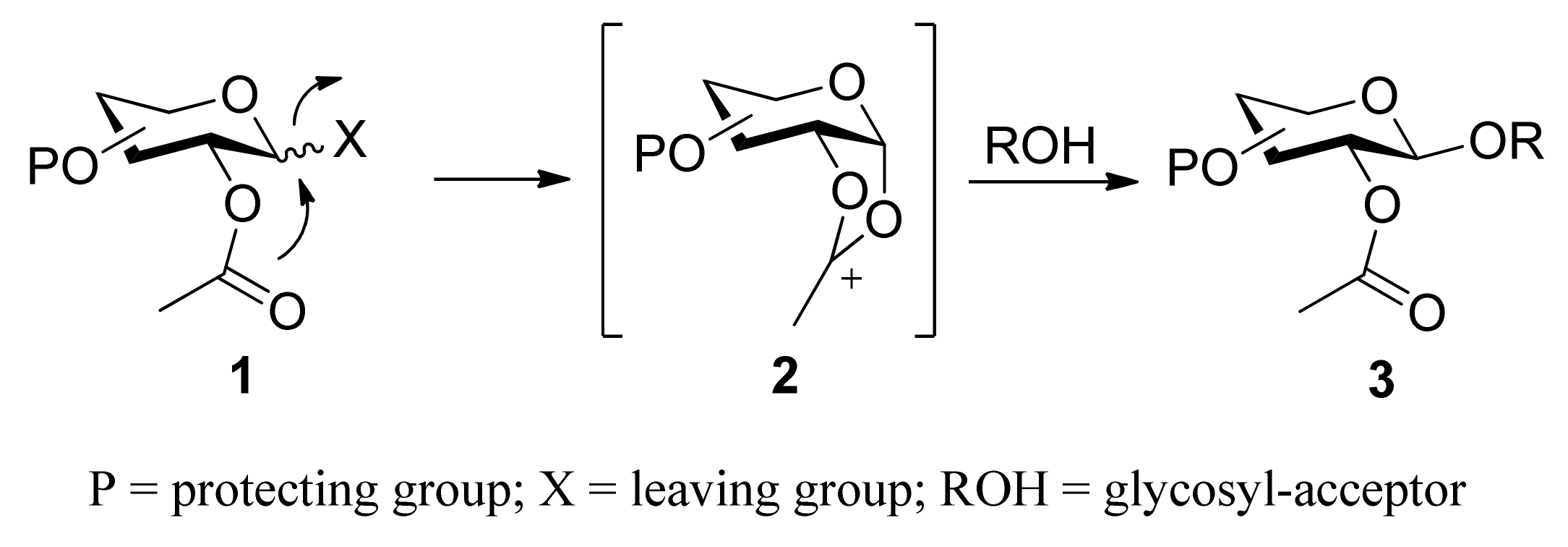{kind=link}
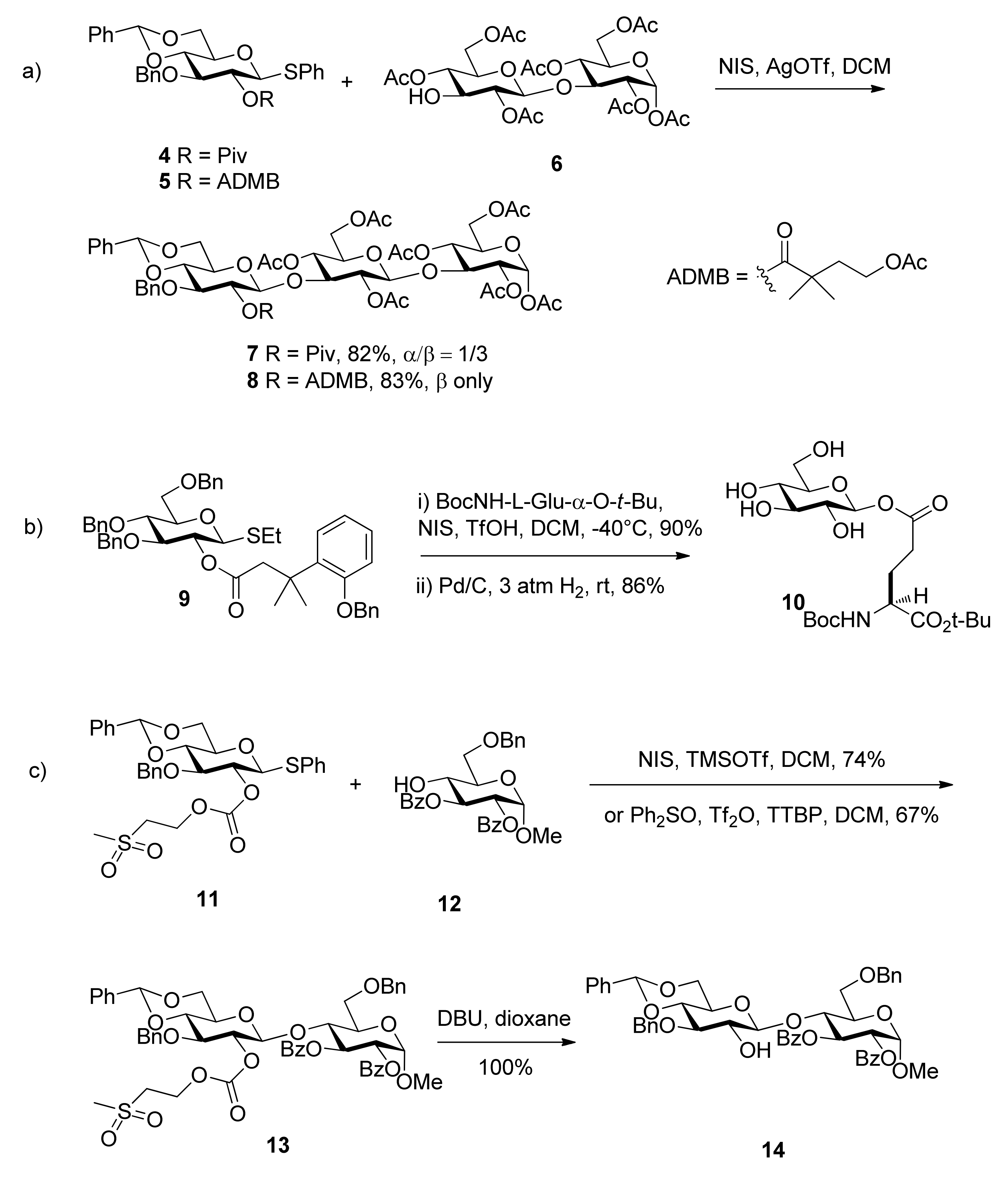{kind=link}
{kind=link}
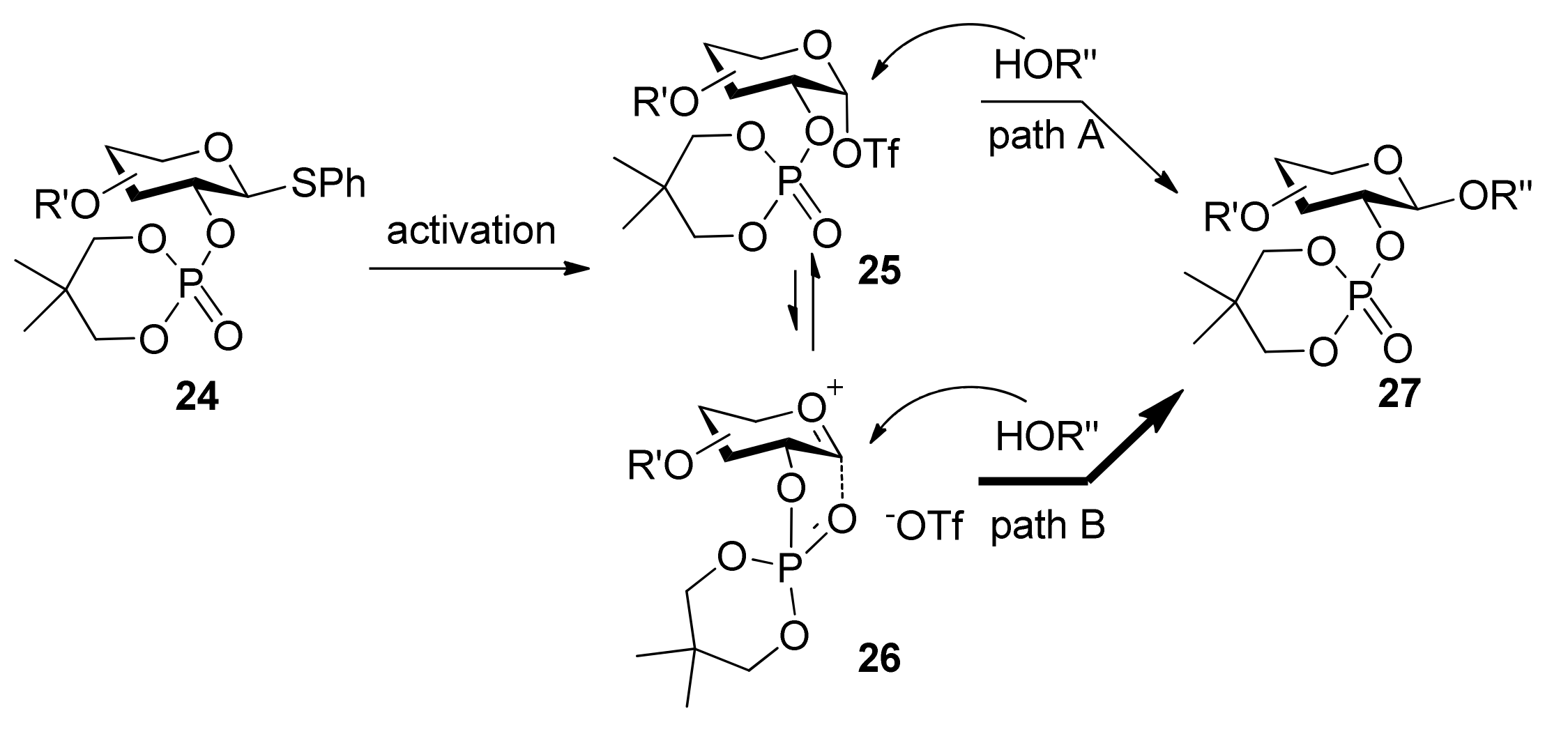{kind=link}
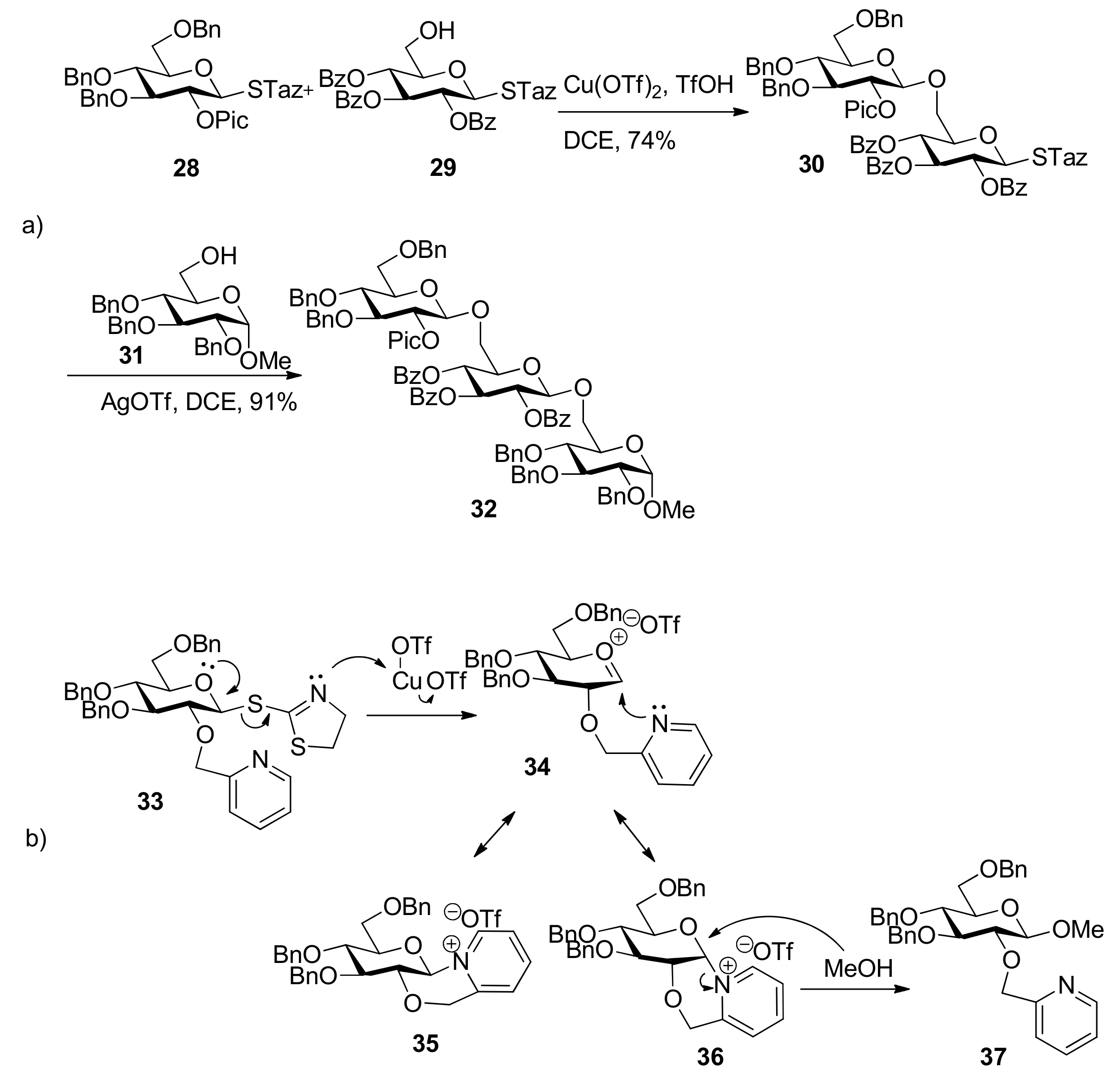{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
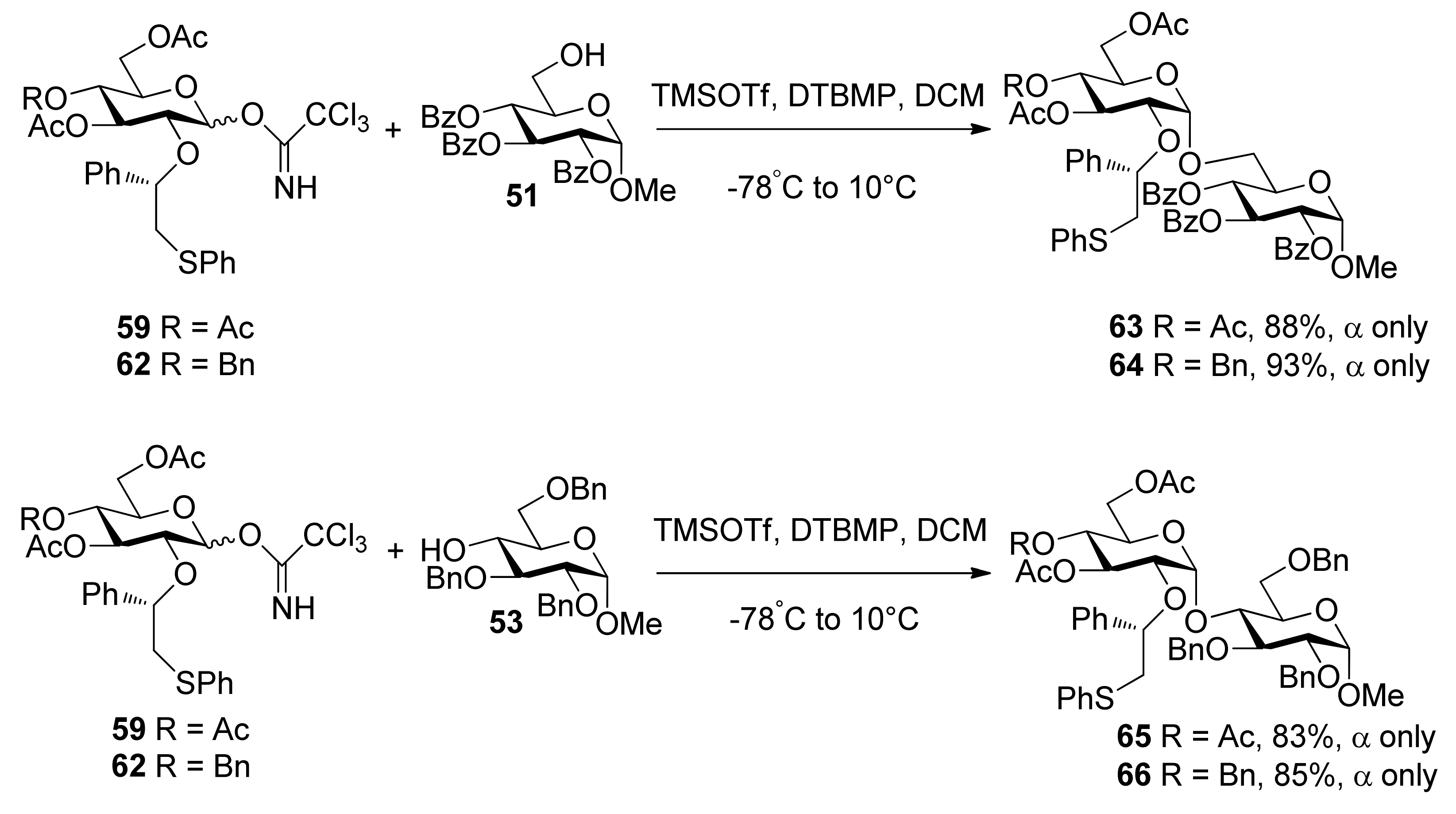{kind=link}
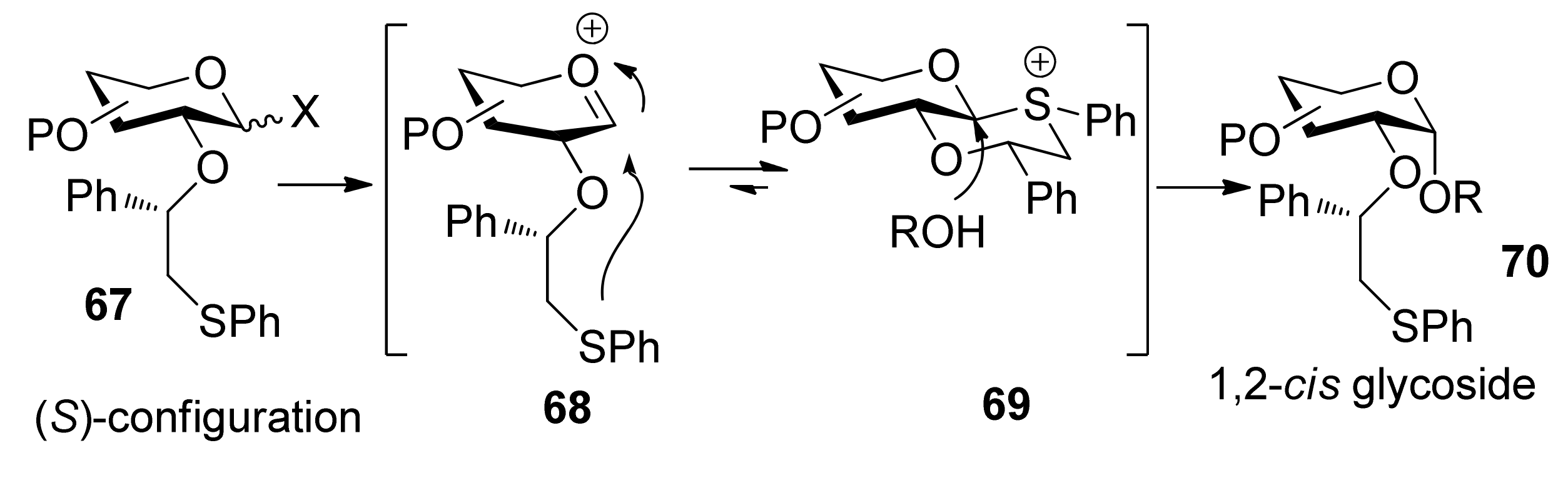{kind=link}
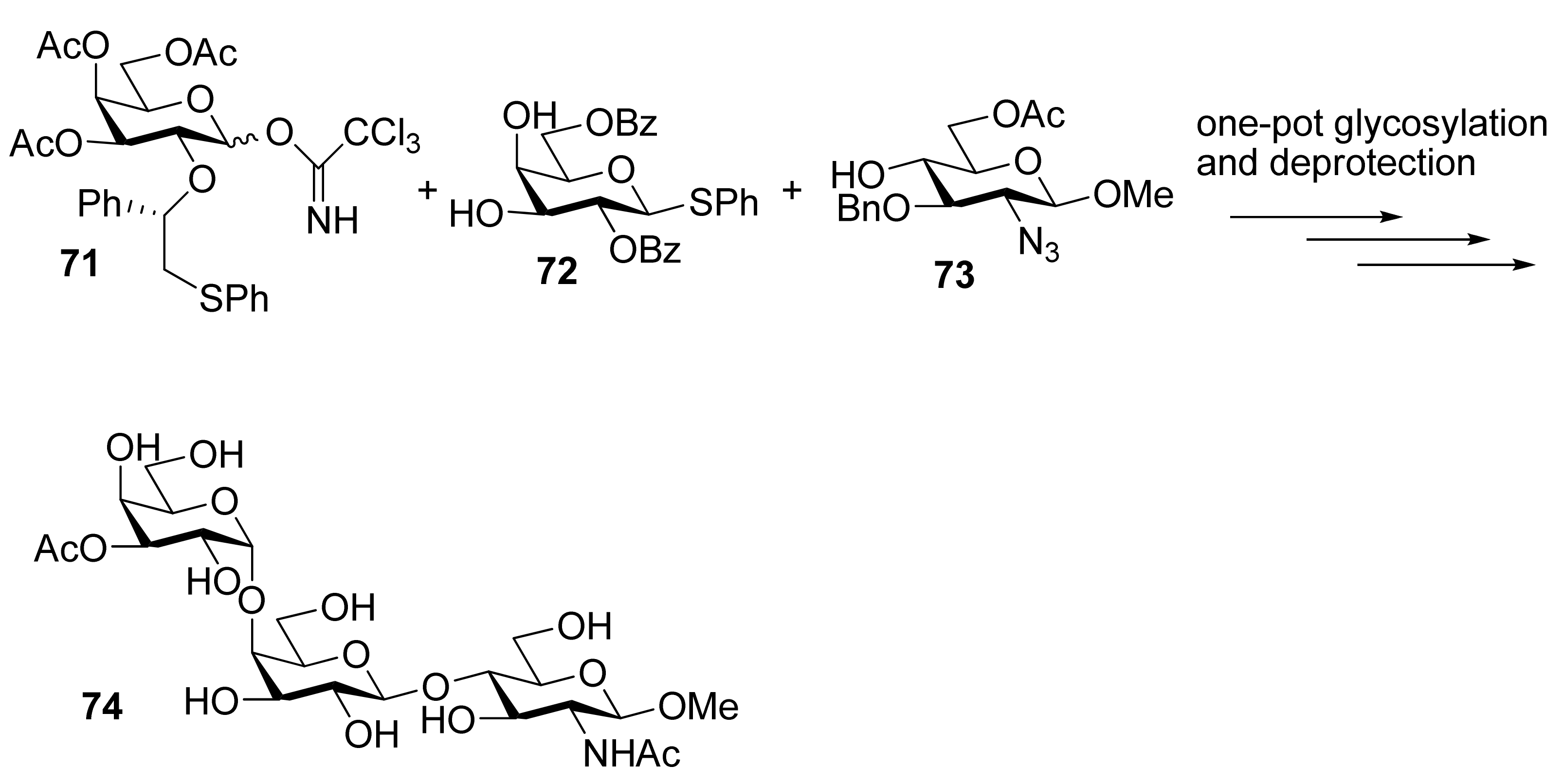{kind=link}
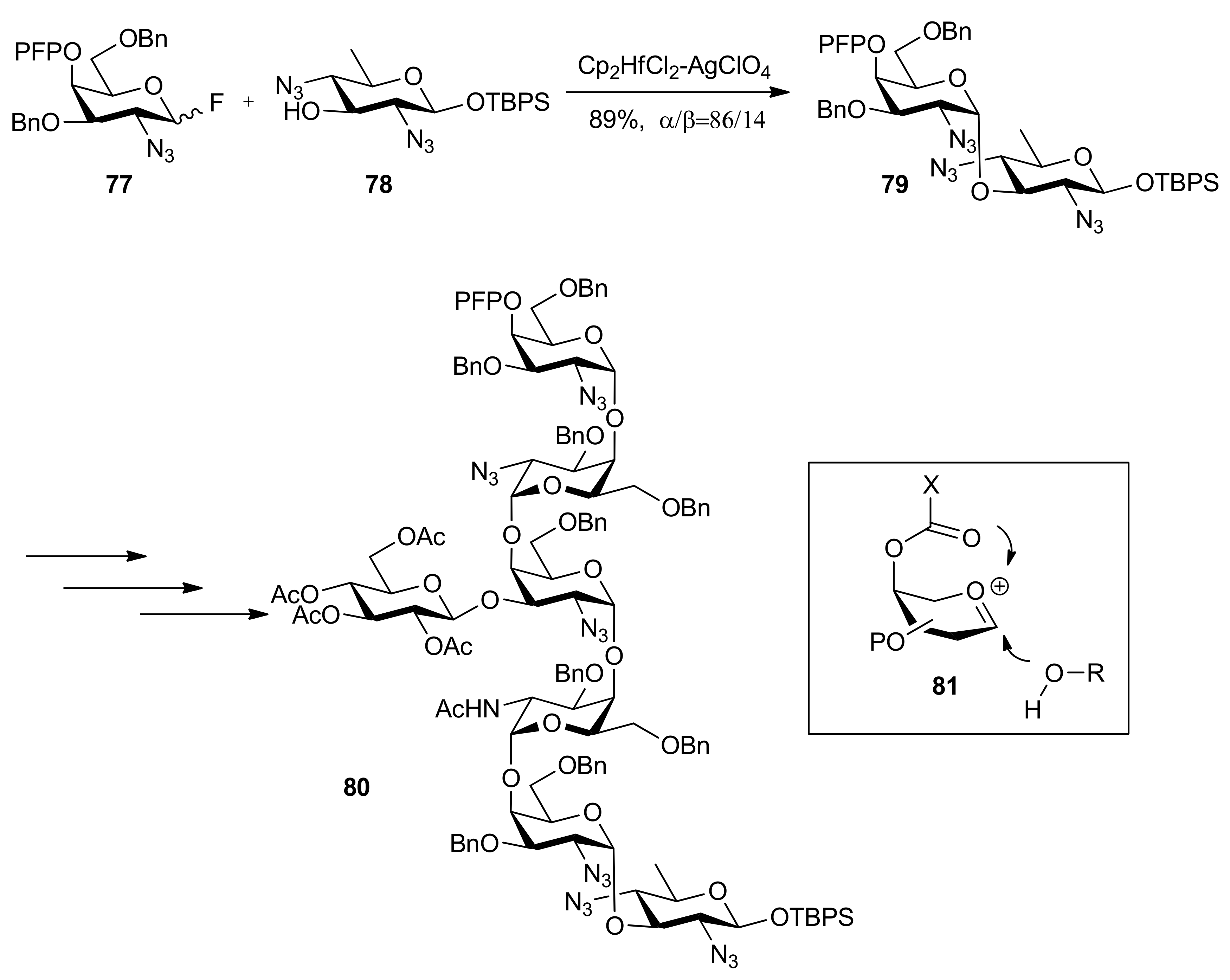{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
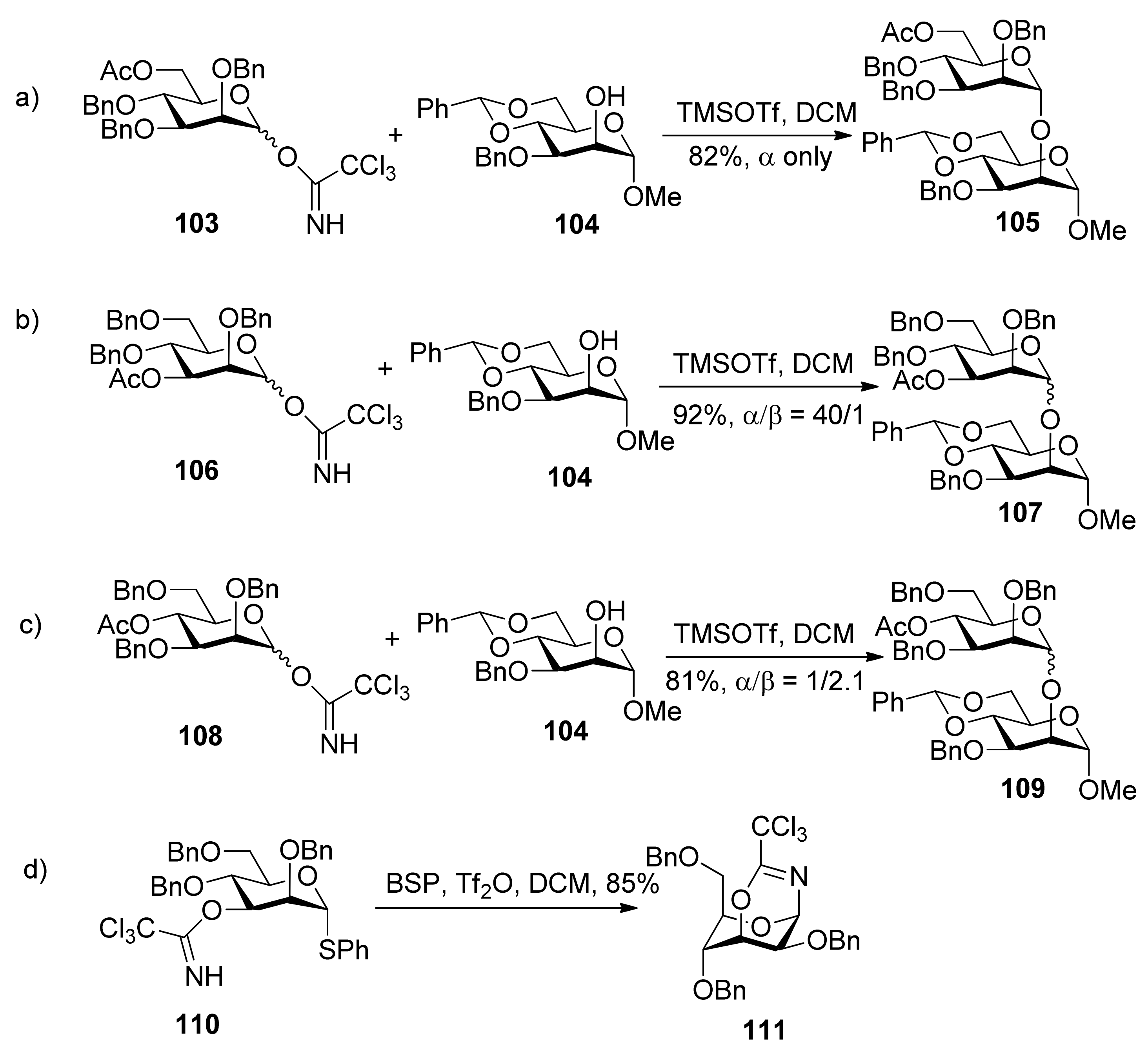{kind=link}
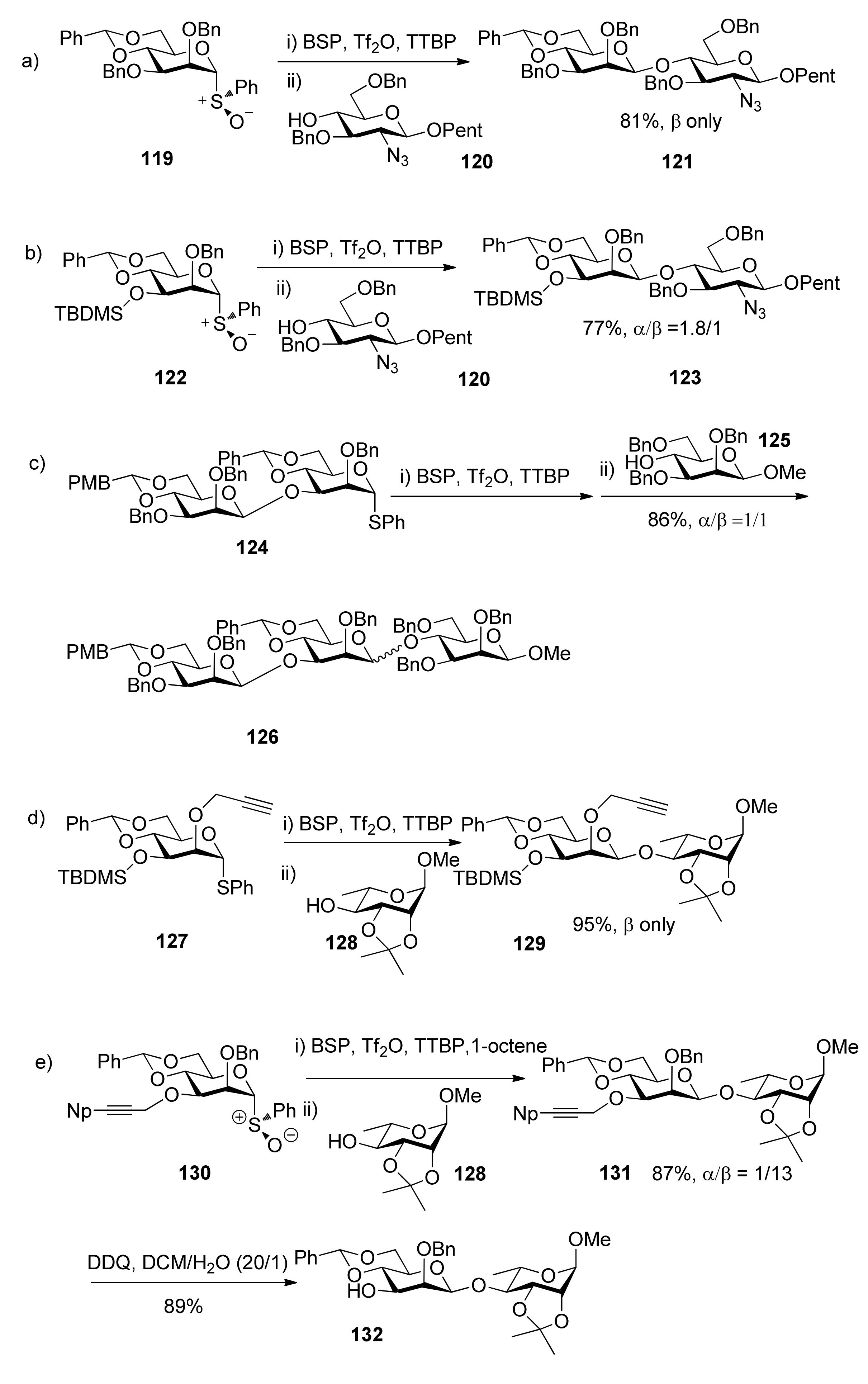{kind=link}
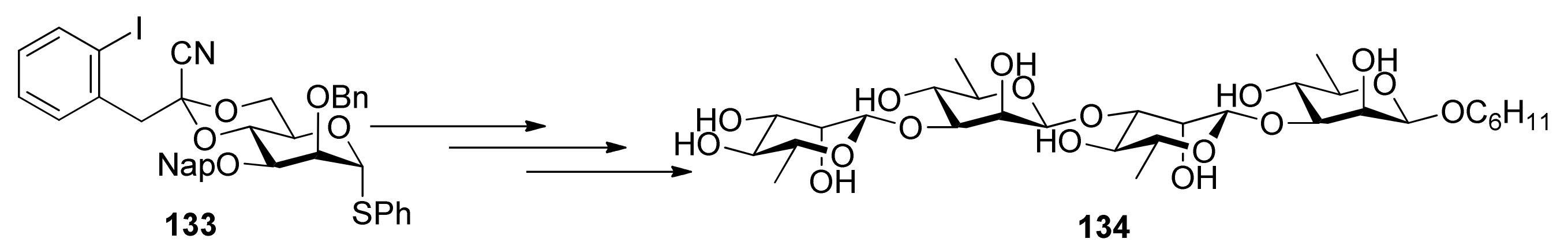{kind=link}
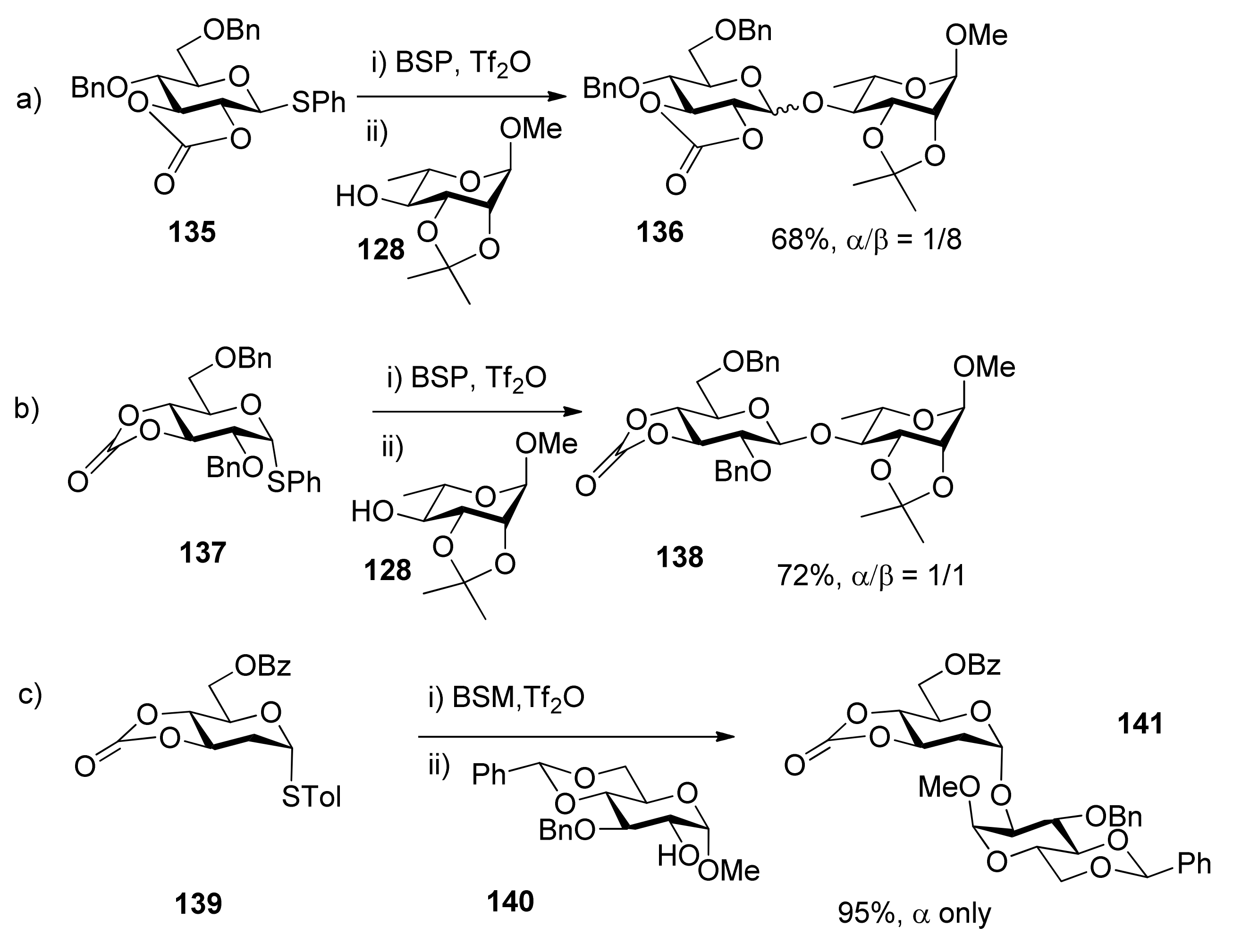{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| Ac | acetyl |
| ADMB | 4-acetoxy-2,2-dimethylbutanoyl |
| Bac | bacillosamine |
| Bn | benzyl |
| BSP | 1-benzenesulfinylpiperidine |
| Boc | tert-butoxycarbonyl |
| Bz | benzoyl |
| DCM | dichloromethane |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DMBPP | 3-(2-hydroxyphenyl)-3,3-dimethylpropanoate |
| DMTM | 2,2-dimethyltrimethylene |
| DTBMP | 2,6-di-tert-butyl-4-methylpyridine |
| DTBS | di-tert-butylsilylene |
| FPsc | fluorous propylsulfonylethoxycarbonyl |
| Hex | hexyl |
| MP | p-methoxyphenyl |
| Msc | methylsulfonylethoxycarbonyl |
| NAP | 2-napthylmethyl |
| NIS | N-iodosuccinimide |
| Pent | n-pentenol |
| Phth | phthalimido |
| Pic | 2-Pyridylmethyl |
| PMB | p-methoxybenzyl |
| STaz | S-thiazolyl |
| TIPDS | 3,5-O-tetraisopropyldisiloxanylidene |
| TBDMS | tert-butyldimethylsilyl |
| TCP | N-tetrachlorophthalimido |
| TMBPP | 3-(2-hydroxy-4,6-dimethylphenyl)-3,3-dimethylpropanoate |
| Tf | trifluoromethanesulfonyl |
| TMS | trimethylsilyl |
| Tol | toluene |
| Troc | 2,2,2-trichloroethoxycarbonyl |
| TTBP | 2,4,6-tri-tert-butylpyrimidine |
1. Introduction
2. Participating-Type Protecting Groups

2.1. Improved ester groups

2.2. Dialkyl phosphates


2.3. 2-Pyridylmethyl group

2.4. Chiral auxiliary groups








2.5. Remote participation groups







3. Conformation-Constraining Protecting Groups
3.1. Benzylidene group



3.2. Carbonate and oxazolidinone groups






3.3. Cyclic silyl groups





3.4. Other conformation-constraining protecting groups



4. Conclusions
Acknowledgements
References
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef]
- Murrey, H.E.; Hsieh-Wilson, L.C. The chemical neurobiology of carbohydrates. Chem. Rev. 2008, 108, 1708–1731. [Google Scholar] [CrossRef]
- Walker-Nasir, E.; Kaleem, A.; Hoessli, D.C.; Khurshid, A.; Nasir-ud-Din. Galactose: a specifically recognized, terminal carbohydrate moiety in biological processes. Curr. Org. Chem. 2008, 12, 940–956. [Google Scholar] [CrossRef]
- Chen, S.; Fukuda, M. Cell type-specific roles of carbohydrates in tumor metastasis. Methods Enzymol. 2006, 416, 371–380. [Google Scholar] [CrossRef]
- Paulsen, H. Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem. Int. Ed. 1982, 21, 155–173. [Google Scholar] [CrossRef]
