General
1H-,
13C-, and
31P-NMR spectra were recorded at 500, 126 and 203 MHz, respectively. The chemical shifts were measured from tetramethylsilane for
1H NMR spectra, CDCl
3 (77 ppm) for
13C-NMR spectra and 85% phosphoric acid (0 ppm) for
31P NMR spectra. UV spectra were recorded on a U-2000 spectrometer. Column chromatography was performed with silica gel C-200 and a minipump for a goldfish bowl was conveniently used to attain sufficient pressure for rapid chromatographic separation. HPLC was performed using the following systems. Reversed-phase HPLC was done on a system with a 3D UV detector and a C18 column (4.6 x 150 mm). A linear gradient (0-30%) of solvent I [0.03 M ammonium acetate buffer (pH 7.0)] in solvent II (CH3CN) was used at 30 °C at a rate of 1.0 mL/min for 30 min. Anion-exchange HPLC was done on an apparatus with a 3D UV detector and a FAX column (Waters, 4.6 × 100 mm). A linear gradient (0-60%) of Solvent III [1 M NaCl and 25 mM phosphate buffer, 10% CH3CN (v/v)] in solvent IV [25 mM phosphate buffer, 10% CH3CN (v/v)] was used at 50 °C at a flow rate of 1.0 mL/min for 45 min. ESI mass was performed by use of Mariner
TM (PerSeptive Biosystems Inc.). MALDI-TOF mass was performed by use of Bruker Daltonics [Matrix: 3-hydroxypicolinic acid (100 mg/ml) in H
2O-diammonium hydrogen citrate (100 mg/ml) in H
2O (10:1, v/v)]. Compounds
1 and
21 were purchased from GeneACT, Inc. Compounds
5,
7 [
31],
9 [
40],
14 [26],and
16 [41] were prepared according to the published procedure. Common protected phosphoramidites were purchased from Glen Research Corporation.
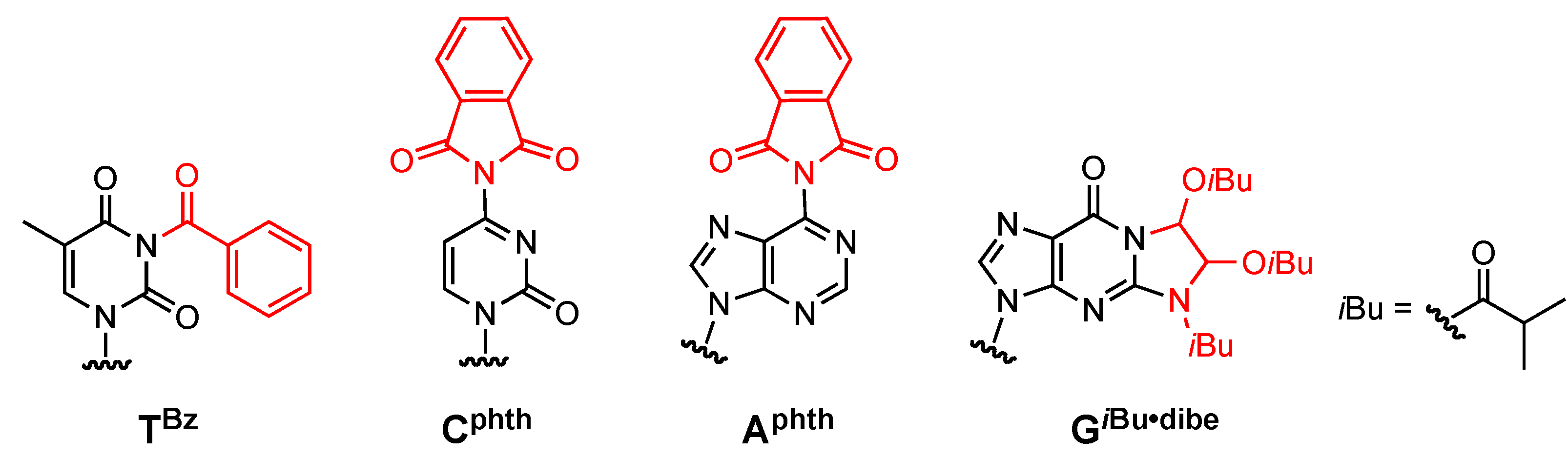
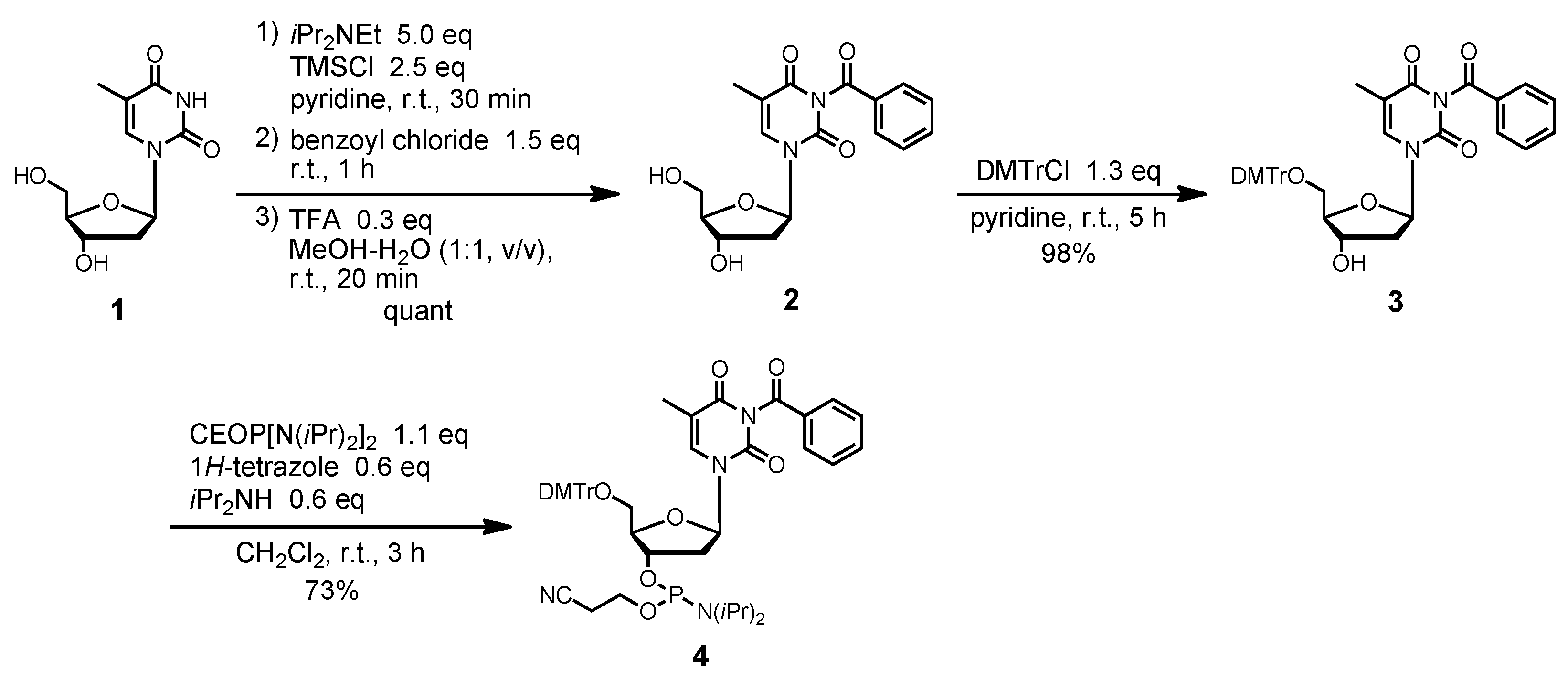
N3-Benzoyl-5´-O-(4, 4'-dimethoxytrityl)thymidine (3). Compound 2 (2.42 g, 7.0 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (3 mL × 1) and dissolved in dry pyridine (70 mL). To the solution was added DMTrCl (3.08 g, 9.1 mmol) and the mixture was stirred at room temperature for 5 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 3. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 3 (4.45 g, 98%). 1H-NMR (CDCl3) δ 1.50 (s, 3H), 2.36–2.42 (m, 2H), 3.40 (dd, 1H, J = 2.2 Hz, J = 9.5 Hz), 3.52 (dd, 1H, J = 2.7 Hz, J = 9.2 Hz), 3.81 (s, 6H), 4.05–4.06 (m, 1H), 4.60 (m, 1H), 6.39 (t, 1H, J = 6.6 Hz), 6.85–6.87 (m, 4H), 7.27–7.32 (m, 7H), 7.41 (d, 2H, J = 7.6 Hz), 7.49 (t, 2H, J = 7.6 Hz), 7.64 (t, 1H, J = 7.6 Hz), 7.70 (s, 1H), 7.94 (d, 1H, J = 7.6 Hz); 13C-NMR (CDCl3) δ 12.1, 41.4, 55.5, 63.7, 72.7, 85.2, 86.4, 87.3, 111.5, 113,57, 113.59, 127.5, 128.30, 128.37, 129.3, 130.3, 130.7, 131.9, 135.2, 135.50, 135.57, 135.63, 144.5, 149.5, 159.0, 163.1, 169.3. HRMS (ESI) m/z (M+Na)+: calcd for C38H36N2NaO8+ 671.2364; found, 671.2364.
