Selenol Protecting Groups in Organic Chemistry: Special Emphasis on Selenocysteine Se-Protection in Solid Phase Peptide Synthesis

Abstract
:
1. Introduction



{kind=link}
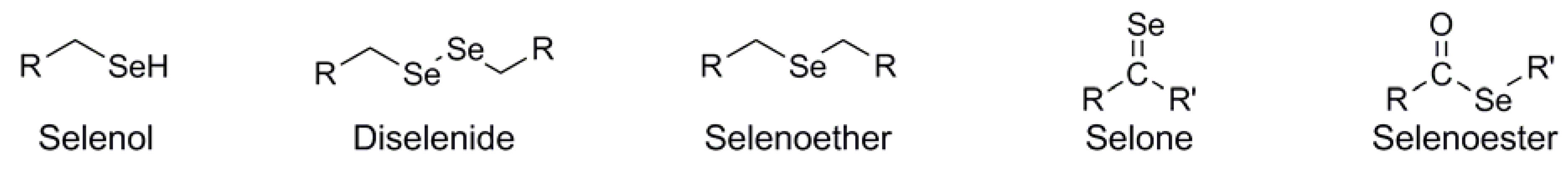{kind=link}
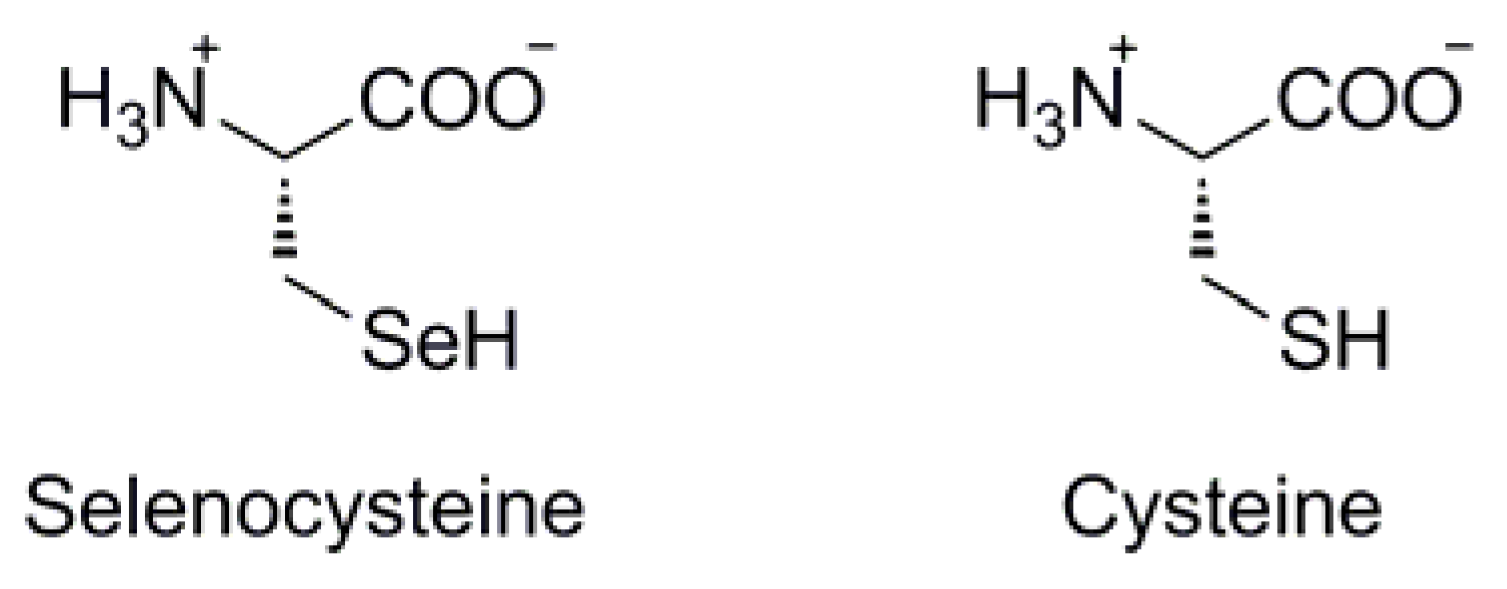{kind=link}
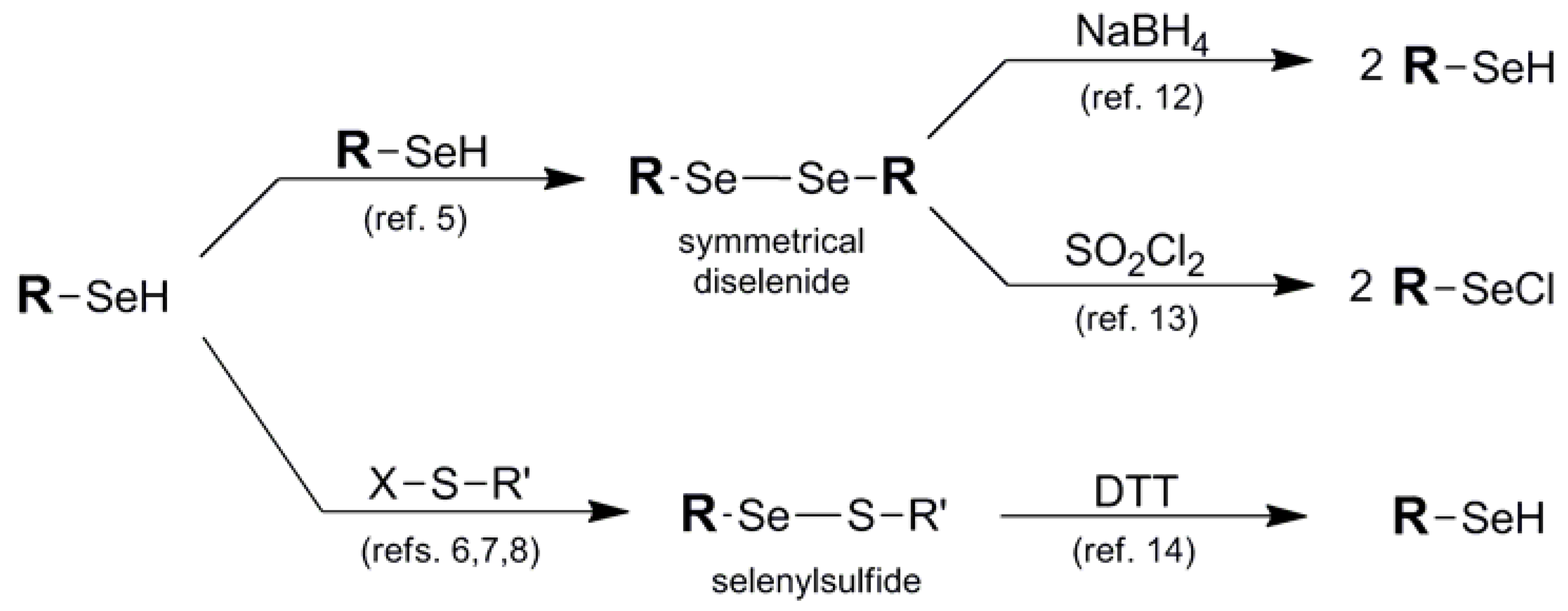{kind=link}
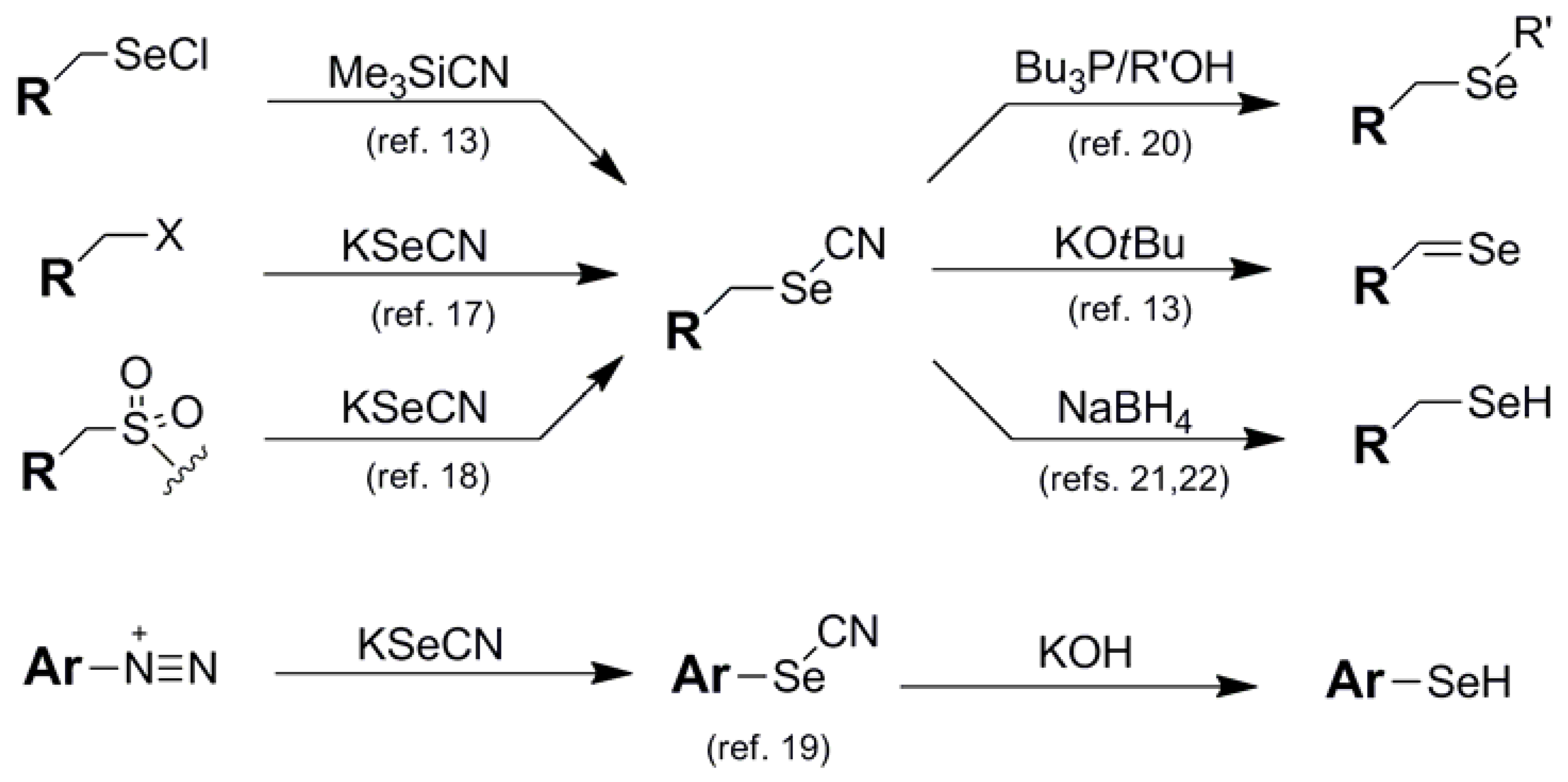{kind=link}
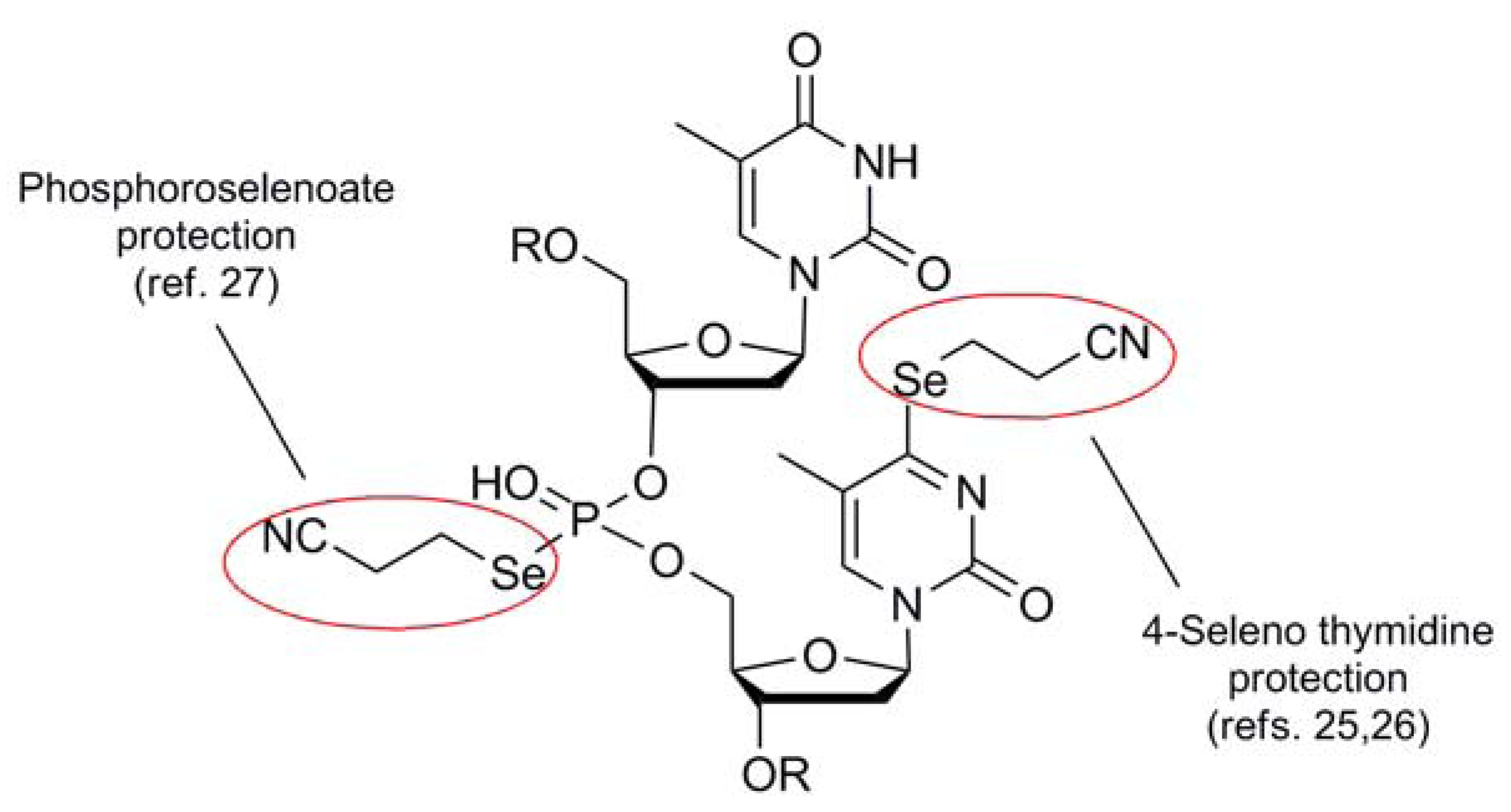{kind=link}
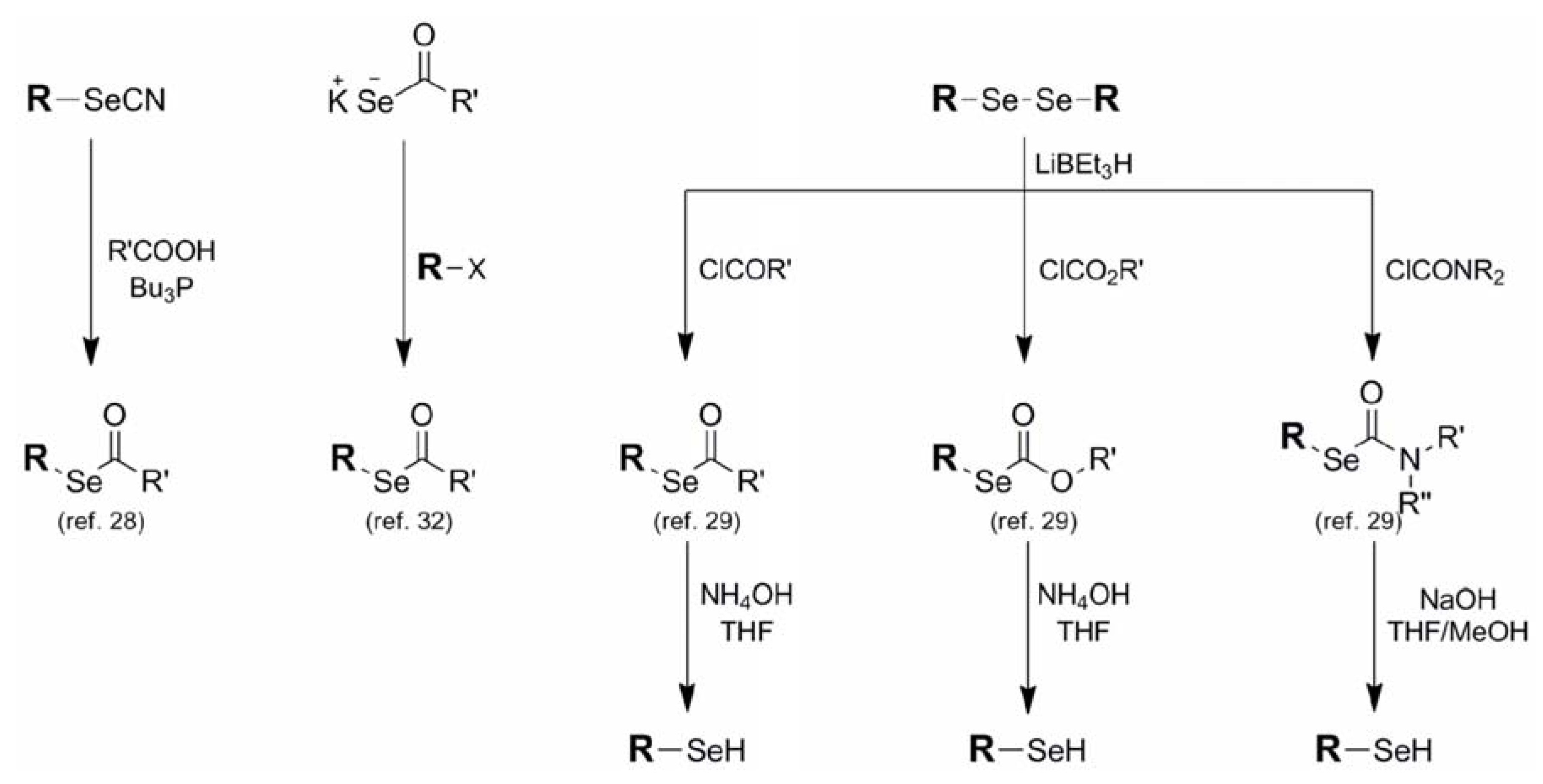{kind=link}
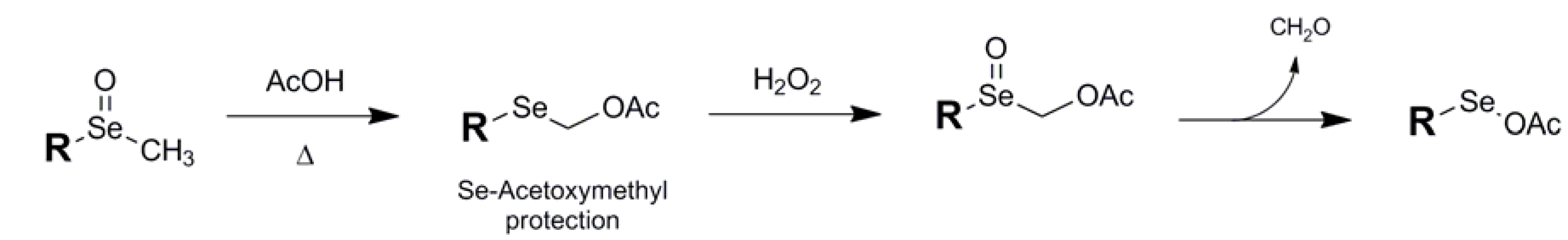{kind=link}
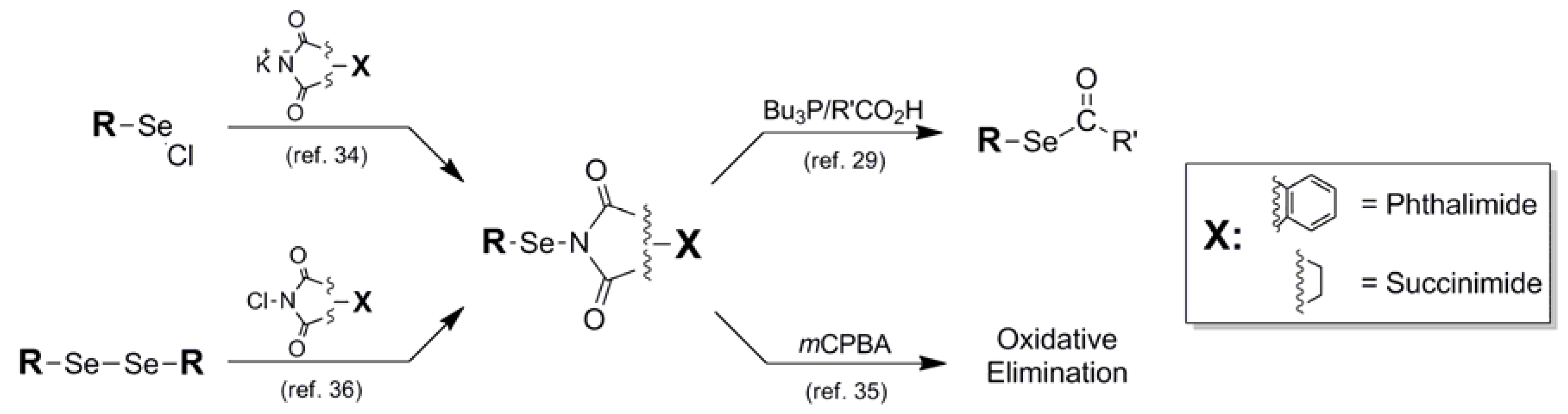{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Method of Introduction | Ref | DeprotectionConditions | Ref |
|---|---|---|---|---|---|
| Diselenide (1) |  | RSe- with RS-X (Se-S) | 6,7,8 | NaBH4 (Se-Se) | 12 |
| SO2Cl2 (Se-Se) | 13 | ||||
| Selenylsulfide (2) | DTT (Se-S) | 14 | |||
| Thiuocyanate (3) |  | KSCN nucleophile | 15 | - - - | - - |
| Cyano (4) |  | KSeCN nucleophile | 17,18 | NaBH4/LiBEt3H | 21,22 |
| Me3SiCN nucleophile | |||||
| 13 | KOH | 19 | |||
| 2-Cyanoethyl (5) |  | NC(CH2)2Se nucleophile | 25,26 | K2CO3/MeOH | 25,26 |
| NC(CH2)2Se phthalamide | 27 | DBU | 27 | ||
| Acetate (6) |  | AcCl electrophile | 29 | NH4OH/THF | 29 |
| 32 | KOH/MeOH- | ||||
| KSeAc nucleophile | |||||
| [R=CH3] | |||||
| 32 | |||||
| RSeCN/Bu3P-RCOOH | |||||
| 28 | DCM | ||||
| Carbonate (7) | ClCO2R electrophile | 29 | NH4OH/THF | 29 | |
| [R=OR] | |||||
| Carbamate (8) | ClCONR2 electrophile | 29 | NaOH/THF-MeOH | 29 | |
| [R=NR2] | |||||
| Acetoxymethyl (9) |  | RSe(O)CH3/AcOH | 33 | H2O2 | 33 |
| Phthalimide (10) |  | Potassium phthalimide nucleophile | 34 | - - - | - - |
| Succinimide (11) | N-Chloro Succinimide electrophile | 36 | - - - | - - | |
| Methyl (12) |  | Methyl electrophile | 13,38 | Br2 | 41 |
| (CH3Se)2 electrophile | 39 | ||||
| CH3Se- nucleophile | 40 | ||||
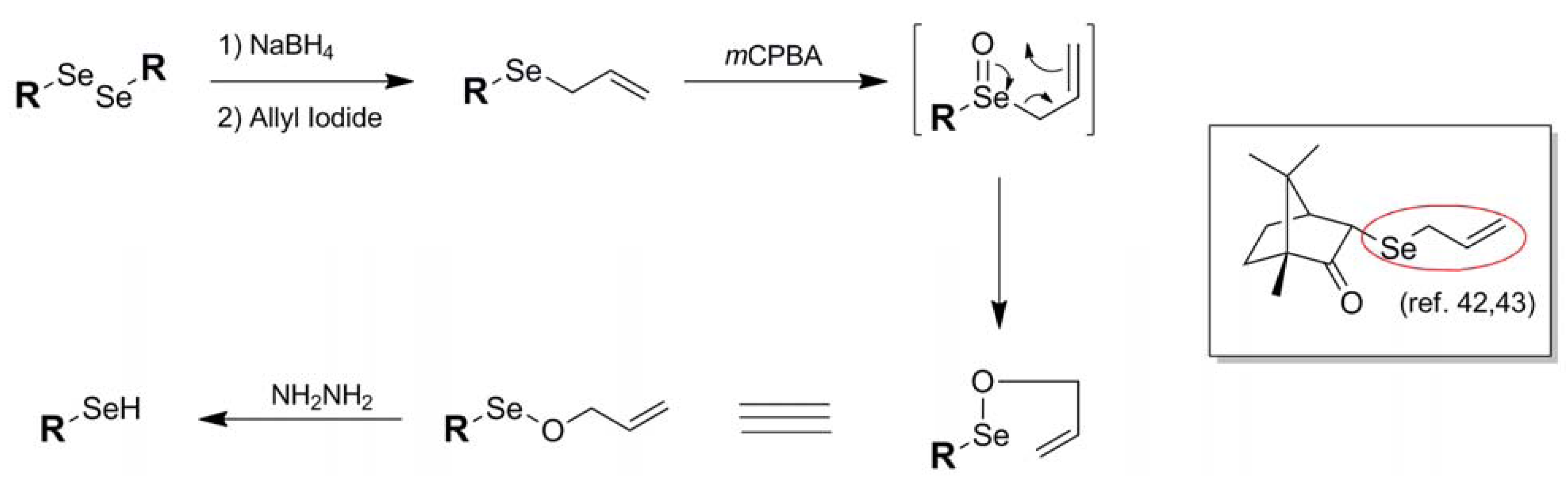
| Allyl (13) |  | Allyl electrophile | 42,43 | m-CPBA/NH2NH2 | 42,43 |
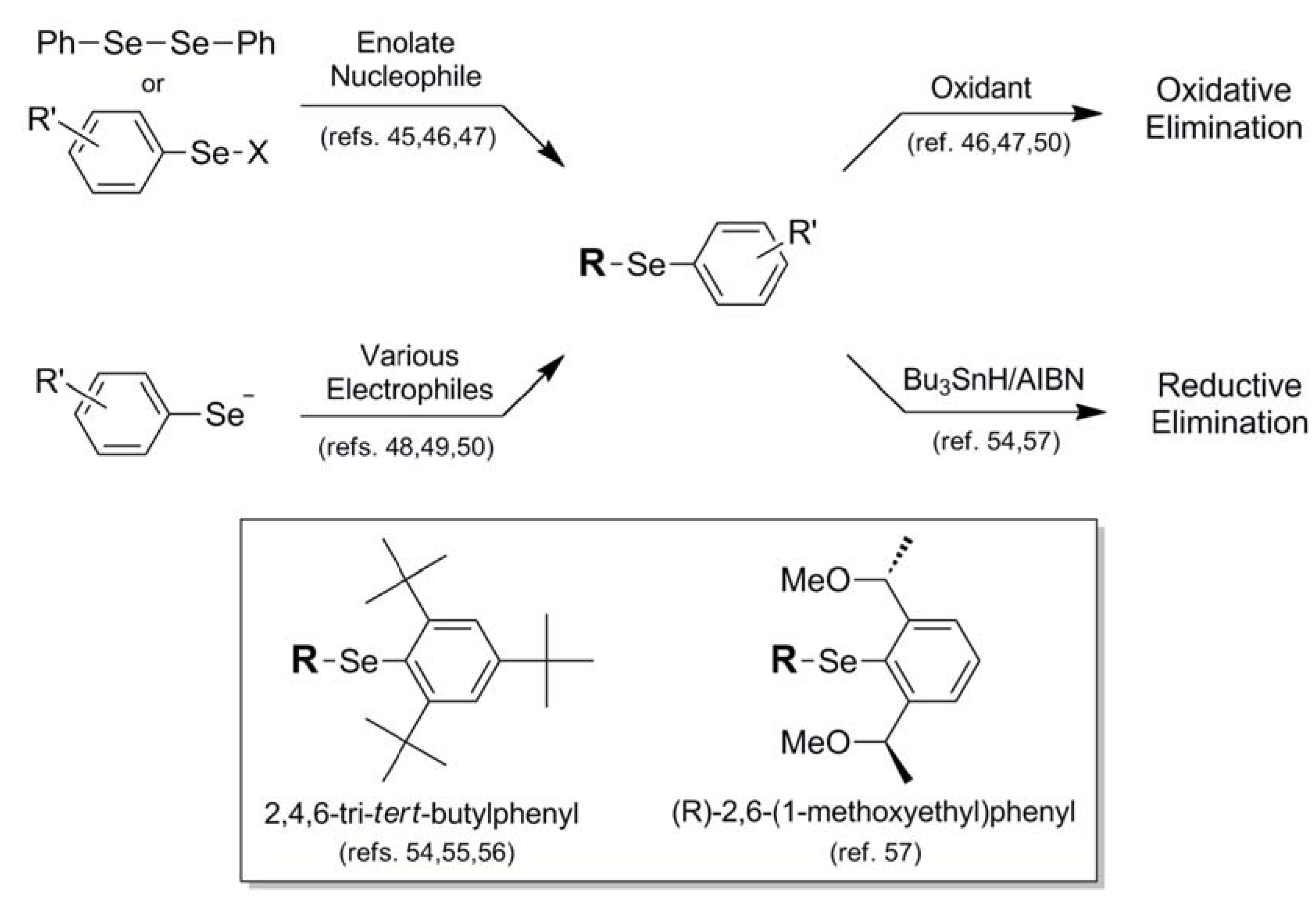
| Phenyl (14) |  | Enolate α- selenation | 45 | H2O2 | 46 |
| PhSeX electrophile | 46,47 | NaIO4 | 50 | ||
| PhSe- nucleophile | 48,49 | O3 | 47 | ||
| [R1,R2,R3=H] | |||||
| 2,4,6-tri-tert-Butylphenyl (15) | ArSe nucleophile | 54,55 | Bu3SnH/AIBN | 54 | |
| [R1,R2,R3=t-Bu] | |||||
| 2,6-(1-methoxyethyl)Phenyl (16) | Ar*SeOTf electrophile | 57 | Bu3SnH/AIBN | 57 | |