- Sears, P.; Wong, C.H. Toward automated synthesis of oligosaccharides and glycoproteins. Science 2001, 291, 2344–2350. [Google Scholar] [CrossRef]
- Wang, Y.H.; Ye, X.S.; Zhang, L.H. Oligosaccharide assembly by one-pot multi-step strategy. Org. Biomol. Chem. 2007, 5, 2189–2200. [Google Scholar] [CrossRef]
- Dhanawat, M.; Shrivastava, S.K. Solid-phase synthesis of oligosaccharide drugs: a review. Mini-Rev. Med. Chem. 2009, 9, 169–185. [Google Scholar] [CrossRef]
- Smoot, J.T.; Demchenko, A.V. Oligosaccharide synthesis: from conventional methods to modern expeditious strategies. Adv. Carbohydr. Chem. Biochem. 2009, 62, 161–250. [Google Scholar] [CrossRef]
- Castagner, B.; Seeberger, P.H. Automated solid phase oligosaccharide synthesis. In Combinatorial Chemistry on Solid Supports; Braese, S., Ed.; Springer-Verlag: Berlin, Germany, 2007; Volume 278, pp. 289–309. [Google Scholar]
- Ojeda, R.; Terenti, O.; de Paz, J.L.; Martin-Lomas, M. Synthesis of heparin-like oligosaccharides on polymer supports. Glycoconjugate J. 2004, 21, 179–195. [Google Scholar] [CrossRef]
- Boltje, T.J.; Buskas, T.; Boons, G.J. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat. Chem. 2009, 1, 611–622. [Google Scholar] [CrossRef]
- Zhu, X.M.; Schmidt, R.R. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Rashed, N.; Ibrahim, E.S.I. Strategies of synthetic methodologies for constructing β-mannosidic linkage. Curr. Org. Synth. 2005, 2, 175–213. [Google Scholar] [CrossRef]
- Bongat, A.; Demchenko, A.V. Recent trends in the synthesis of O-glycosides of 2-amino-2-deoxysugars. Carbohydr. Res. 2007, 342, 374–406. [Google Scholar] [CrossRef]
- De Meo, C.; Priyadarshani, U. C-5 modifications in N-acetyl-neuraminic acid: scope and limitations. Carbohydr. Res. 2008, 343, 1540–1552. [Google Scholar] [CrossRef]
- Ress, D.K.; Linhardt, R.J. Sialic acid donors: Chemical synthesis and glycosylation. Curr. Org. Synth. 2004, 1, 31–46. [Google Scholar] [CrossRef]
- Liang, F.F.; Chen, L.; Xing, G.W. Recent advances in sialylation. Chin. J. Org. Chem. 2009, 29, 1317–1324. [Google Scholar]
- Boons, G.J.; Demchenko, A.V. Recent advances in O-sialylation. Chem. Rev. 2000, 100, 4539–4566. [Google Scholar] [CrossRef]
- Zeng, Y.; Ning, J.; Kong, F. Remote control of α- or β-stereoselectivity in (1→3)-glucosylations in the presence of a C-2 ester capable of neighboring-group participation. Carbohydr. Res. 2003, 338, 307–311. [Google Scholar] [CrossRef]
- Zeng, Y.; Ning, J.; Kong, F. Pure α-linked products can be obtained in high yields in glycosylation with glucosyl trichloroacetimidate donors with a C2 ester capable of neighboring group participation. Tetrahedron Lett. 2002, 43, 3729–3733. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Eckhardt, M.; Gutteridge, C.E.; Danishefsky, S.J. Coupling of glycal derived thioethyl glycosyl donors with glycal acceptors. An advance in the scope of the glycal assembly. J. Am. Chem. Soc. 1997, 119, 10064–10072. [Google Scholar] [CrossRef]
- Yu, H.; Williams, D.L.; Ensley, H.E. 4-Acetoxy-2,2-dimethylbutanoate: a useful carbohydrate protecting group for the selective formation of β-(1→3)-D-glucans. Tetrahedron Lett. 2005, 46, 3417–3421. [Google Scholar] [CrossRef]
