From Polymer to Small Organic Molecules: A Tight Relationship between Radical Chemistry and Solid-Phase Organic Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
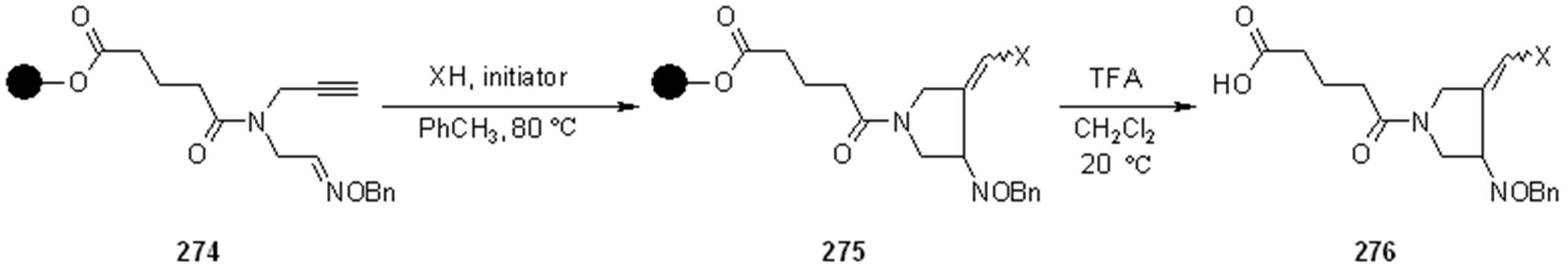{kind=link}
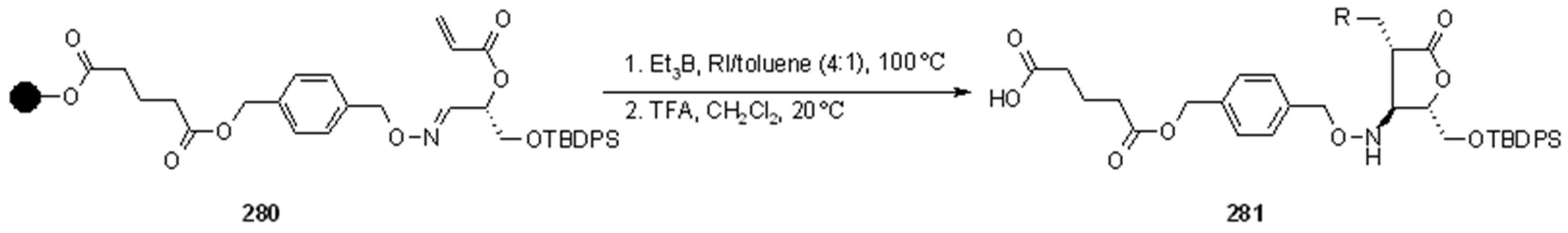
{kind=link}
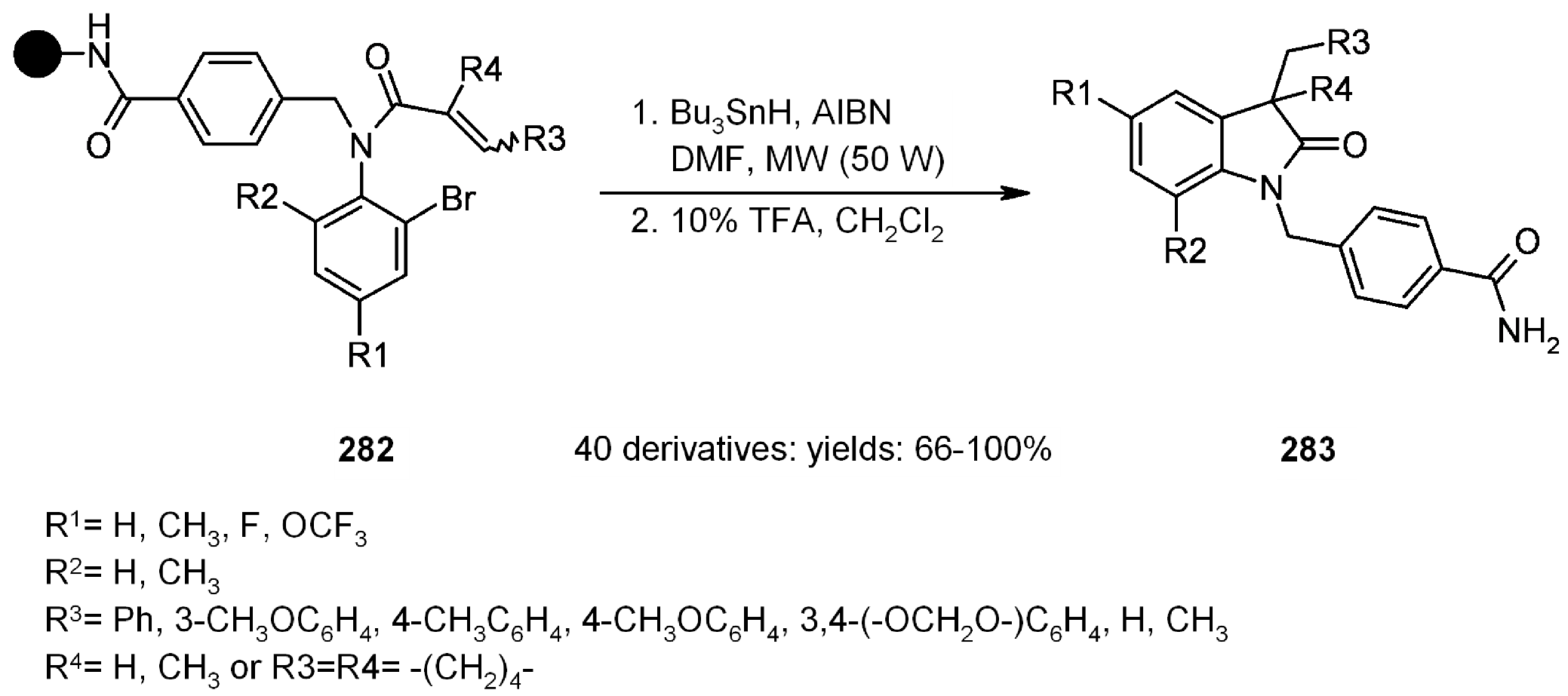