N3-Benzoyl-5´-O-(4,4'-dimethoxytrityl)thymidine 3'-(2-cyanoethyl N,N-diisopropylphosphor-amidite) (4). Compound 3 (380 mg, 0.59 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1) and dissolved in dry CH2Cl2 (6 mL). To the solution was added bis(diisopropylamino)(2-cyanoethoxy)phosphine (205 g, 0.65 mmol), 1H-tetrazole (25 mg, 0.35 mmol), diisopropylamine (50 μL, 0.35 mmol) and the mixture was stirred at room temperature for 3 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 4. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 4 (338 mg, 73%). 1H-NMR (CDCl3) δ 1.07 (d, 3H, J = 6.5 Hz), 1.16 (d, 9H, J = 5.6 Hz), 1.48 (s, 3H), 2.39 (t, 1H, J = 6.0 Hz), 2.48 (m, 1H), 2.55 (t, 1H, J = 6.0 Hz), 2.61 (m, 1H), 3.40 (t, 1H, J = 10.1 Hz), 3.54–3.71 (m, 5H), 3.76 (s, 6H), 4.18, 4.24 (2s, 1H), 4.75 (m, 1H), 6.42–6.45 (m, 1H), 6.86–6.89 (m, 4H), 7.24 (m, 1H), 7.32–7.37 (m, 6H), 7.43–7.48 (m, 4H), 7.59 (t, 1H, J = 7.3 Hz), 7.79, 7.84 (2s, 1H), 7.94 (d, 1H, J = 7.3 Hz); 13C-NMR (CDCl3) δ 12.1, 12.1, 20.4, 20.5, 20.6, 20.6, 24.8, 40.5, 43.4, 43.5, 43.5, 43.6, 55.5, 55.5, 55.5, 58.3, 58.4, 58.6, 63.3, 63.4, 73.7, 74.0, 85.2, 85.8, 86.1, 87.3, 111.4, 111.4, 113.5, 113.6, 117.9, 118.1, 127.5, 128.3, 128.5, 128.5, 129.4, 130.4, 130.7, 131.9, 135.3, 135.5, 135.5, 135.6, 135.6, 135.9, 144.6, 144.6, 149.6, 149.7, 159.1, 163.1, 169.5; 31P-NMR (CDCl3) δ 149.5, 149.9 (2s). HRMS (ESI) m/z (M+Na)+: calcd for C47H53N4NaO9P+ 871.3442; found, 871.3441.
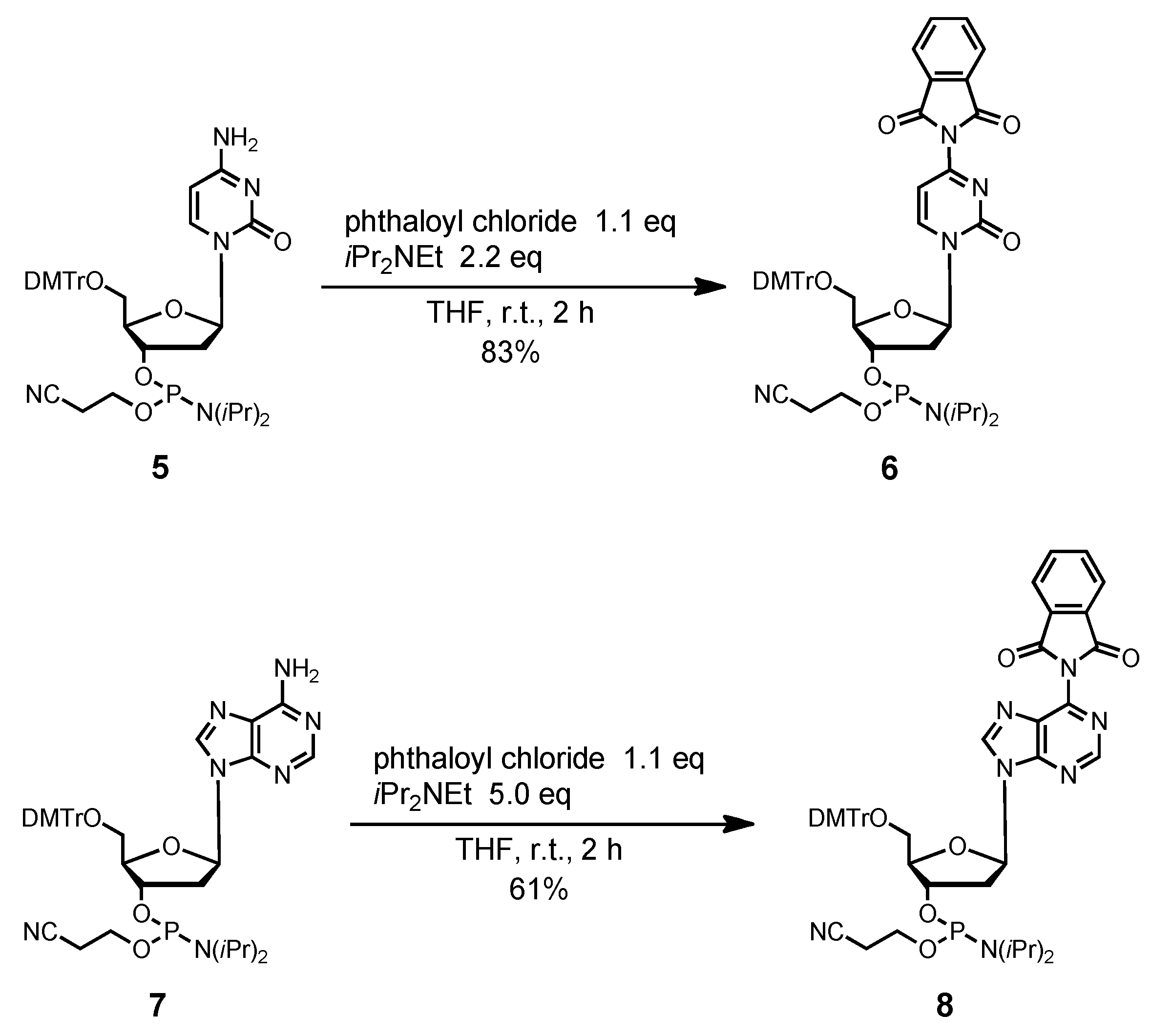
4-N-Phthaloyl-5´-O-(4,4'-dimethoxytrityl)-2´-deoxycytidine 3'-(2-cyanoethyl N,N-diisopropylphos-phoramidite) (6). Compound 5 (869 mg, 1.19 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry THF (12 mL). To the solution was added phthaloyl chloride (189 μL, 1.31 mmol), diisopropylethylamine (456 μL, 2.62 mmol) at 0 °C and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of H2O. The mixture was partitioned between ethyl acetate and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (40:60–30:70, v/v) containing 1% pyridine to give the fractions containing 6. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of pyridine to give 6 (846 mg, 83%). 1H-NMR (CDCl3) δ 1.04–1.17 (m, 12H), 2.40–2.45 (m, 2H), 2.62 (t, 1H, J = 6.3 Hz), 2.72–2.82 (m, 1H), 3.40–3.61 (m, 6H), 3.78 (s, 6H), 4.20–4.21 (m, 1H), 4.59–4.69 (m, 1H), 6.22–6.32 (m, 2H), 6.81–6.86 (m, 4H), 7.26–7.39 (m, 9H), 7.79–7.82 (m, 2H), 7.94–7.97 (m, 2H), 8.51, 8.60 (2d, 1H, J = 7.3 Hz); 13C-NMR (CDCl3) δ 20.1, 20.2, 20.4, 24.4, 24.5, 24.6, 40.7, 41.1, 43.1, 43.2, 43.3, 53.4, 55.2, 58.1, 58.4, 61.5, 61.9, 70.9, 71.2, 71.8, 72.1, 85.8, 86.9, 87.5, 87.6, 100.5, 113.2, 117.3, 117.5, 124.3, 127.1, 128.0, 128.1, 128.2, 130.07, 130.13, 131.4, 135.1, 135.3, 144.0, 145.2, 154.6, 158.6, 159.1, 164.9; 31P-NMR (CDCl3) δ 149.6, 150.2 (2s). HRMS (ESI) m/z (M+H)+: calcd for C47H51N5O9P+ 860.3419; found, 860.3411.