- Crich, D.; Cai, F. Stereocontrolled glycoside and glycosyl ester synthesis. Neighboring group participation and hydrogenolysis of 3-(2’-Benzyloxyphenyl)-3,3-dimethylpropanoates. Org. Lett. 2007, 9, 1613–1615. [Google Scholar] [CrossRef]
- Ali, A.; van den Berg, R.; Overkleeft, H.S.; Filippov, D.V.; van der Marel, G.A.; Codee, J. Methylsulfonylethoxycarbonyl (Msc) and fluorous propylsulfonylethoxycarbonyl (Fpsc) as hydroxy-protecting groups in carbohydrate chemistry. Tetrahedron Lett. 2009, 50, 2185–2188. [Google Scholar] [CrossRef]
- Yamada, T.; Takemura, K.; Yoshida, J.; Yamago, S. Dialkylphosphates as stereodirecting protecting groups in oligosaccharide synthesis. Angew. Chem. Int. Ed. 2006, 45, 7575–7578. [Google Scholar] [CrossRef]
- Crich, D.; Sun, S. Are glycosyl triflates intermediates in the sulfoxide glycosylation method? A chemical and 1H, 13C, and 19F NMR spectroscopic investigation. J. Am. Chem. Soc. 1997, 119, 11217–11223. [Google Scholar] [CrossRef]
- Mootoo, D.R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. Armed and disarmed n-pentenyl glycosides in saccharide couplings leading to oligosaccharides. J. Am. Chem. Soc. 1988, 110, 5583–5584. [Google Scholar]
- Fraser-Reid, B.; Udodong, U.E.; Wu, Z.; Ottosson, H.; Merritt, J.R.; Rao, C.S.; Roberts, C.; Madsen, R. n-Pentenyl glycosides in organic chemistry: a contemporary example of serendipity. Synlett 1992, 927–942. [Google Scholar]
- Smoot, J.T.; Pornsuriyasak, P.; Demchenko, A.V. Development of an arming participating group for stereoselective glycosylation and chemoselective oligosaccharide synthesis. Angew. Chem. Int. Ed. 2005, 44, 7123–7126. [Google Scholar] [CrossRef]
- Kim, J.H.; Yang, H.; Boons, G.J. Stereoselective glycosylation reactions with chiral auxiliaries. Angew. Chem. Int. Ed. 2005, 44, 947–949. [Google Scholar]
- Kim, J.H.; Yang, H.; Boons, G.J. Stereoselective glycosylations using chiral auxiliaries. In Frontiers in Modern Carbohydrate Chemistry; Demchenko, A.V., Ed.; American Chemical Society: Washington, DC, USA, 2007; Volume 960, pp. 73–90. [Google Scholar]
- Kim, J.; Yang, H.; Park, J.; Boons, G. A general strategy for stereoselective glycosylations. J. Am. Chem. Soc. 2005, 127, 12090–12097. [Google Scholar]
- Galili, U.; Macher, B.A.; Buehler, J.; Shohet, S.B. Human natural anti-α-galactosyl IgG. II. The specific recognition of α-(1→3)-linked galactose residues. J. Exp. Med. 1985, 162, 573–582. [Google Scholar] [CrossRef]
- Boltje, T.J.; Kim, J.; Park, J.; Boons, G.J. Chiral-auxiliary-mediated 1,2-cis-glycosylations for the solid-supported synthesis of a biologically important branched α-glucan. Nat. Chem. 2010, 2, 552–557. [Google Scholar] [CrossRef]
- Zhao, C.; Li, M.; Luo, Y.; Wu, W. Isolation and structural characterization of an immune-stimulating polysaccharide from fuzi, Aconitum carmichaeli. Carbohydr. Res. 2006, 341, 485–491. [Google Scholar] [CrossRef]
- Amin, M.N.; Ishiwata, A.; Ito, Y. Synthesis of N-linked glycan derived from Gram-negative bacterium, Campylobacter jejuni. Tetrahedron 2007, 63, 8181–8198. [Google Scholar] [CrossRef]