| [R1,R3=CH(OCH3)CH3; R2=H] | |||||
| Benzyl (17) |  | (BnSe)2 electrophile | 44 | Br2/NH2NH2 | 44,58 |
| BnSeCH2Br electrophile | 58 |
2. Discussion
2.1. Heteroatom-Containing Se-Protection






2.2. Hydrocarbon-based Se-Protection


2.3. Selenocysteine Se-Protection
 | |||||
| P1 | P2 | Method of Introduction | Ref | P2 DeprotectionConditions | Ref |
| Z |  | BnSe- nucleophile | 61 | Na/NH3 | 59,60 |
| Boc | -- | -- | -- | -- | |
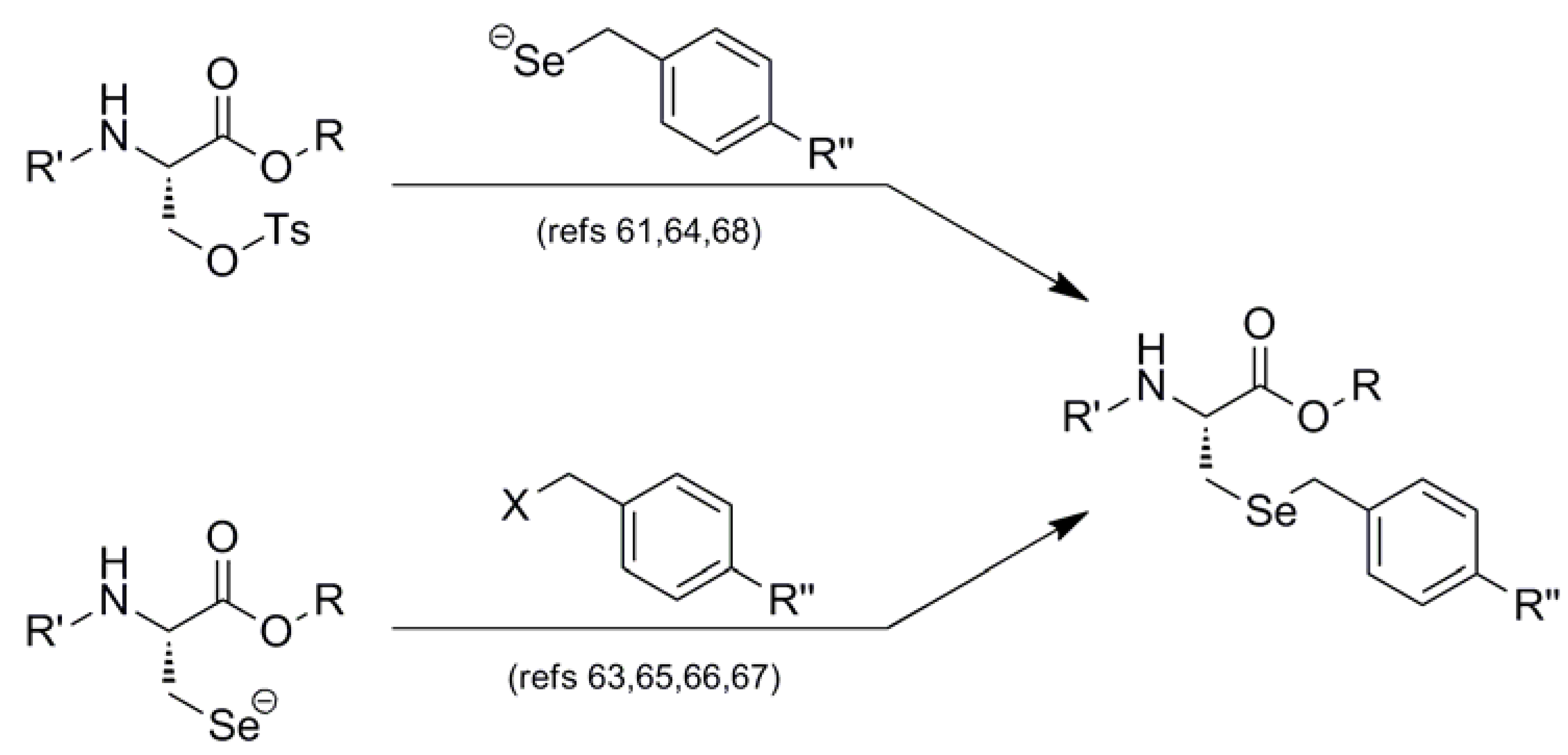
| Boc |  | Meb-Br electrophile | 63 | HF | 63,64 |
| MebSe- nucleophile | 64 | ||||
| Z |  | Mob-Cl electrophile | 65 | TFMSA/TFA | 65 |
| Boc | Mob-Cl electrophile | 66,69 | TMSBr/TFA | 66,69 | |
| Fmoc | Mob-Cl electrophile | 67 | I2 | 67,68 | |
| DMSO/TFA | 67 | ||||
| MobSe- nucleophile | 68 | ||||
| DTNP/TFA | 70 | ||||
| Boc |  | pNb-Br electrophile | 72 | Zn, then I2 | 72 |
| SnCl2, then I2 | 72 | ||||
| Boc |  | Acetamidomethanol/H+ | 72 | I2 | 72 |

3. Conclusions
References and Notes
- Gladyshev, V.N.; Jeang, K.T.; Stadtman, T.C. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. USA 1996, 93, 6146–6151. [Google Scholar]
- Epp, O.; Ladensteine, R.; Wendel, A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983, 133, 51–69. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Protection for the Thiol Group. In Protective Groups in Organic Synthesis, 4th ed; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2007; pp. 647–687. [Google Scholar]
- Prigol, M.; Wilhelm, E.A.; Schneider, C.C.; Nogueira, C.W. Protective effect of unsymmetrical dichalcogenide, a novel antioxident agent, in vitro and an in vivo model of brain oxidative damage. Chem. Biol. Interact. 2008, 176, 129–136. [Google Scholar]
- Nauser, T.; Dockheer, S.; Kissner, R.; Koppenol, W.H. Catalysis of electron transfer by selenocysteine. Biochemistry 2006, 45, 6038–6043. [Google Scholar]
- Schneider, C.C.; Godoi, B; Prigol, M.; Nogueira, C.W.; Zeni, G. Highly stereoselective one-pot procedure to prepare unsymmetrical bis- and tris-chalcogenide alkenes via addition of chalcogens to alkynes. Organometallics 2007, 26, 4252–4256. [Google Scholar]
- Zeni, G. Universidade Federal de Santa Maria, Camobi-Santa Maria, RS, Brazil, Personal Communication.
- Flemer, S., Jr.; Lacey, B.M.; Hondal, R.J. Synthesis of peptide substrates for mammalian thioredoxin reductase. J. Pep. Sci. 2007, 14, 637–647. [Google Scholar]