6-N-Phthaloyl-5´-O-(4,4'-dimethoxytrityl)-2´-deoxyadenosine 3'-(2-cyanoethyl N,N-diisopropyl-phosphoramidite) (8). Compound 7 (1.06 g, 1.40 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1) and dissolved in dry THF (14 mL). To the solution was added phthaloyl chloride (221.7 μL, 1.54 mmol), diisopropylethylamine (1.22 mL, 7.0 mmol) at 0 °C and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of H2O. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (40:60–30:70, v/v) containing 1% pyridine to give the fractions containing 8. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of pyridine to give 8 (761 mg, 61%). 1H-NMR (CDCl3) δ 1.13–1.22 (m, 12H), 2.48 (t, 1H, J = 6.3 Hz), 2.62–2.68 (m, 3H), 2.72–2.75 (m, 1H), 3.35–3.37 (m, 1H), 3.59–3.77 (m, 9H), 4.33–4.35 (m, 1H), 4.77–4.78 (m, 1H), 6.55 (t, 1H, J = 5.7 Hz), 6.79–6.81 (m, 4H), 7.18–7.30 (m, 7H), 7.40 (d, 2H, J = 7.3 Hz), 7.82–7.83 (m, 2H), 8.01–8.02 (m, 2H), 8.34, 8.36 (2s, 1H), 8.96 (d, 1H, J = 3.7 Hz); 13C-NMR (CDCl3) δ 20.4, 20.6, 24.65, 24.70, 24.76, 24.8, 39.7, 39.8, 43.4, 43.5, 55.35, 55.36, 58.4, 58.5, 63.3, 63.5, 73.6, 73.7, 74.3, 74.4, 85.11, 85.13, 86.7, 113.3, 117.5, 117.6, 124.5, 127.08, 127.11, 128.0, 128.2, 128.3, 130.11, 130.15, 130.19, 130.23, 130.27, 132.1, 134.9, 135.6, 135.7, 135.8, 144.44, 144.48, 144.5, 152.5, 152.5, 153.5, 153.6, 158.7, 165.7; 31P-NMR (CDCl3) δ 149.9, 150.2 (2s). HRMS (ESI) m/z (M+H)+: calcd for C48H51N7O8P+ 884.3531; found, 884.3533.
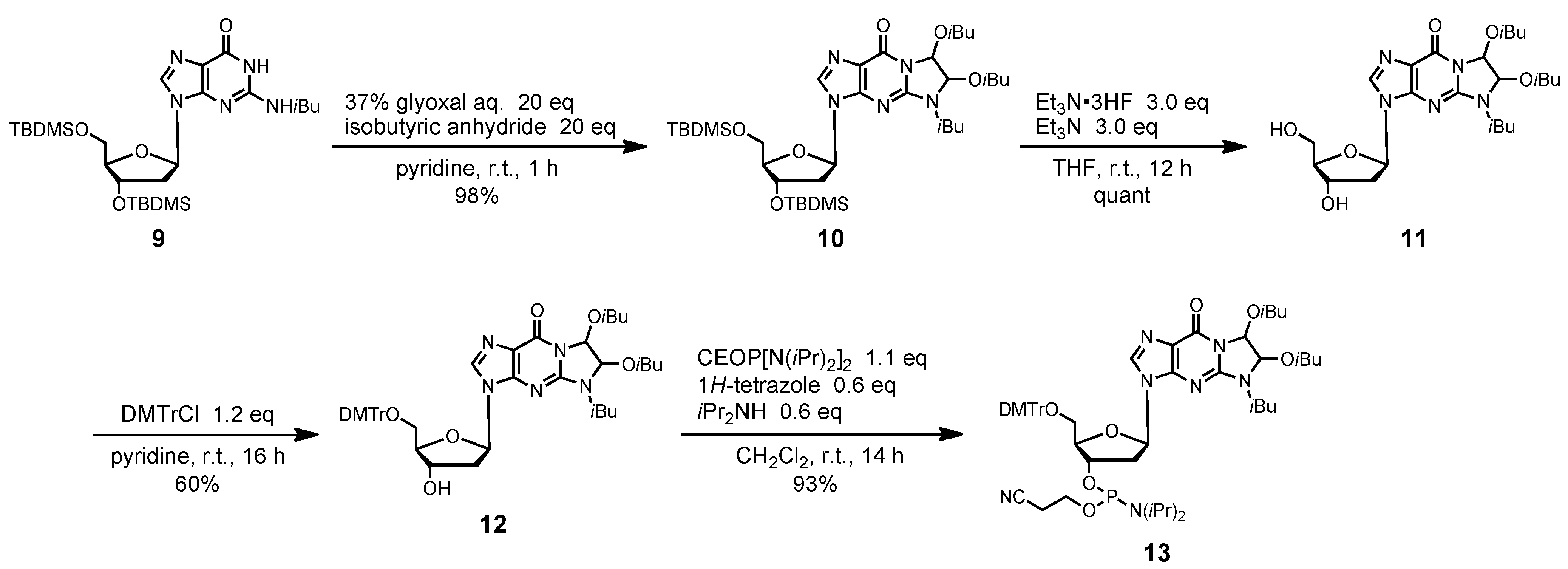
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxy-guanosine (10). 37% Glyoxal (5 mL, 40 mmol) was added to compound 9 (1.26 g, 2.0 mmol) at room temperature. The mixture was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL x1) and dissolved in dry pyridine (20 mL). After the solution was stirred at room temperature for 3 h, isobutyric anhydride (6.5 mL, 40 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (90:10–70:30, v/v) to give the fractions containing 10. The fractions were collected and evaporated under reduced pressure to give 10 (1.51 g, 98%). 1H-NMR (CDCl3) δ 0.11 (s, 12H), 0.92 (s, 18H), 1.15–1.19 (m, 12H), 1.25–1.31 (m, 6H), 2.35–2.38 (m, 2H), 2.54–2.59 (m, 2H), 3.80 (m, 2H), 4.00–4.04 (m, 2H), 4.51 (m, 1H), 6.29 (t, 1H, J = 6.5 Hz), 6.78 (d, 1H, J = 10.0 Hz), 6.83 (d, 1H, J = 6.3 Hz), 8.016, 8.021 (2s, 1H); 13C-NMR (CDCl3) δ -5.4, -5.3, -4.65, -4.63, 18.0, 18.46, 18.50, 18.53, 18.55, 18.57, 18.6, 18.7, 18.80, 18.83, 18.9, 19.0, 25.8, 26.1, 33.85, 33.94, 33.97, 42.6, 63.1, 72.3, 72.4, 78.6, 81.6, 84.3, 88.3, 120.6, 137.1, 148.1, 148.3, 153.7, 174.2, 174.4, 175.6. HRMS (ESI) m/z (M+H)+: calcd for C36H62N5O9Si2+ 764.4081; found, 764.4021.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-2´-deoxyguanosine (11). Compound 10 (1.0 g, 1.3 mmol) was dissolved in dry THF (6 mL). To the solution was added triethylamine (544 μL, 3.9 mmol) and Et3N·3HF (635 μL, 3.9 mmol) and the mixture was stirred at room temperature for 12 h. The mixture was partitioned between CHCl3-iPrOH (3:1, v/v) and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–95:5, v/v) to give the fractions containing 11. The fractions were collected and evaporated under reduced pressure to give 11 quantitatively. 