- Tokimoto, H.; Fujimoto, Y.; Fukase, K.; Kusumotot, S. Stereoselective glycosylation using the long-range effect of a [2-(4-phenylbenzyl)oxycarbonyl]benzoyl group. Tetrahedron: Asymmetry 2005, 16, 441–447. [Google Scholar] [CrossRef]
- De Meo, C.; Kamat, M.N.; Demchenko, A.V. Remote participation-assisted synthesis of β-mannosides. Eur. J. Org. Chem. 2005, 706–711. [Google Scholar]
- Demchenko, A.V.; Rousson, E.; Boons, G.J. Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar]
- Mukaiyama, T.; Ishikawa, T.; Uchiro, H. Highly stereoselective synthesis of 2’-deoxy-α-ribonucleosides and 2-deoxy-α-C-ribofuranosides by remote stereocontrolled glycosylation. Chem. Lett. 1997, 26, 389–390. [Google Scholar]
- Demchenko, A.V.; Rousson, E.; Boons, G.-J. Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar]
- Takatani, M.; Matsuo, I.; Ito, Y. Pentafluoropropionyl and trifluoroacetyl groups for temporary hydroxyl group protection in oligomannoside synthesis. Carbohydr. Res. 2003, 338, 1073–1081. [Google Scholar] [CrossRef]
- Park, J.; Boltje, T.J.; Boons, G.J. Direct and stereoselective synthesis of α-linked 2-deoxyglycosides. Org. Lett. 2008, 10, 4367–4370. [Google Scholar] [CrossRef]
- Crich, D.; Yao, Q.L. Benzylidene acetal fragmentation route to 6-deoxy sugars: Direct reductive cleavage in the presence of ether protecting groups, permitting the efficient, highly stereocontrolled synthesis of β-D-rhamnosides from D-mannosyl glycosyl donors. Total synthesis of α-D-Gal-(1→3)-α-D-Rha-(1→3)-β-D-Rha-(1→4)-β-D-Glu-OMe, the repeating unit of the antigenic lipopolysaccharide from Escherichia hermannii ATCC 33650 and 33652. J. Am. Chem. Soc. 2004, 126, 8232–8236. [Google Scholar] [CrossRef]
- Crich, D.; Li, H. Synthesis of the salmonella type E1 core trisaccharide as a probe for the generality of 1-(benzenesulfinyl)piperidine/triflic anhydride combination for glycosidic bond formation from thioglycosides. J. Org. Chem. 2002, 67, 4640–4646. [Google Scholar] [CrossRef]
- Crich, D.; Sun, S. Direct synthesis of β-mannopyranosides by the sulfoxide method. J. Org. Chem. 1997, 62, 1198–1199. [Google Scholar] [CrossRef]
- Crich, D.; Sun, S. Formation of β-mannopyranosides of primary alcohols using the sulfoxide method. J. Org. Chem. 1996, 61, 4506–4507. [Google Scholar] [CrossRef]
- Crich, D.; Cai, W.L.; Dai, Z.M. Highly diastereoselective α-mannopyranosylation in the absence of participating protecting groups. J. Org. Chem. 2000, 65, 1291–1297. [Google Scholar] [CrossRef]
- Crich, D.; Xu, H.D. Direct stereocontrolled synthesis of 3-amino-3-deoxy-β-mannopyranosides: Importance of the nitrogen protecting group on stereoselectivity. J. Org. Chem. 2007, 72, 5183–5192. [Google Scholar] [CrossRef]
- Crich, D.; Hu, T.S.; Cai, F. Does neighboring group participation by non-vicinal esters play a role in glycosylation reactions? Effective probes for the detection of bridging intermediates. J. Org. Chem. 2008, 73, 8942–8953. [Google Scholar] [CrossRef]