- Clark, E.R.; Al-Turaihi, M.A.S. The reaction of o-nitro- and p-nitro-phenyl selenocyanates with arylthiols. J. Organometallic. Chem. 1977, 134, 181–187. [Google Scholar]
- Baldwin, J.E.; Haber, S.B.; Kitchin, J. Dehydropeptides related to β-lactam antibiotics: a scheme for the biosynthesis of penicillins and cephalosporins. J. Chem. Soc. Chem. Comm. 1973, 790–791. [Google Scholar]
- Besse, D.; Budisa, N.; Karnbrock, W.; Minks, C.; Musiol, H.J.; Pegoraro, S.; Siedler, F.; Weyher, E.; Moroder, L. Chalcogen-analogs of amino acids: Their use in X-ray crystallographic and folding studies of peptides and proteins. Biol. Chem. 1997, 378, 211–218. [Google Scholar]
- Hondal, R.J.; Nilsson, B.L.; Raines, R.T. Selenocysteine in native chemical ligation and expressed protein ligation. J. Am. Chem. Soc. 2001, 123, 5140–5141. [Google Scholar]
- Back, T.G.; Dyck, B.P.; Parvez, M. 1,3-diselenetanes and 1,3-dithietanes derived from camphor. Formation, structure, stereochemistry, and oxidation to selenoxide and sulfoxide products. J. Org. Chem. 1995, 60, 703–710. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Rocha, J.B.T. Diphenyl diselenide; a Janus-faced molecule. J. Braz. Chem. Soc. 2010, 21, 2055–2071. [Google Scholar] [CrossRef]
- Parr, W.J.E.; Crafts, R.C. The electrophilic addition of selenenyl thiocyanates to olefins. Tetrahedron Lett. 1981, 22, 1371–1372. [Google Scholar]
- Rheinboldt, H.; Perrier, M. Thiocyanates d'acides sélénéniques aromatiques. II. Condensation avec l'acétone. 1950, 17, 759–763. [Google Scholar]
- van Ende, D.; Krief, A. Stereoselective isomerisations of disubstituted olefins via seleniranes and thiiranes (1). Tetrahedron Lett. 1975, 31, 2709–2712. [Google Scholar] [CrossRef]
- Baig, N.B.R.; Chandrakala, R.N.; Sudhir, V.S.; Chandrasekaran, S. Synthesis of unnatural selenocystines and β-aminosiselenides via regioselective ring-opening of sulfamaidates using a sequential, one-pot, multistep strategy. J. Org. Chem. 2010, 75, 2910–2921. [Google Scholar]
- Yavuz, S.; Disli, A.; Yildirir, Y.; Turker, L. The syntheses of some novel (naphthanen-1-yl-selenyl)acetic acid derivatives. Molecules 2005, 10, 1000–1004. [Google Scholar]
- Grieco, P.A.; Gilman, S.; Nishizawa, M. Organoselenium chemistry. A facile one-step synthesis of alkyl aryl selenides from alcohols. J. Org. Chem. 1976, 41, 1485–1486. [Google Scholar] [CrossRef]