1H-NMR (DMSO) δ1.05–1.11 (m, 12H), 1.17–1.22 (m, 6H), 2.36–2.40 (m, 1H), 2.59–2.64 (m, 3H), 3.52–3.57 (m, 2H), 3.87 (m, 1H), 3.96 (m, 1H), 4.36 (m, 1H), 4.95–4.96 (m, 1H), 5.36 (d, 1H, J = 4.2 Hz), 6.23–6.24 (m, 1H), 6.70 (s, 1H), 6.77 (s, 1H), 8.29, 8.30, 8.31 (3s, 1H); 13C-NMR (DMSO) δ 18.7, 18.8, 18.8, 19.1, 19.1, 19.4, 19.5, 33.7, 33.8, 40.8, 61.9, 62.0, 70.9, 71.0, 78.6, 79.7, 82.2, 84.6, 84.6, 88.4, 120.4, 120.5, 139.2, 139.4, 148.2, 148.3, 148.7, 153.8, 174.8, 175.0, 175.0, 176.0. HRMS (ESI) m/z (M+H)+: calcd for C24H34N5O9+ 536.2351; found, 536.2348.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-(4,4'-dimethoxytrityl)-2´-deoxy-guanosine (12). Compound 11 (696 mg, 1.3 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), and dissolved in dry pyridine (13 mL). To the solution was added DMTrCl (533 mg, 1.6 mmol), and the mixture was stirred at room temperature for 16 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 12. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 12 (657 mg, 60%). 1H-NMR (CDCl3) δ 1.14–1.19 (m, 12H), 1.26–1.31 (m, 6H), 2.55–2.59 (m, 4H), 3.80 (s, 6H), 3.92–4.03 (m, 3H), 4.74–4.75 (m, 1H), 6.28 (d, 1H, J = 5.4 Hz), 6.77, 6.78 (2s, 1H), 6.81–6.84 (m, 4H), 7.17 (d, 1H, J = 8.8 Hz), 7.22–7.32 (m, 9H), 8.60, 8.64 (2s, 1H); 13C -NMR (CDCl3) δ 18.6, 18.7, 18.9, 19.0, 19.1, 34.0, 34.1, 41.3, 41.6, 55.4, 64.2, 72.3, 78.4, 78.5, 81.8, 81.9, 84.5, 86.7, 86.8, 86.9, 113.4, 120.7, 120.8, 127.2, 128.1, 128.3, 130.2, 130.3, 130.3, 135.7, 135.8, 135.9, 137.7, 137.9, 144.7, 148.3, 148.4, 148.5, 153.9, 158.8, 174.4, 175.8, 175.9. HRMS (ESI) m/z (M+H)+: calcd for C45H52N5O11+ 838.3658; found, 838.3659.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-(4,4'-dimethoxytrityl)-2´-deoxy-guanosine 3'-(2-cyanoethyl N,N-diisopropylphosphramidite) (13). Compound 12 (210 mg, 0.25 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry CH2Cl2 (2.5 mL). To the solution was added bis(diisopropylamino)(2-cyanoethoxy)phosphine (89 μL, 0.28 mmol), 1H-tetrazole (11 mg, 0.15 mmol), diisopropylamine (21 μL, 0.15 mmol), and the mixture was stirred at room temperature for 14 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 13. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 13 (239 mg, 93%). 1H-NMR (CDCl3) δ 1.11–1.29 (m, 30H), 2.42 (m, 1H), 2.53–2.62 (m, 5H), 3.32–3.35 (m, 2H), 3.57–3.76 (m, 8H), 3.81–3.99 (m, 2H), 4.29–4.32 (m, 1H), 4.64 (m, 1H), 6.28 (m, 1H), 6.79–6.84 (m, 6H), 7.18–7.20 (m, 1H), 7.24–7.30 (m, 6H), 7.38–7.40 (m, 2H), 7.85, 7.90 (2s, 1H); 13C-NMR (CDCl3) δ 18.6, 18.7, 18.9, 19.1, 19.2, 20.4, 20.6, 24.8, 24.9, 34.0, 40.7, 40.9, 41.2, 43.5, 43.6, 55.5, 58.3, 58.4, 63.6, 63.7, 74.2, 74.3, 74.6, 78.5, 78.6, 81.7, 81.8, 84.3, 84.4, 86.0, 86.4, 86.9, 113.5, 117.6, 117.7, 120.9, 121.0, 127.2, 128.2, 128.4, 130.2, 130.3, 135.6, 135.7, 135.8, 137.2, 137.3, 144.6, 148.4, 148.5, 153.8, 158.9, 174.3, 174.4, 175.7, 175.8; 31P-NMR (CDCl3) 150.0, 150.4, 150.5 (3s). HRMS (ESI) m/z (M+H)+: calcd for C54H69N7O12P+ 1038.4736; found, 1038.4064.