- Baek, J.Y.; Lee, B.; Jo, M.G.; Kim, K.S. Directing effect of electron-withdrawing groups at O-3,O-4, and O-6 positions and α-directing effect by remote participation of 3-O-acyl and 6-O-acetyl groups of donors in Mannopyranosylations. J. Am. Chem. Soc. 2009, 131, 17705–17713. [Google Scholar] [CrossRef]
- Litjens, R.; van den Bos, L.J.; Codee, J.; Overkleeft, H.S.; van der Marel, G.A. The use of cyclic bifunctional protecting groups in oligosaccharide synthesis - an overview. Carbohydr. Res. 2007, 342, 419–429. [Google Scholar] [CrossRef]
- Crich, D.; Chandrasekera, N.S. Mechanism of 4,6-O-benzylidene-directed β-mannosylation as determined by α-deuterium kinetic isotope effects. Angew. Chem. Int. Ed. 2004, 43, 5386–5389. [Google Scholar] [CrossRef]
- Jensen, H.H.; Nordstrøm, L.U.; Bols, M. The disarming effect of the 4,6-acetal group on glycoside reactivity: torsional or electronic? J. Am. Chem. Soc. 2004, 126, 9205–9213. [Google Scholar] [CrossRef]
- Huang, X.; Huang, L.; Wang, H.; Ye, X.S. Iterative one-pot synthesis of oligosaccharides. Angew. Chem. Int. Ed. 2004, 43, 5221–5224. [Google Scholar] [CrossRef]
- Crich, D. Mechanism of a chemical glycosylation reaction. Acc. Chem. Res. 2010, 43, 1144–1153. [Google Scholar] [CrossRef]
- Crich, D.; Wu, B. 1-Naphthylpropargyl ether group: A readily cleaved and sterically minimal protecting system for stereoselective glycosylation. Org. Lett. 2006, 8, 4879–4882. [Google Scholar] [CrossRef]
- Crich, D.; Jayalath, P.; Hutton, T.K. Enhanced diastereoselectivity in β-mannopyranosylation through the use of sterically minimal propargyl ether protecting groups. J. Org. Chem. 2006, 71, 3064–3070. [Google Scholar] [CrossRef]
- Codée, J.D.C.; Hossain, L.H.; Seeberger, P.H. Efficient installation of β-mannosides using a dehydrative coupling strategy. Org. Lett. 2005, 7, 3251–3254. [Google Scholar] [CrossRef]
- El-Badri, M.H.; Willenbring, D.; Tantillo, D.J.; Gervay-Hague, J. Mechanistic studies on the stereoselective formation of β-mannosides from mannosyl iodides using α-deuterium kinetic isotope effects. J. Org. Chem. 2007, 72, 4663–4672. [Google Scholar] [CrossRef]
- Tsuda, T.; Arihara, R.; Sato, S.; Koshiba, M.; Nakamura, S.; Hashimoto, S. Direct and stereoselective synthesis of β-D-mannosides using 4,6-O-benzylidene-protected mannosyl diethyl phosphite as a donor. Tetrahedron 2005, 61, 10719–10733. [Google Scholar] [CrossRef]
- Baek, J.Y.; Choi, T.J.; Jeon, H.B.; Kim, K.S. A highly reactive and stereoselective β-manno-pyranosylation system: mannosyl 4-pentenoate/PhSeOTf. Angew. Chem. Int. Ed. 2006, 45, 7436–7440. [Google Scholar] [CrossRef]
- Crich, D.; Dudkin, V. An unusual example of steric buttressing in glycosylation. Tetrahedron Lett. 2000, 41, 5643–5646. [Google Scholar] [CrossRef]
- Crich, D.; Jayalath, P. 2-O-Propargyl Ethers: Readily cleavable, minimally intrusive protecting groups for β-mannosyl donors. Org. Lett. 2005, 7, 2277–2280. [Google Scholar] [CrossRef]
- Crich, D.; Karatholuvhu, M.S. Application of the 4-trifluoromethylbenzenepropargyl ether group as an unhindered, electron deficient protecting group for stereoselective glycosylation. J. Org. Chem. 2008, 73, 5173–5176. [Google Scholar] [CrossRef]