- Muller, J.; Terfort, A. Synthesis of pure aromatic, aliphatic, and araliphatic diselenides. Inorg. Chim. Acta 2006, 359, 4821–4827. [Google Scholar] [CrossRef]
- Ie, Y.; Hirose, T.; Yao, A.; Yamada, T.; Takagi, N.; Kawai, M.; Aso, Y. Synthesis of tripodal anchor units beariung selenium functional groups and their adsorptoin behavior on gold. Phys. Chem. Chem. Phys. 2009, 11, 4949–4951. [Google Scholar]
- Logan, G.; Igunbor, C.; Chen, G.X.; Davis, H.; Simon, A.; Salon, J.; Huang, Z. A simple strategy for incorporation, protection, and deprotection of selenium functionality. Synlett 2006, 10, 1554–1558. [Google Scholar]
- Ohtsuka, Y.; Oishi, T. A synthetic approach to taxane diterpenes. A synthesis of the bicyclo[5.3.1]undecenone ring system. Tetrahedron Lett. 1986, 27, 203–206. [Google Scholar] [CrossRef]
- Salon, J.; Sheng, J.; Jiang, J.; Chen, G.; Caton-Williams, J.; Huang, Z. Oxygen replacement with selenium at the thymidine 4-position for the Se base pairing and crystal structure studies. J. Am. Chem. Soc. 2007, 129, 4862–4863. [Google Scholar]
- Salon, J.; Jiang, J.; Sheng, J.; Gerlits, O.O.; Huang, Z. Derivatization of DNAs with selenium at 6-position of guanine for function and crystal structure studies. Nucleic Acids Res. 2008, 36, 7009–7018. [Google Scholar] [CrossRef]
- Tram, K.; Wang, X.; Yan, H. Facile synthesis of oligonucleotide phosphoroselenoates. Org. Lett. 2007, 9, 5103–5106. [Google Scholar]
- Grieco, P.A.; Yokoyama, Y.; Williams, E. Aryl selenocyanates and aryl thiocyanates: reagents for the preparation of acivated esters. J. Org. Chem. 1978, 43, 1283–1285. [Google Scholar]
- Reinerth, W.A.; Tour, J.M. Protecting groups for organoselenium compounds. J. Org. Chem. 1997, 63, 2397–2400. [Google Scholar]
- La Groia, A.; Feroci, M.; Inesi, A.; Rossi, L. Electrochemical synthesis of selenocarbonates. Lett. Org. Chem. 2006, 3, 854–856. [Google Scholar] [CrossRef]
- Maeda, H.; Tanabe, T.; Hotta, K.; Mizuno, K. Synthesis of Se-arylmethyl selenoformates by reaction of aluminum arylmethaneselenoates with formates. Tetrahedron Lett. 2005, 46, 2015–2019. [Google Scholar] [CrossRef]
- Balakumar, A.; Lysenko, A.B.; Carcel, C.; Malinovskii, V.L.; Gryko, D.T.; Schweikart, K.K.H.; Loewe, R.S.; Yasseri, A.A.; Liu, Z.; Bocian, D.F.; Lindsay, J.S. Diverse redox-active molecules bearing O-, S-, or Se-terminated tethers for attachement to silicon in studies of molecular information storage. J. Org. Chem. 2003, 69, 1435–1443. [Google Scholar]