{kind=link}
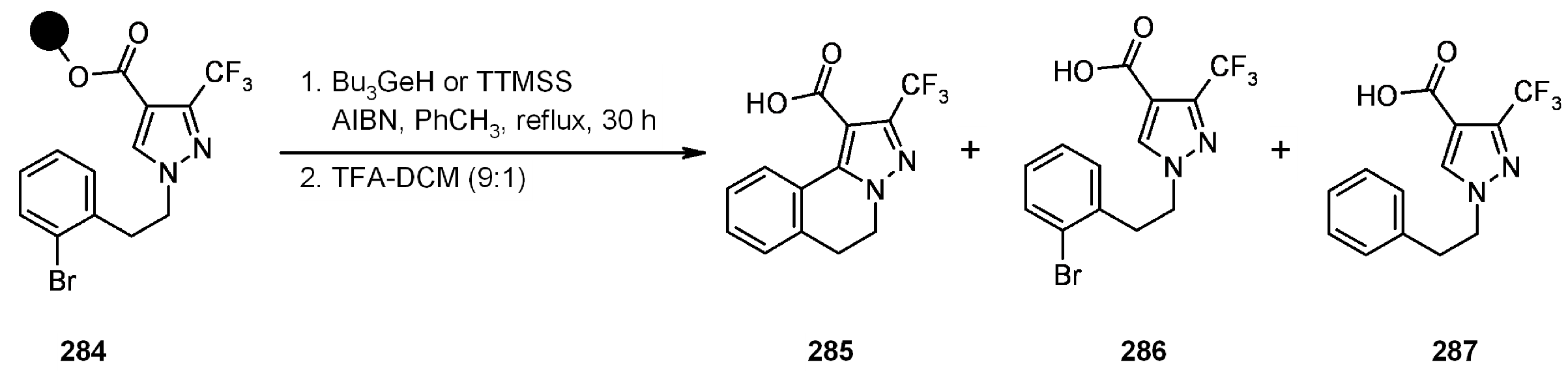
{kind=link}
{kind=link}
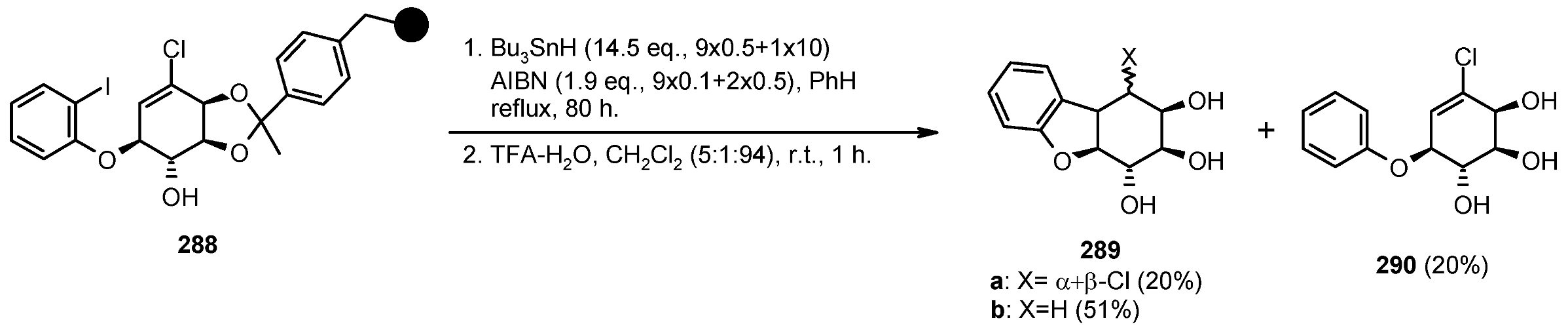{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