- Crich, D.; Bowers, A.A. Synthesis of a β-(1→3)-D-rhamnotetraose by a one-pot, multiple radical fragmentation. Org. Lett. 2006, 8, 4327–4330. [Google Scholar] [CrossRef]
- Crich, D.; Yao, Q.J. The 4,6-O-[α-(2-(2-iodophenyl)ethylthiocarbonyl)benzylidene] protecting group: stereoselective glycosylation, reductive radical fragmentation, and synthesis of β-D-rhamnopyranosides and other deoxy sugars. Org. Lett. 2003, 5, 2189–2191. [Google Scholar] [CrossRef]
- Crich, D.; Cai, W. Chemistry of 4,6-O-benzylidene-D-glycopyranosyl triflates: contrasting behavior between the gluco and manno series. J. Org. Chem. 1999, 64, 4926–4930. [Google Scholar] [CrossRef]
- Crich, D.; Vinogradova, O. On the Influence of the C2−O2 and C3−O3 bonds in 4,6-O-benzylidene-directed β-mannopyranosylation and α-glucopyranosylation. J. Org. Chem. 2006, 71, 8473–8480. [Google Scholar] [CrossRef]
- Chen, L.; Kong, F. Unusual α-glycosylation with galactosyl donors with a C2 ester capable of neighboring group participation. Tetrahedron Lett. 2003, 44, 3691–3695. [Google Scholar] [CrossRef]
- Crich, D.; Jayalath, P. Stereocontrolled formation of β-glucosides and related linkages in the absence of neighboring group participation: Influence of a trans-fused 2,3-O-carbonate group. J. Org. Chem. 2005, 70, 7252–7259. [Google Scholar] [CrossRef]
- Lu, Y.S.; Li, Q.; Wang, Y.H.; Ye, X.S. Highly direct α-selective glycosylations of 3,4-O-carbonate-protected 2-deoxy- and 2,6-dideoxythioglycosides by preactivation protocol. Org. Lett. 2008, 10, 3445–3448. [Google Scholar] [CrossRef]
- Lu, Y.S.; Li, Q.; Wang, Y.H.; Ye, X.S. Direct α-selective glycosylations of acetyl-protected 2-deoxy- and 2,6-dideoxythioglycosides by preactivation protocol. Synlett 2010, 1519–1524. [Google Scholar]
- Benakli, K.; Zha, C.; Kerns, R.J. Oxazolidinone protected 2-amino-2-deoxy-d-glucose derivatives as versatile intermediates in stereoselective oligosaccharide synthesis and the formation of α-linked glycosides. J. Am. Chem. Soc. 2001, 123, 9461–9462. [Google Scholar] [CrossRef]
- Boysen, M.; Gemma, E.; Lahmann, M.; Oscarson, S. Ethyl 2-acetamido-4,6-di-O-benzyl-2,3-N,O-carbonyl-2-deoxy-1-thio-β-D-glycopyranoside as a versatile GlcNAc donor. Chem. Commun. 2005, 3044–3046. [Google Scholar]
- Kerns, R.J.; Zha, C.X.; Benakli, K.; Liang, Y.Z. Extended applications and potential limitations of ring-fused 2,3-oxazolidinone thioglycosides in glycoconjugate synthesis. Tetrahedron Lett. 2003, 44, 8069–8072. [Google Scholar] [CrossRef]
- Wei, P.; Kerns, R.J. Factors affecting stereocontrol during glycosidation of 2,3-oxazolidinone-protected 1-tolylthio-N-acetyl-D-glucosamine. J. Org. Chem. 2005, 70, 4195–4198. [Google Scholar] [CrossRef]
- Geng, Y.Q.; Zhang, L.H.; Ye, X.S. Pre-activation protocol leading to highly stereoselectivity-controllable glycosylations of oxazolidinone protected glucosamines. Chem. Commun. 2008, 597–599. [Google Scholar]