- Miyoshi, N.; Murai, S.; Sonoda, N. Oxyselenation: reaction of acetoxymethyl methyl selenide with olefins in the presence of hydrogen peroxide. Tetrahedron Lett. 1977, 10, 851–854. [Google Scholar]
- Nicolaou, K.C.; Claremon, D.A.; Barnette, W.E.; Seitz, S.P. N-Phenylselenophthalimide (N-PSP) and N-Phenylselenosuccinimide (N-PSS). Two versatile carriers of the phenylseleno group. Oxyselenation of olefins and a selenium-based macrolide synthesis. J. Am. Chem. Soc. 1979, 101, 3704–3706. [Google Scholar] [CrossRef]
- Liu, P.S.; Marquez, V.E.; Kelley, J.A.; Driscoll, J.S. Synthesis of 1,3-diazepin-2-one nucleosides as transition-state inhibitors of cytidine deaminase. J. Org. Chem. 1980, 45, 5225–5227. [Google Scholar]
- Hori, T.; Sharpless, K.B. Conversion of allylic phenylselenides to the rearranged allylic chlorides by N-chlorosuccinimide. Mechanism of selenium-catalyzed allylic chlorination of β-pinene. J. Org. Chem. 1979, 44, 4208–4210. [Google Scholar] [CrossRef]
- Grieco, P.A.; Jaw, J.Y. N-Phenylselenophthalimide. A useful reagent for the facile transformation of (1) carboxylic acids into either selenol esters or amides and (2) alcohols into alkyl phenyl selenides. J. Org. Chem. 1981, 46, 1215–1217. [Google Scholar] [CrossRef]
- Liotta, D.; Saindane, M.; Barnum, C. Reactions involving selenium metal. 2. A general procedure for the preparation of unsaturated β-carbonyl compounds. Tetrahedron Lett. 1981, 22, 3043–3046. [Google Scholar] [CrossRef]
- Gol'dfarb, Y.L.; Lifvinov, L.; Mortikov, V.P.; Yu, V. Condensed heteroaromatic systems including a thiophene ring. 36. New complex-forming and chelate compounds of the benzothiophene series with selenium as the donor. Khim. Geterotsikl. Soedin. 1979, 7, 898–904. [Google Scholar]
- Du, Q.; Carrasco, N.; Teplova, M.; Wilds, C.J.; Egli, M.; Huang, Z. Internal derivatization of oligonucleotides with selenium for X-ray crystalography using MAD. J. Am. Chem. Soc. 2001, 124, 24–25. [Google Scholar]
- Weber, R.; Renson, M. Transformation of 3-benzisoselenazolinones to benz[β]selenophene derivatives. Bull. Soc. R. Sci. Liege. 1979, 48, 146–151. [Google Scholar]
- Back, T.G.; Dyck, B.P. Asymmetric cyclization of unsaturated alcohols and carboxylic acids with camphor-based selenium electrophiles. Chem. Commun. 1996, 2567–2568. [Google Scholar]