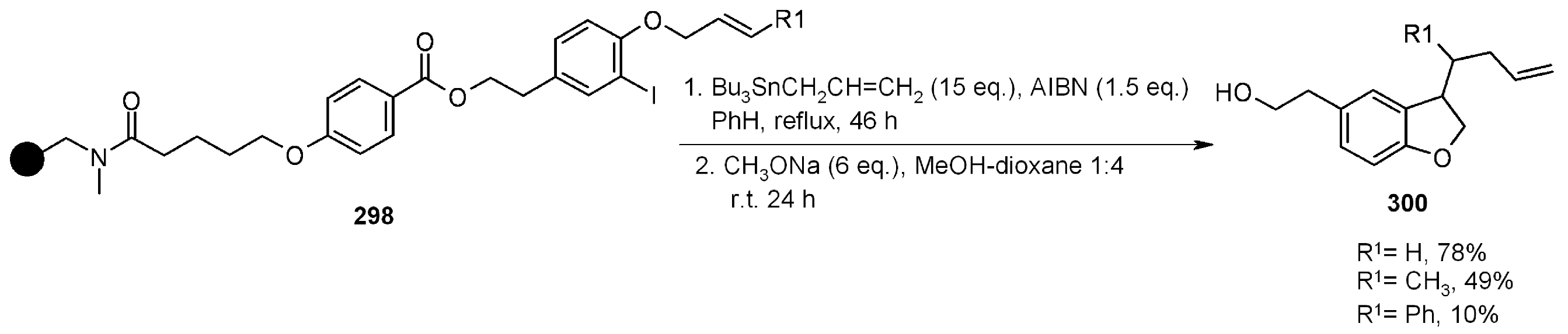{kind=link}
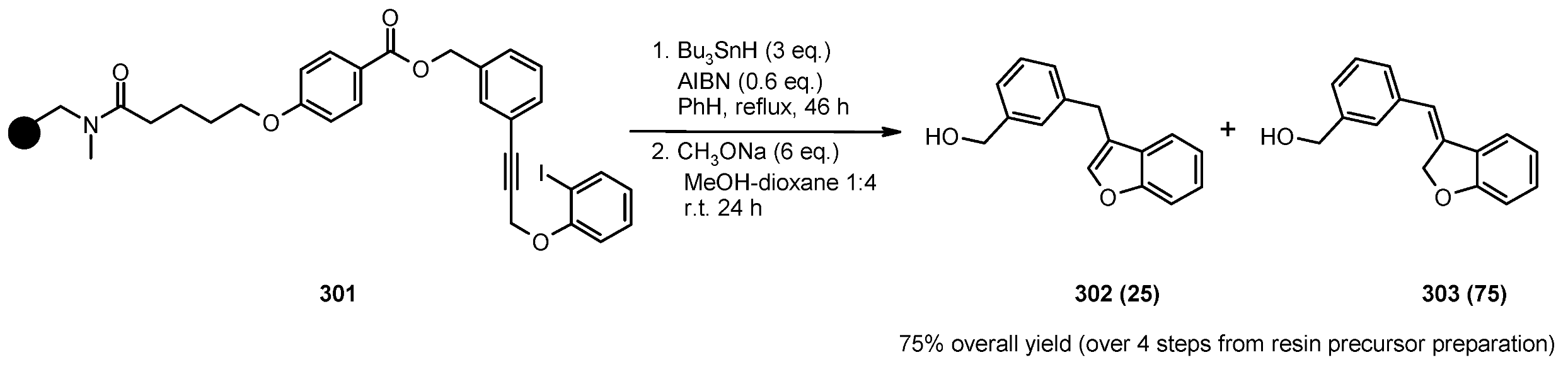{kind=link}
{kind=link}