- Geng, Y.Q.; Zhang, L.H.; Ye, X.S. Stereoselectivity investigation on glycosylation of oxazolidinone protected 2-amino-2-deoxy-D-glucose donors based on pre-activation protocol. Tetrahedron 2008, 64, 4949–4958. [Google Scholar] [CrossRef]
- Olsson, J.D.M.; Eriksson, L.; Lahmann, M.; Oscarson, S. Investigations of glycosylation reactions with 2-N-acetyl-2N,3O-oxazolidinone-protected glucosamine donors. J. Org. Chem. 2008, 73, 7181–7188. [Google Scholar] [CrossRef]
- Yang, L.; Ye, X.S. A highly α-selective glycosylation for the convenient synthesis of repeating α-(1→4)-linked N-acetyl-galactosamine units. Carbohydr. Res. 2010, 345, 1713–1721. [Google Scholar] [CrossRef]
- Manabe, S.; Ishii, K.; Ito, Y. N-Benzyl-2,3-oxazolidinone as a glycosyl donor for selective α-glycosylation and one-pot oligosaccharide synthesis involving 1,2-cis-glycosylation. J. Am. Chem. Soc. 2006, 128, 10666–10667. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishiura, Y.; Takahashi, T. Stereoselective synthesis of oligo-α-(2,8)-sialic acids. J. Am. Chem. Soc. 2006, 128, 7124–7125. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishiura, Y.; Takahashi, T. Stereoselective synthesis of α(2,9) di- to tetrasialic acids, using a 5,4-N,O-carbonyl protected thiosialoside. J. Org. Chem. 2009, 74, 4383–4386. [Google Scholar] [CrossRef]
- Crich, D.; Li, W. O-Sialylation with N-acetyl-5-N,4-O-carbonyl-protected thiosialoside donors in dichloromethane: facile and selective cleavage of the oxazolidinone ring. J. Org. Chem. 2007, 72, 2387–2391. [Google Scholar] [CrossRef]
- Hanashima, S.; Sato, K.; Ito, Y.; Yamaguchi, Y. Silylene/oxazolidinone double-locked sialic acid building blocks for efficient sialylation reactions in dichloromethane. Eur. J. Org. Chem. 2009, 4215–4220. [Google Scholar]
- Ando, H.; Koike, Y.; Ishida, H.; Kiso, M. Extending the possibility of an N-Troc-protected sialic acid donor toward variant sialo-glycoside synthesis. Tetrahedron Lett. 2003, 44, 6883–6886. [Google Scholar] [CrossRef]
- Adachi, M.; Tanaka, H.; Takahashi, T. An effective sialylation method using N-Troc- and N-Fmoc-protected β-thiophenyl sialosides and application to the one-pot two-step synthesis of 2,6-sialyl-T antigen. Synlett 2004, 609–614. [Google Scholar]
- Imamura, A.; Ando, H.; Korogi, S.; Tanabe, G.; Muraoka, O.; Ishida, H.; Kiso, M. Di-tert-butylsilylene (DTBS) group-directed α-selective galactosylation unaffected by C-2 participating functionalities. Tetrahedron Lett. 2003, 44, 6725–6728. [Google Scholar]
- Imamura, A.; Kimura, A.; Ando, H.; Ishida, H.; Kiso, M. Extended applications of di-tert-butylsilylene-directed α-predominant galactosylation compatible with C2-participating groups toward the assembly of various glycosides. Chem. Eur. J. 2006, 12, 8862–8870. [Google Scholar] [CrossRef]
- Miljkovic, M.; Yeagley, D.; Deslongchamps, P.; Dory, Y. Experimental and theoretical evidence of through-space electrostatic stabilization of the incipient oxocarbenium ion by an axially oriented electronegative substituent during glycopyranoside acetolysis. J. Org. Chem. 1997, 62, 7597–7604. [Google Scholar] [CrossRef]
- Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. DTBS (di-tert-butylsilylene)-directed α-galactosylation for the synthesis of biologically relevant glycans. Curr. Org. Chem. 2008, 12, 675–689. [Google Scholar] [CrossRef]
- Komori, T.; Ando, T.; Imamura, A.; Li, Y.T.; Ishida, H.; Kiso, M. Design and efficient synthesis of novel GM2 analogues with respect to the elucidation of the function of GM2 activator. Glycoconjugate J. 2008, 25, 647–661. [Google Scholar] [CrossRef]
- Zhu, X.; Kawatkar, S.; Rao, Y.; Boons, G. Practical approach for the stereoselective introduction of β-arabinofuranosides. J. Am. Chem. Soc. 2006, 128, 11948–11957. [Google Scholar]
- Gadikota, R.R.; Callam, C.S.; Wagner, T.; Del Fraino, B.; Lowary, T.L. 2,3-Anhydro sugars in glycoside bond synthesis. Highly stereoselective syntheses of oligosaccharides containing α- and β-arabinofuranosyl linkages. J. Am. Chem. Soc. 2003, 125, 4155–4165. [Google Scholar] [CrossRef]
- Joe, M.; Bai, Y.; Nacario, R.C.; Lowary, T.L. Synthesis of the docosanasaccharide arabinan domain of mycobacterial arabinogalactan and a proposed octadecasaccharide biosynthetic precursor. J. Am. Chem. Soc. 2007, 129, 9885–9901. [Google Scholar] [CrossRef]
- Ishiwata, A.; Akao, H.; Ito, Y. Stereoselective synthesis of a fragment of mycobacterial arabinan. Org. Lett. 2006, 8, 5525–5528. [Google Scholar] [CrossRef]
- Ishiwata, A.; Akao, H.; Ito, Y.; Sunagawa, M.; Kusunose, N.; Kashiwazaki, Y. Synthesis and TNF-α inducing activities of mycoloyl-arabinan motif of mycobacterial cell wall components. Bioorg. Med. Chem. 2006, 14, 3049–3061. [Google Scholar] [CrossRef]
- Smith, D.M.; Tran, M.B.; Woerpel, K.A. Nucleophilic additions to fused bicyclic five-membered ring oxocarbenium ions: evidence for preferential attack on the inside face. J. Am. Chem. Soc. 2003, 125, 14149–14152. [Google Scholar] [CrossRef]
- Crich, D.; Subramanian, V.; Hutton, T.K. β-Selective glucosylation in the absence of neighboring group participation: influence of the 3,4-O-bisacetal protecting system. Tetrahedron 2007, 63, 5042–5049. [Google Scholar] [CrossRef]
- Imamura, A.; Lowary, T.L. Selective arabinofuranosylation using a 2,3-O-xylylene-protected donor. Org. Lett. 2010, 12, 3686–3689. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, J.; Ye, X.-S. Protecting Groups in Carbohydrate Chemistry: Influence on Stereoselectivity of Glycosylations. Molecules 2010, 15, 7235-7265. https://doi.org/10.3390/molecules15107235
Guo J, Ye X-S. Protecting Groups in Carbohydrate Chemistry: Influence on Stereoselectivity of Glycosylations. Molecules. 2010; 15(10):7235-7265. https://doi.org/10.3390/molecules15107235
Chicago/Turabian StyleGuo, Jian, and Xin-Shan Ye. 2010. "Protecting Groups in Carbohydrate Chemistry: Influence on Stereoselectivity of Glycosylations" Molecules 15, no. 10: 7235-7265. https://doi.org/10.3390/molecules15107235
APA StyleGuo, J., & Ye, X. -S. (2010). Protecting Groups in Carbohydrate Chemistry: Influence on Stereoselectivity of Glycosylations. Molecules, 15(10), 7235-7265. https://doi.org/10.3390/molecules15107235