- Back, T.G.; Dyck, B.P.; Nan, S. Asymmetric electrophilic methoxyselenylations and cyclizations with 3-camphorseleno derivatives. Tetrahedron 1999, 55, 3191–3208. [Google Scholar] [CrossRef]
- Reich, H.J.; Yelm, K.E. Asymmetric induction in the oxidation of [2,2]paracyclophane-substituted selenides. Application of chirality transfer in the selenoxide [2,3] sigmatropic rearrangement. J. Org. Chem. 1991, 56, 5672–5679. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Koguma, Y.; Tanaka, T.; Umeda, R. Cesium carbonate-catalyzed α-phenylchalcogenation of carbonyl compounds with diphenyl dichalcogenide. Molecules 2009, 14, 3367–3375. [Google Scholar]
- Miyano, M.; Smith, J.N.; Dorn, C.R. A synthesis of 11-homo-aldosterone. Tetrahedron 1982, 38, 3447–3455. [Google Scholar] [CrossRef]
- Zaidi, J.H.; Waring, A.J. Synthesis of 3,4-dihydro-3,3,8a-trimethylnaphthalene-1,6(2H, 8aH)-dione, a 4-acylcyclohexa-2,5-dienone. J. Chem. Soc. Chem. Comm. 1980, 618–619. [Google Scholar]
- Salmond, W.G.; Barta, M.A.; Cain, A.M.; Sobala, M.C. Alternative modes of decomposition of allylic selenoxides diastereomeric at selenium. Tetrahedron Lett. 1977, 20, 1683–1686. [Google Scholar]
- Gilman, H.; Cason, L.F. Some addition reactions of chalcones. II. The preparation of some χ-ketoselenides. J. Am. Chem. Soc. 1951, 73, 1074–1076. [Google Scholar] [CrossRef]
- Stevens, R.V.; Albizati, K.F. Synthetic approach to the amphilectane diterpenes: the use of nitriles as terminators of carbocation-olefin cyclizations. J. Org. Chem. 1984, 50, 632–640. [Google Scholar]
- Okeley, N.M.; Zhu, Y.; van der Donk, W.A. Facile chemoselective synthesis of dehydroalanine-containing peptides. Org. Lett. 2000, 2, 3603–3606. [Google Scholar] [CrossRef]
- Mairanovsky, V.G. Electro-deprotection: electrochemical removal of protecting groups. Angew. Chem. Int. Ed. Engl. 1976, 15, 281–292. [Google Scholar]
- Chung, M.K.; Schlaf, M. A catalytic synthesis of thiosilanes and silthianes: palladium nanoparticle-mediated cross-coupling of silanes with thio phenyl and thio vinyl ethers through selective carbon-sulfur bond activation. J. Am. Chem. Soc. 2004, 126, 7386–7392. [Google Scholar] [CrossRef]
- Toshimitsu, A.; Nakano, K.; Mukai, T.; Tamao, K. Steric protection of the selenium atom of the episelenonium ion intermediate to prevent both the racemization of the chiral carbon and the selenophilic attack of carbon nucleophiles. J. Am. Chem. Soc. 1996, 118, 2756–2757. [Google Scholar] [CrossRef]