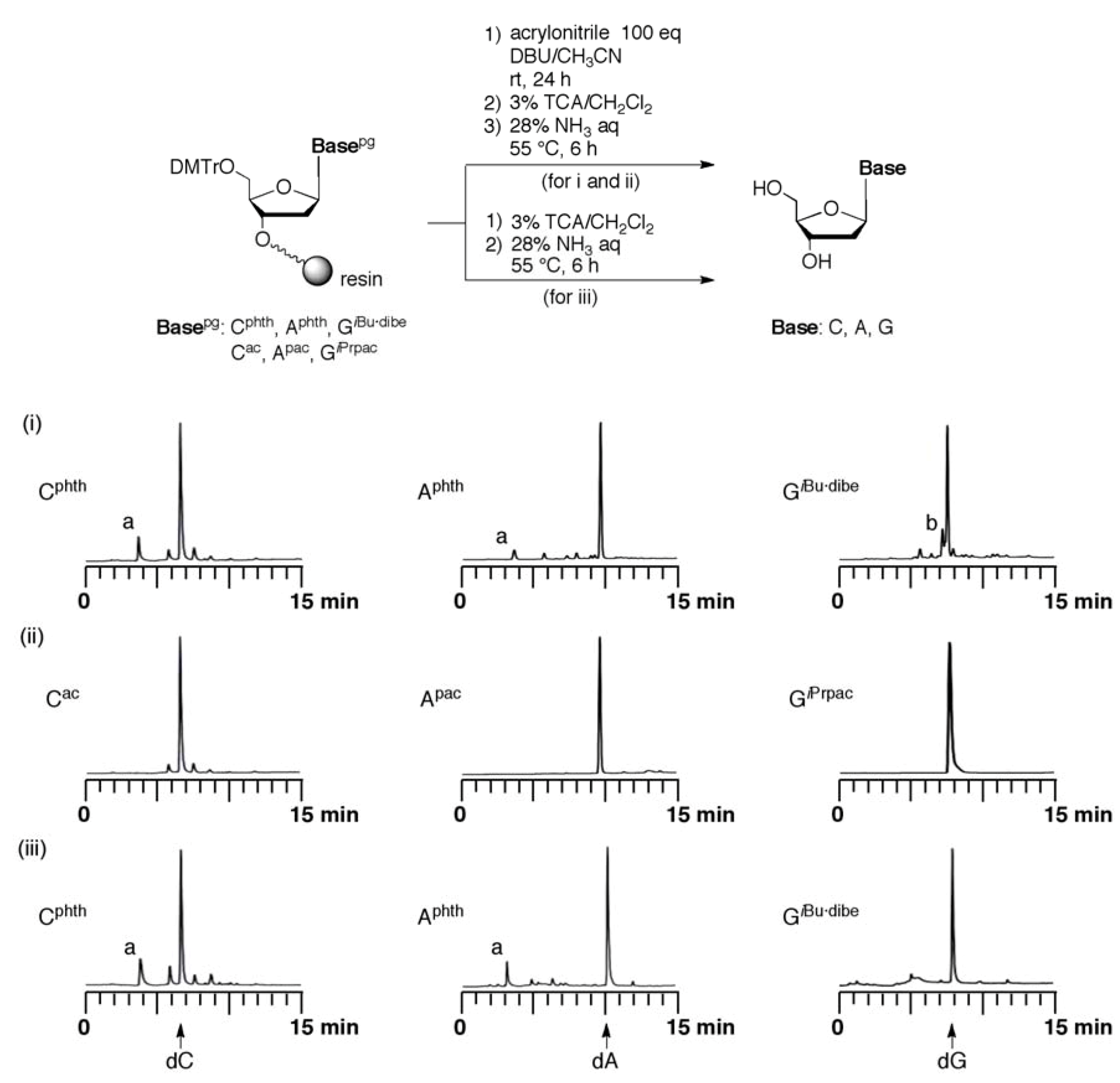
Evaluation of the resistance for addition of acrylonitrile to protected monomers. Fully protected monomers and common protected monomers were introduced into UnyLinker™ NittoPhase® by use of the ABI 392 DNA synthesizer. The protected monomer (125 nmol) loading UnyLinker™ NittoPhase® were treated with acrylonitrile (0.82 μL, 12.5 μmol) in 4 mM DBU-CH3CN (200 μL). After the reaction for appropriate time, the reaction solution was removed by filtration. The DMTr group was removed by treatment with 3% trichloroacetic acid in CH2Cl2 (1 mL) for 1 min, and the resin was washed with CH2Cl2 (1 mL × 3), and CH3CN (1 mL × 3). The monomer was deprotected and released from UnyLinker™ NittoPhase® by treatment with concentrated NH3 aq (500 µL) at 55 °C for 6 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and purified by reversed-phase HPLC.
6-N-Phenoxyacetyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (15). 3´,5´-O-Bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (960 mg, 2.0 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1) and dissolved in dry pyridine (10 mL). To the solution was added phenoxyacetic anhydride (1.37 mg, 4.8 mmol), and the mixture was stirred at room temperature for 3 h. To the reaction was added concentrated NH3 aq (5 mL), and the mixture was stirred at room temperature for 10 min. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0, v/v) to give the fractions containing 15. The fractions were collected and evaporated under reduced pressure to give 15 (831 mg, 68%). 1H NMR (CDCl3) δ 1H NMR (CDCl3) δ 0.10 (s, 12H), 0.91 (s, 18H), 2.45–2.49 (m, 1H), 2.65–2.70 (m, 1H), 3.78 (dd, 1H, J = 2.9 Hz, J = 9.8 Hz), 3.87 (dd, 1H, J = 4.2 Hz, J = 9.2 Hz), 4.037–4.043 (m, 1H), 4.62–4.63 (m, 1H), 4.87 (s, 2H), 6.50 (t, 1H, J = 6.3 Hz), 7.08 (d, 3H, J = 8.3 Hz), 7.35 (t, 2H, J = 7.9 Hz), 8.33 (s, 1H), 8.79 (s, 1H), 9.41 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.6, -4.5, 18.1, 18.5, 26.0, 26.1, 40.9, 62.8, 68.6, 72.1, 84.9, 88.1, 115.0, 117.6, 122.1, 123.0, 129.8, 142.5, 148.7, 151.5, 152.3, 157.5, 167.9. HRMS (ESI) m/z (M+H)+: calcd for C30H48N5O5Si2+ 614.3188; found, 614.3186.
General procedure for evaluation of cyanoethylation of compounds 14-17. An appropriate compound (0.30 mmol) was dissolved in DBU-CH
3CN [3 mL, (1:9, v/v)]. To the solution was added acrylonitrile (1.96 mL, 30 mmol). After being stirred at room temperature for appropriate time, as shown in
Table 1, the mixture was partitioned between CHCl
3 and H
2O. The organic phase was collected, dried over Na
2SO
4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl
3-MeOH (100:0–99:1, v/v) to give the fractions containing the cyanoethylated compound. The fractions were collected and evaporated under reduced pressure to give the cyanoethylated compound. Thus, the following compounds
18–
20 were obtained.
4-N-Acetyl-4-N-2-cyanoethyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxycytidine (18). The compound 18 was obtained (25.3 mg, 15%). 1H NMR (CDCl3) δ 0.11 (s, 12H), 0.90 (s, 18H), 2.13–2.18 (m, 1H), 2.47 (s, 3H), 2.52–2.57 (m, 1H), 2.84–2.93 (m, 2H), 3.78 (dd, 1H, J = 2.6 Hz, J = 10.8 Hz), 3.95–3.97 (m, 2H), 4.26 (t, 2H, J = 6.6 Hz), 4.38–4.41 (m, 1H), 6.61–6.23 (m, 1H), 6.88 (d, 1H, J = 7.3 Hz), 8.39 (d, 1H, J = 7.6 Hz); 13C-NMR (CDCl3) δ -5.4, -5.3, -4.8, -4.4, 17.5, 18.1, 18.5, 22.6, 25.8, 26.0, 42.3, 42.4, 61.8, 70.0, 87.1, 88.0, 99.3, 144.0, 154.9, 164.2, 172.1. HRMS (ESI) m/z (M+H)+: calcd for C26H47N4O5Si2+ 551.3080; found, 551.3079.