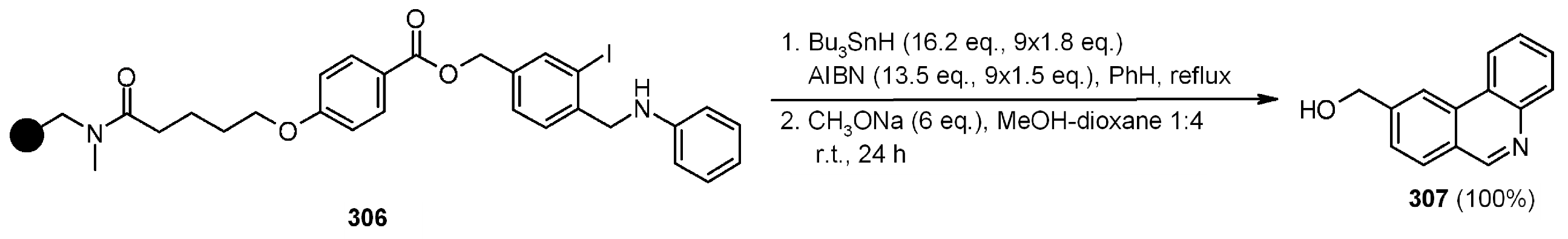{kind=link}
{kind=link}
{kind=link}
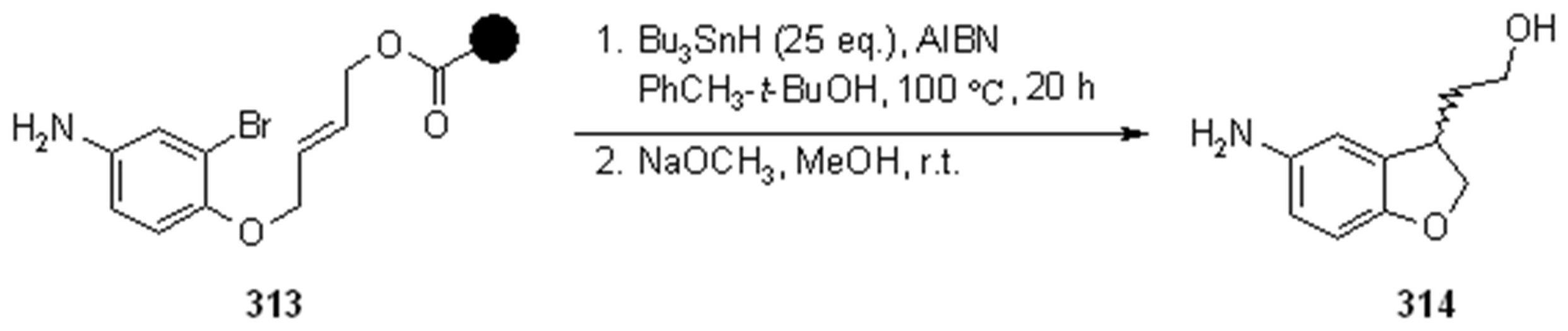{kind=link}
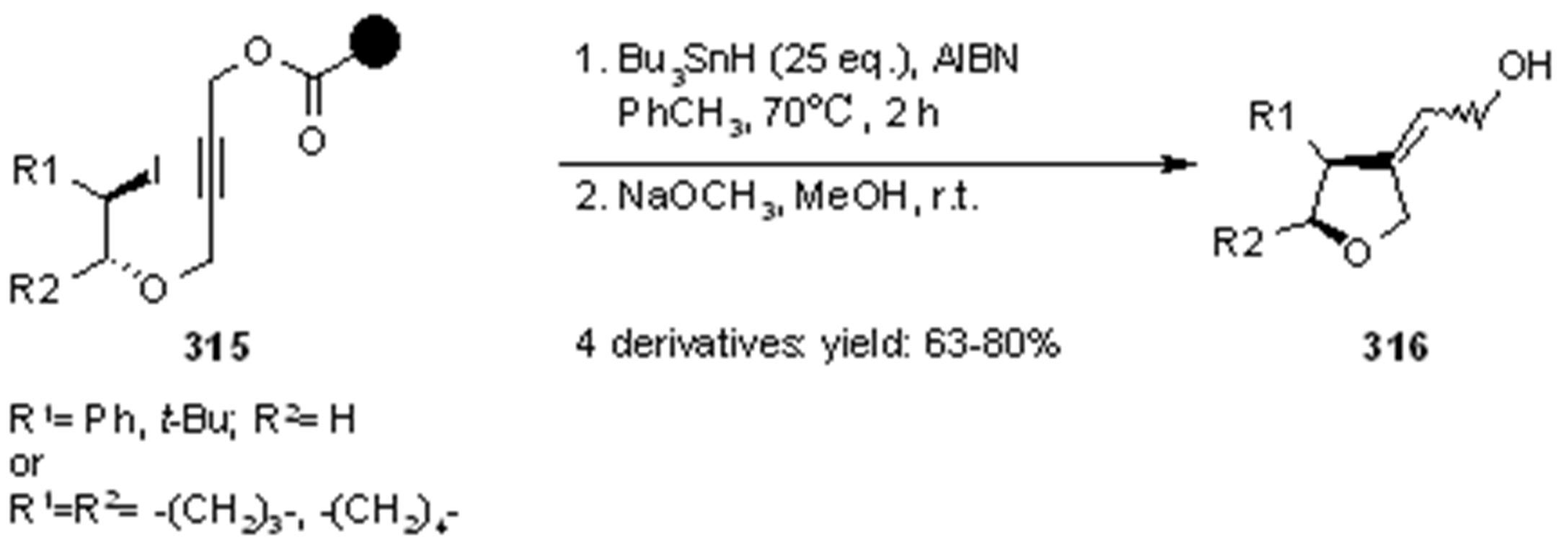{kind=link}