- Toshimitsu, A.; Terada, M.; Tamao, K. Intramolecular cyclization reaction via a sterically protected episelenonium ion intermediate. Chem. Lett. 1997, 733–734. [Google Scholar]
- Toshimitsu, A.; Hirosawa, C.; Nakano, K.; Mukai, T.; Tamao, K. Stereospecific transformations of chiral compounds using anchimeric assistance of arylthio and arylseleno group. Phosphorus Sulfur Silicon 1997, 120/121, 355–356. [Google Scholar]
- Okamoto, K.; Nishibayashi, Y.; Uemura, S.; Toshimitsu, A. Asymmetric carboselenylation reaction of alkenes with aromatic compounds. Angew. Chem. Int. Ed. 2005, 44, 3588–3591. [Google Scholar]
- Reich, H.J.; Jasperse, C.P.; Renga, J.M. Organoselenium chemistry. Alkylation of acid, ester, amide, and ketone enolates with bromomethyl benzyl selenide and sulfide: preparation of selenocysteine derivatives. J. Org. Chem. 1986, 51, 2981–2988. [Google Scholar] [CrossRef]
- Walter, R.; du Vigneaud, V. 6-hemi-L-selenocystine-oxytocin and 1-deamino-6-hemi-L-selenocystine-oxytocin, highly potent isologs of oxytocin and 1-deamino-oxytocin. J. Am. Chem. Soc. 1965, 87, 4192–4193. [Google Scholar]
- Walter, R.; Chan, W.Y. Syntheses and pharmacological properties of selenium analogs of oxytocin and demaino-oxytocin. J. Am. Chem. Soc. 1967, 89, 3892–3898. [Google Scholar]
- Theodoropoulos, D.; Schwartz, I.L.; Walter, R. Synthesis of selenium-containing peptides. Biochemistry 1967, 6, 3927–3932. [Google Scholar] [CrossRef]
- Soda, K.; Nobuyoshi, E. Glutathione derivative and medicine containing the same as active ingredient. Jpn Patent 04-066567, 1992. [Google Scholar]
- Oikawa, T.; Esaki, N.; Tanaka, H.; Soda, K. Metalloselenonein, the selenium analogue of metallothionein: synthesis and characterization of its complex with copper ions. Proc. Natl. Acad. Sci. USA 1991, 88, 3057–3059. [Google Scholar]
- Metanis, N.; Keinan, E.; Dawson, P.E. Synthetic seleno-glutaredoxin 3 analogues and highly reducing oxidoreductases with enhanced catalytic efficiency. J. Am. Chem. Soc. 2006, 128, 16684–16691. [Google Scholar]
- Tamura, T.; Oikawa, T.; Ohtaka, A.; Fujii, N.; Esaki, N.; Soda, K. Synthesis and characterization of the selenium analog of glutathione disulfide. Anal. Biochem. 1993, 208, 151–154. [Google Scholar]
- Casi, G.; Roelfes, G.; Hilvert, D. Selenoglutaredoxin as a glutathione peroxidase mimic. Chembiochem 2008, 9, 1623–1631. [Google Scholar]