6-N-2-Cyanoethyl-6-N-phenoxyacetyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (19). The compound 19 was obtained (3.4 mg, 2%). 1H NMR (CDCl3) δ 0.12 (s, 12H), 0.92 (s, 18H), 2.46–2.51 (m, 1H), 2.57–2.62 (m, 1H), 2.93 (t, 2H, J = 7.3 Hz), 3.79 (dd, 1H, J = 2.7 Hz, J = 9.9 Hz), 3.89 (dd, 1H, J = 3.9 Hz, J = 9.3 Hz), 4.05–4.06 (m, 1H), 4.57–4.60 (m, 2H), 4.62–4.64 (m, 1H), 5.17 (d, 2H, J = 3.9 Hz), 6.52 (t, 1H, J = 6.2 Hz), 6.67 (d, 2H, J = 8.1 Hz), 6.93 (t, 1H, J = 7.3 Hz), 7.20 (t, 2H, J = 7.9 Hz), 8.42 (s, 1H), 8.70 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.6, -4.5, 17.3, 18.2, 18.6, 25.9, 26.1, 41.7, 43.6, 62.8, 68.9, 71.9, 84.9, 88.3, 114.6, 117.6, 121.7, 125.5, 129.6, 142.7, 151.4, 151.6, 152.7, 157.7, 170.8. HRMS (ESI) m/z (M+H)+: calcd for C33H51N6O5Si2+ 667.3454; found, 667.3454.
N3-(2-Cyanoethyl)-3´,5´-O-bis(tert-butyldimethylsilyl)thymidine (20). The compound 20 was obtained (127.3 mg, 83%). 1H NMR (CDCl3) δ 0.11 (s, 12H), 0.90 (s, 18H), 1.93 (s, 3H), 1.95–2.03 (m, 1H), 2.25–2.28 (m, 1H), 2.73–2.76 (m, 2H), 3.76 (d, 1H, J = 11.0 Hz), 3.86 (d, 1H, J = 11.5 Hz), 3.95 (m, 1H), 4.24–4.29 (m, 2H), 4.39 (m, 1H), 6.34 (t, 1H, J = 6.8 Hz), 7.50 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.7, -4.5, 13.3, 16.2, 18.1, 18.5, 25.9, 26.1, 36.8, 41.6, 63.1, 72.4, 85.8, 88.1, 110.2, 117.3, 134.3, 150.6, 163.1. HRMS (ESI) m/z (M+H)+: calcd for C25H46N3O5Si2+ 524.2971; found, 524.2913.
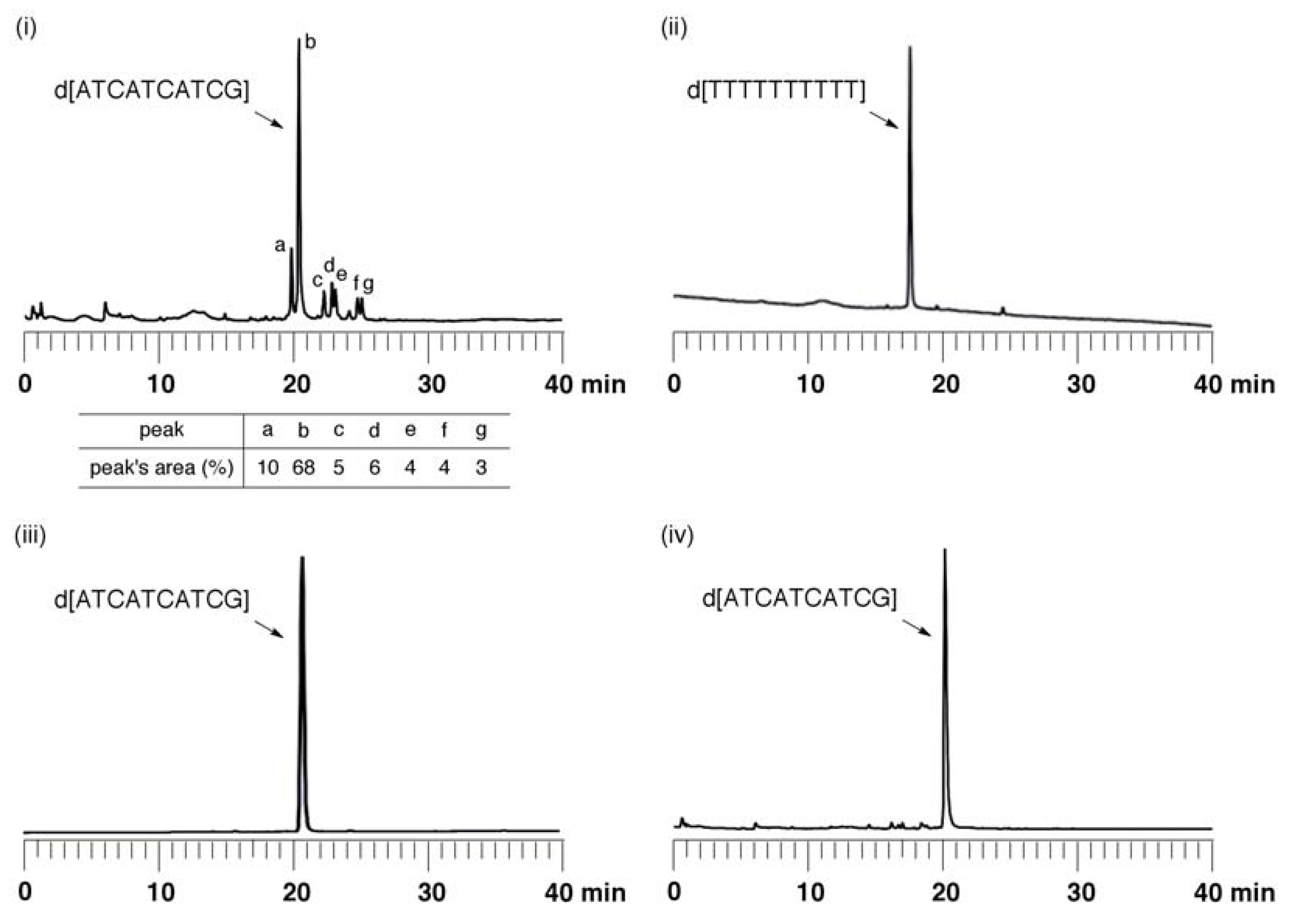
Synthesis of ODN using fully protected monomers: The synthesis of ODN d[ATCATCATCG] by use of an ABI 392 DNA synthesizer was carried out. After chain elongation was finished, the fully protected ODN (125 nmol) on UnyLinker™ NittoPhase® beads was treated with DBU-CH3CN [200 mL, (1:9, v/v)] for 1 min to remove the 2-cyanoethyl groups from the phosphate moieties. The ODN was deprotected and released from the resin by treatment with concentrated NH3 aq (500 µL) at 55 °C for 14 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and purified by anion-exchange HPLC. MALDI-TOF Mass (M+H)+: calcd for C97H125N35O58P9+ 2986.55; found 2987.91.