{kind=link}
{kind=link}
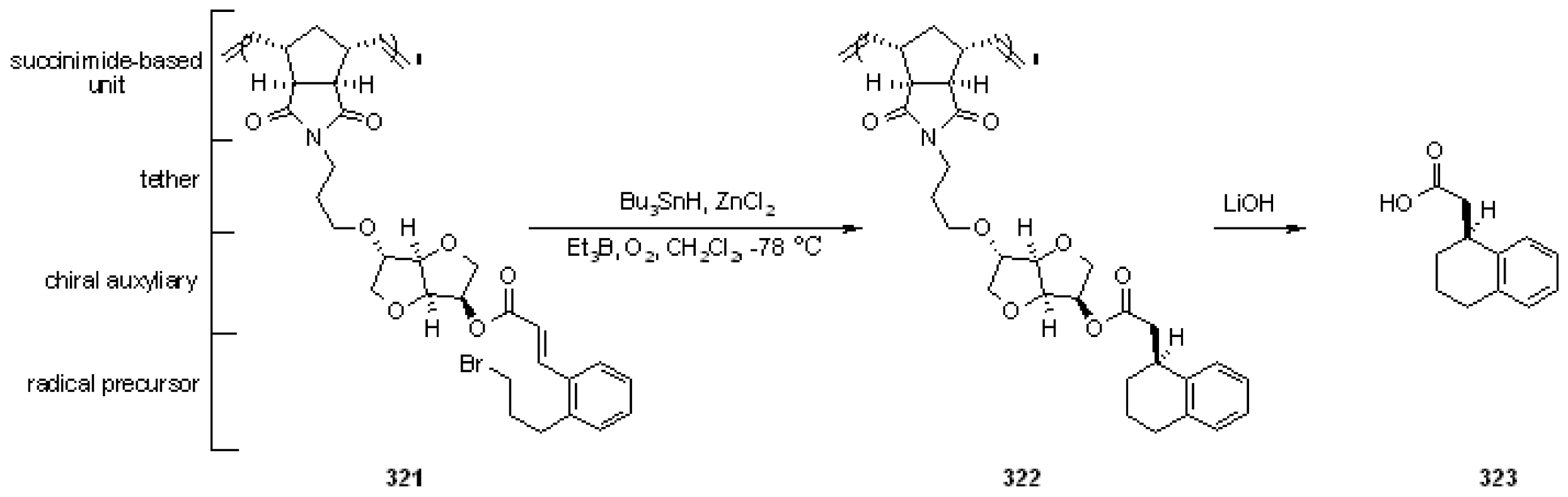{kind=link}
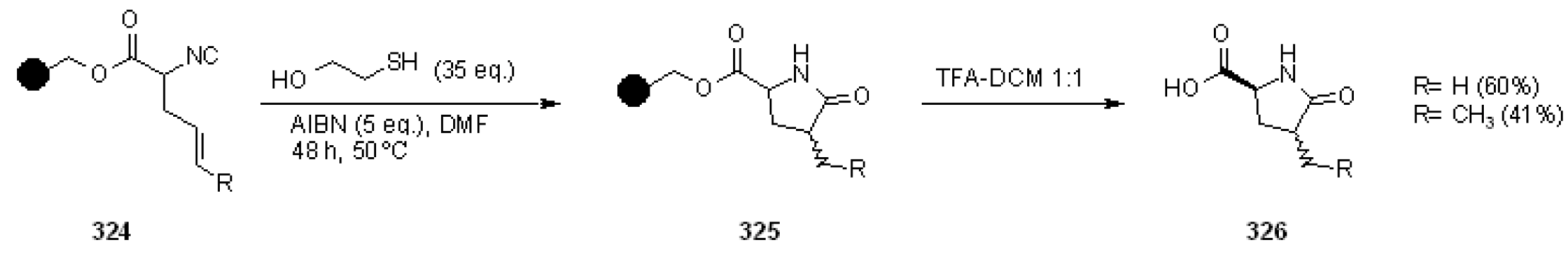{kind=link}