- Koide, T.; Itoh, H.; Otaka, A.; Yasui, H.; Kuroda, M.; Esaki, N.; Soda, K.; Fujii, N. Synthetic study on selenocysteine-containing peptides. Chem. Pharm. Bull. 1993, 41, 502–506. [Google Scholar]
- Gieselman, M.D.; Xie, L.; van der Donk, W.A. Synthesis of a selenocysteine-containing peptide by native chemical ligation. Org. Lett. 2001, 3, 1331–1334. [Google Scholar]
- Flögel, O.; Casi, G.; Hilvert, D.; Seebach, D. Preparation of the β3-homoselenocysteine derivatives Fmoc-β3hSec(PMB)-OH and Boc-β3hSec(PMB)-OH for solution and solid-phase-peptide synthesis and selenoligation. Helv. Chim. Acta 2007, 90, 1651–1666. [Google Scholar]
- Harris, K.M.; Flemer, Jr, S.; Hondal, R.J. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J. Pep. Sci. 2007, 13, 81–93. [Google Scholar]
- Flemer, S., Jr.; Hondal, R.J. DTNP as an Effective and Gentle Deprotectant for Common Selenocysteine Protecting Groups. Unpublished Results.
- Muttenthaler, M.; Ramos, Y.G.; Feytens, D.; de Araujo, A.D.; Alewood, P.F. p-Nitrobenzyl protection for cysteine and selenocysteine: a more stable alternative to the acetamidomethyl group. Biopolymers 2010, 94, 423–432. [Google Scholar]
- Veber, D.F.; Milkowski, J.D.; Varga, S.L.; Denkewalter, R.G.; Hirschmann, R. Acetamidomethyl. A novel thiol protecting group for cysteine. J. Am. Chem. Soc. 1971, 94, 5456–5461. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flemer Jr., S. Selenol Protecting Groups in Organic Chemistry: Special Emphasis on Selenocysteine Se-Protection in Solid Phase Peptide Synthesis. Molecules 2011, 16, 3232-3251. https://doi.org/10.3390/molecules16043232
Flemer Jr. S. Selenol Protecting Groups in Organic Chemistry: Special Emphasis on Selenocysteine Se-Protection in Solid Phase Peptide Synthesis. Molecules. 2011; 16(4):3232-3251. https://doi.org/10.3390/molecules16043232
Chicago/Turabian StyleFlemer Jr., Stevenson. 2011. "Selenol Protecting Groups in Organic Chemistry: Special Emphasis on Selenocysteine Se-Protection in Solid Phase Peptide Synthesis" Molecules 16, no. 4: 3232-3251. https://doi.org/10.3390/molecules16043232
APA StyleFlemer Jr., S. (2011). Selenol Protecting Groups in Organic Chemistry: Special Emphasis on Selenocysteine Se-Protection in Solid Phase Peptide Synthesis. Molecules, 16(4), 3232-3251. https://doi.org/10.3390/molecules16043232