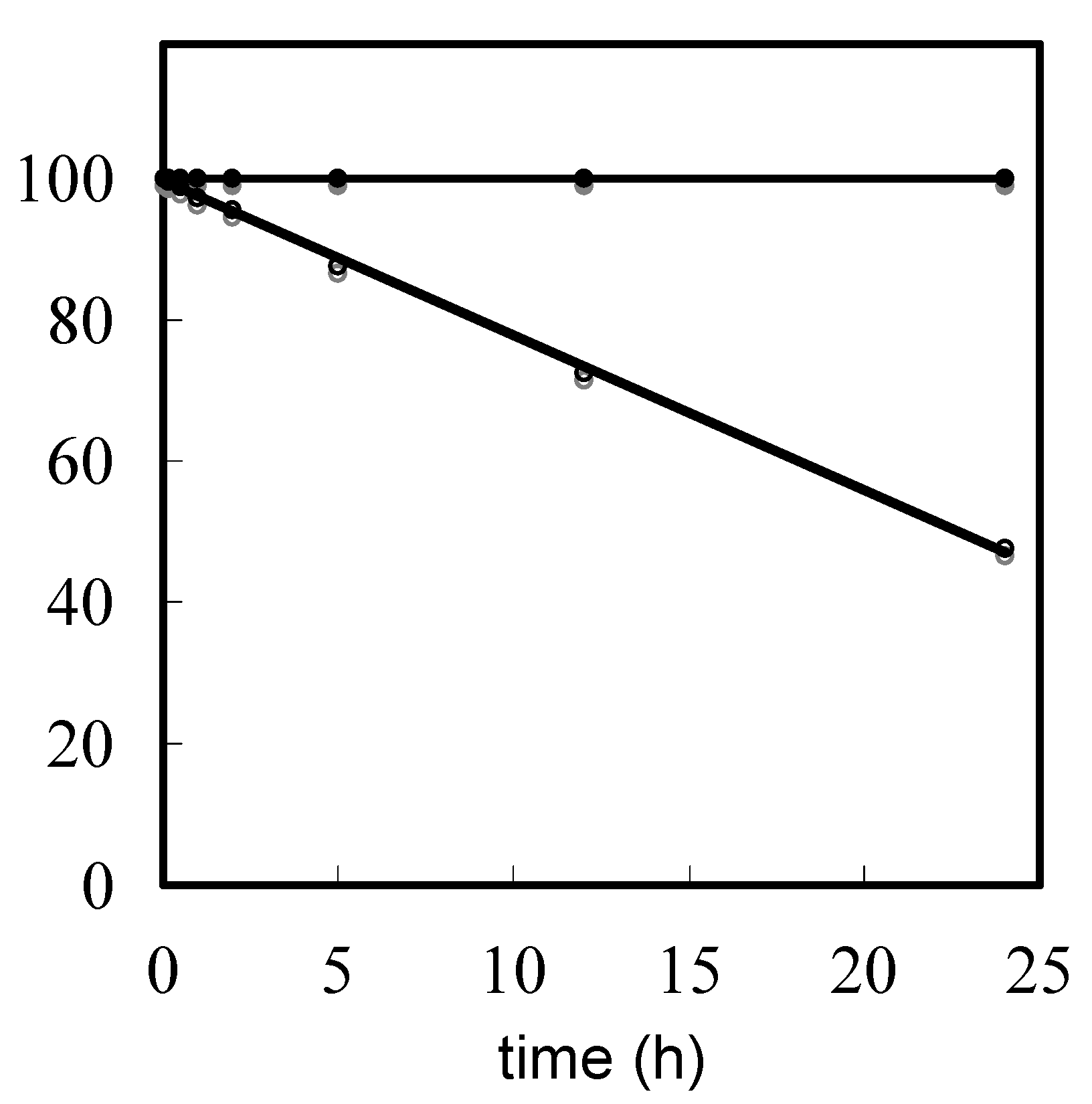
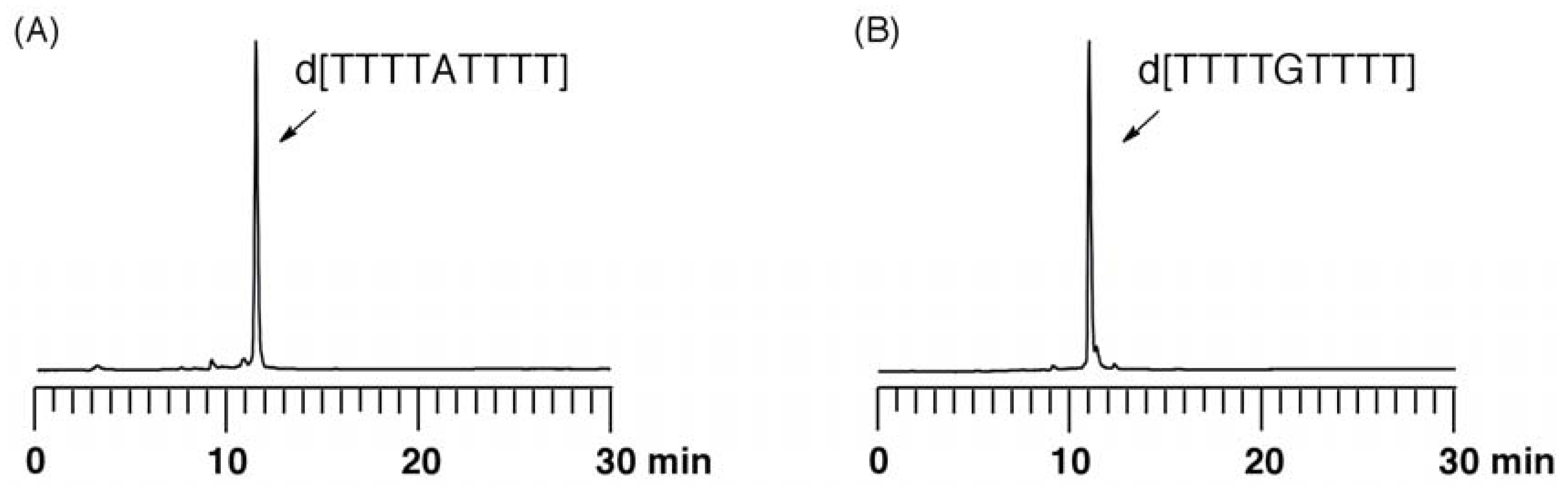
Evaluation of depurination using ODNs containing Aphth or GiBu·dibe. ODN d[TTTTXTTTT] (X = Aphth or GiBu·dibe) (0.5 µmol) on UnyLinker™ NittoPhase® beads was treated with 3% trichloroacetic acid in CH2Cl2 (1 mL) for 24 h. After the filtration of the reaction solution, the resin was washed with CH2Cl2 (1 mL × 3) and CH3CN (1 mL × 3). The ODN was deprotected and released from UnyLinker™ NittoPhase® by treatment with concentrated NH3 aq (500 µL) at 55 °C for 6 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and analyzed by reversed-phase HPLC. d[TTTTAphthTTTT]: MALDI-TOF Mass (M+H)+ calcd for C90H118N21O59P8+ 2684.48; found 2686.58. d[TTTTGiBu·dibeTTTT]: MALDI-TOF Mass (M+H)+ calcd for C90H118N21O60P8+ 2700.47; found 2702.94.
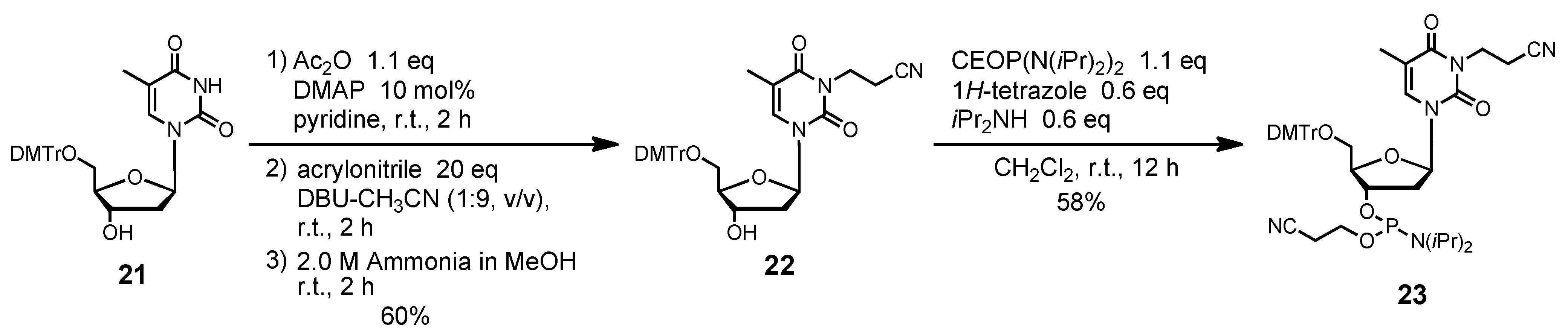
N3-(2-Cyanoethyl)-5´-O-(4, 4'-dimethoxytrityl)thymidine (22). Compound 21 (437 mg, 0.80 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), and dissolved in dry pyridine (8 mL). To the solution was added acetic anhydride (83 μL, 0.88 mmol), N,N-dimethtylaminopyridine (10 mg, 0.08 mmol), and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was dissolved in DBU-CH3CN (8 mL, 1:9, v/v), and to the solution was added acrylonitrile (1.05 mL, 16.0 mmol). After being stirred at room temperature for 2 h, the mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. To the residue was added 2.0 M ammonium in MeOH (10 mL), and the mixture was stirred at room temperature for 2 h. The mixture was evaporated under reduced pressure, chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 22. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 22 (289 mg, 60%). 1H NMR (CDCl3) δ 1.50 (s, 3H), 2.34–2.37 (m, 1H), 2.41–2.42 (m, 1H), 2.76 (t, 2H, J = 7.1 Hz), 3.39 (dd, 1H, J = 2.9 Hz, J = 10.3 Hz), 3.50 (dd, 1H, J = 3.2 Hz, J = 8.9 Hz), 3.80 (s, 6H), 4.05–4.06 (m, 1H), 4.26–4.30 (m, 2H), 4.59 (m, 1H), 6.42 (t, 1H, J = 6.7 Hz), 6.85 (d, 4H, J = 8.5 Hz), 7.25–7.32 (m, 7H), 7.40 (d, 2H, J = 7.3 Hz), 7.61 (s, 1H); 13C-NMR (CDCl3) δ 12.6, 16.2, 36.8, 41.2, 55.4, 63.5, 72.5, 85.5, 86.2, 87.2, 110.6, 113.5, 117.4, 127.3, 128.2, 128.3, 129.2, 130.2, 134.4, 135.41, 135.47, 144.4, 150.7, 158.9, 163.1. HRMS (ESI) m/z (M+Na)+: calcd for C34H35N3NaO7+ 620.2367; found, 620.2368.
N3-(2-Cyanoethyl)-5´-O-(4, 4'-dimethoxytrityl)thymidine 3'-(2-cyanoethyl N, N-diisopropylphosphramidite) (23). Compound 22 (239 mg, 0.40 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry CH2Cl2 (4 mL). To the solution was added bis(diisopropylamino) (2-cyanoethoxy)phosphine (123 μL, 0.44 mmol), 1H-tetrazole (17 mg, 0.24 mmol), diisopropylamine (34 μL, 0.24 mmol), and the mixture was stirred at room temperature for 12 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (70:30–50:50, v/v) containing 1% Et3N to give the fractions containing 23. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 23 (179 mg, 58%). 1H NMR (CDCl3) δ 1.07 (d, 3H, J = 6.8 Hz), 1.17–1.19 (m, 9H), 1.44 (s, 3H), 2.35–2.37 (m, 1H), 2.43 (t, 1H, J = 6.2 Hz), 2.49–2.58 (m, 1H), 2.63 (t, 1H, J = 6.2 Hz), 2.76 (t, 2H, J = 7.1 Hz), 3.32–3.35 (m, 1H), 3.48–3.68 (m, 5H), 3.80, 3.81 (2s, 6H), 4.15–4.19 (m, 1H), 4.26–4.28 (m, 2H), 4.66–4.68 (m, 1H), 6.40–6.44 (m, 1H), 6.83–6.86 (m, 4H), 7.25–7.31 (m, 7H), 7.40-7.42 (m, 2H), 7.65, 7.69 (2s, 1H); 13C-NMR (CDCl3) δ 12.5, 16.2, 20.3, 20.4, 20.55, 20.61, 24.60, 24.66, 24.69, 24.7, 24.8, 36.8, 43.35, 43.43, 43.5, 55.41, 55.43, 58.2, 58.4, 63.1, 63.3, 73.5, 73.6, 73.8, 74.0, 85.6, 85.7, 85.9, 87.1, 110.6, 113.4, 117.4, 117.5, 117.7, 127.32, 127.35, 128.1, 128.3, 128.4, 130.29, 130.31, 130.3, 134.47, 134.51, 135.46, 135.51, 144.4, 150.67, 150.70, 158.9, 163.1; 31P NMR (CDCl3) 149.7, 150.1 (2s). HRMS (ESI) m/z (M+Na)+: calcd for C42H50N5NaO8P+ 820.3446; found, 820.3450.
Enzymes and ODNs. AmpliTaq Gold DNA polymerase was purchased from applied biosystems
TM, and dNTPs were purchased from Takara Bio, Inc. ODNs used in enzyme reactions and
Tm experiments were purchased from Sigma-Aldrich Japan. ODNs 1 and 2 in
Table 2 were synthesized by use of an ABI 392 DNA synthesizer.
Tm experiments. An appropriate ODN (2 μM) and its complementary 2 μM ssDNA 11-mer were dissolved in a buffer consisting of 1 M NaCl, 10 mM sodium phosphate, and 0.1 mM EDTA adjusted to pH 7.0. The solution was maintained at 80 °C for 10 min for complete dissociation of the duplex to single strands and cooled at the rate of UV-1700TM (Shimadzu) by increasing the temperature at the rate of 0.5 °C/min. During this process of annealing and melting, the absorption at 260 nm was recorded and used to draw UV melting curves. The Tm value was calculated as the temperature at which the first derivative of the UV melting curve had a maximum.
Single dNTP Insertion Reaction Using Taq DNA Polymerase. A mixture containing PCR Gold buffer, 1.8 mM MgCl2, and 0.25 unit AmpliTaq Gold DNA polymerase was incubated at 95 °C for 3 min and slowly cooled to room temperature. To the mixture was added 100 nM (final concentration) 5′-FAM-labeled primer/template, and 10 μM (final concentration) dNTP (N = A, C, G, or T). The mixture (10 μL) was incubated at 74 °C for 10 min, and the reactions were terminated by adding 30 μL of stop solution (95% formamide, 20 mM EDTA). After being gently vortexed, the samples were separated by electrophoresis using 20% denaturing polyacrylamide gel containing 7 M urea and visualized by Fujifilm FLA-7000.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