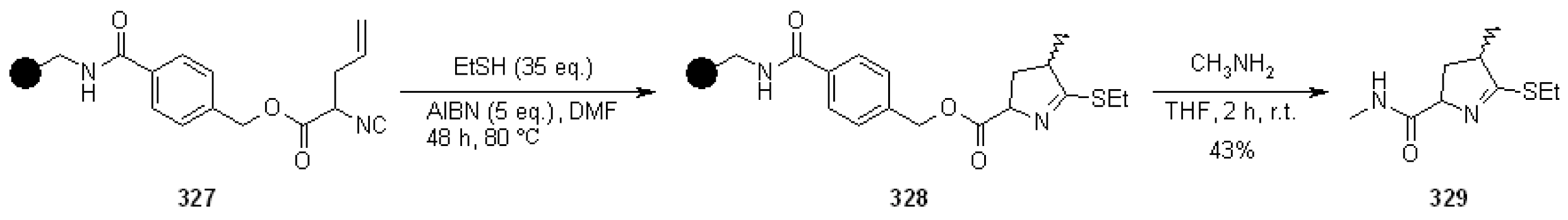{kind=link}
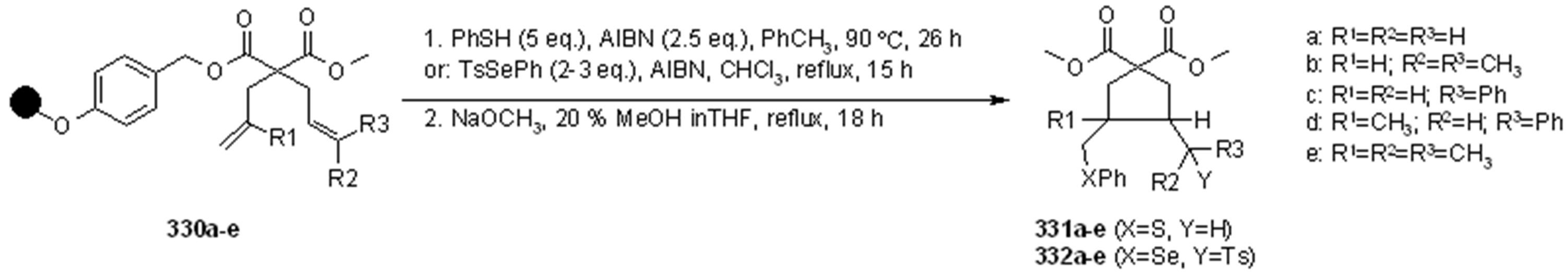{kind=link}
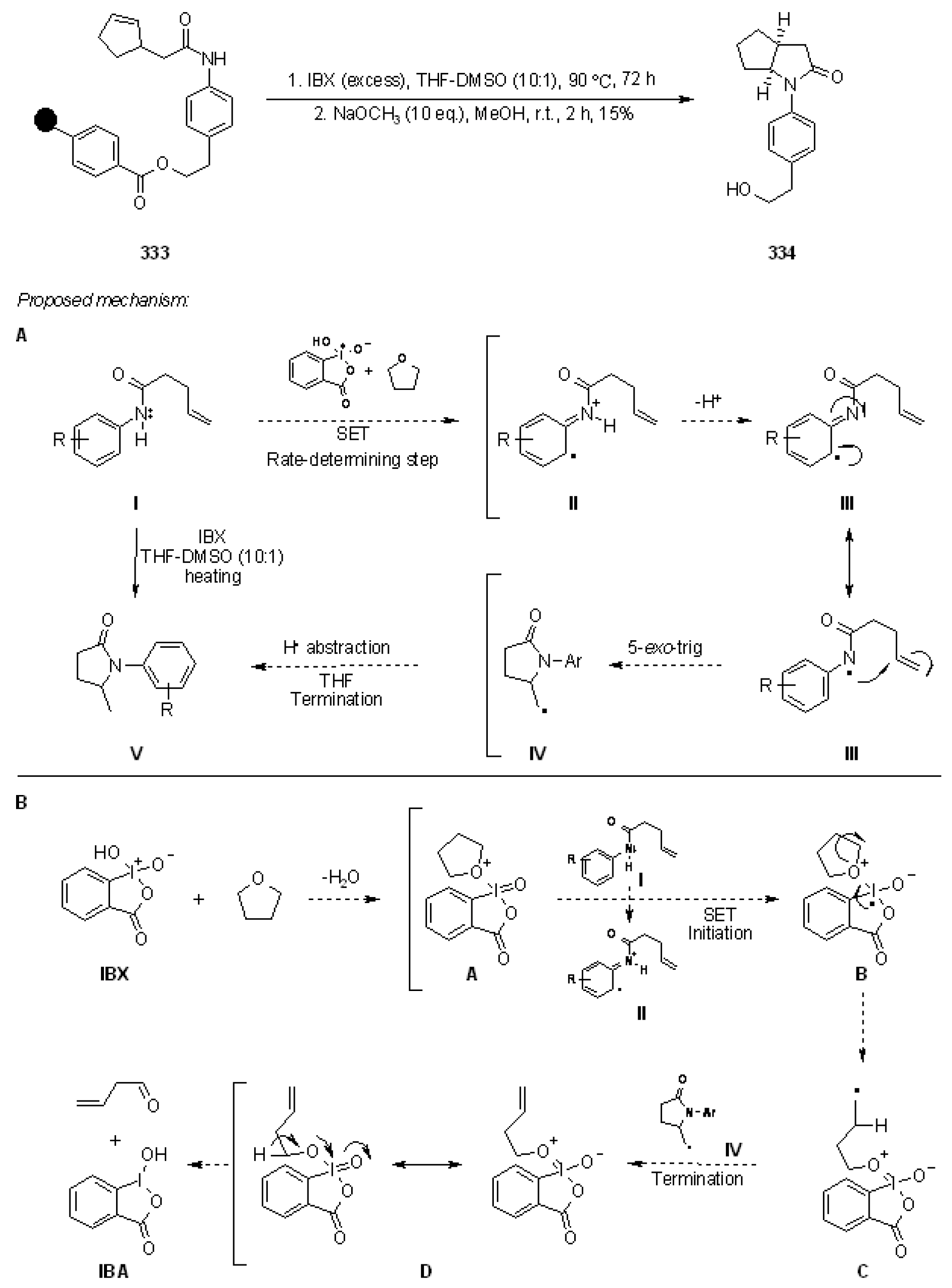{kind=link}
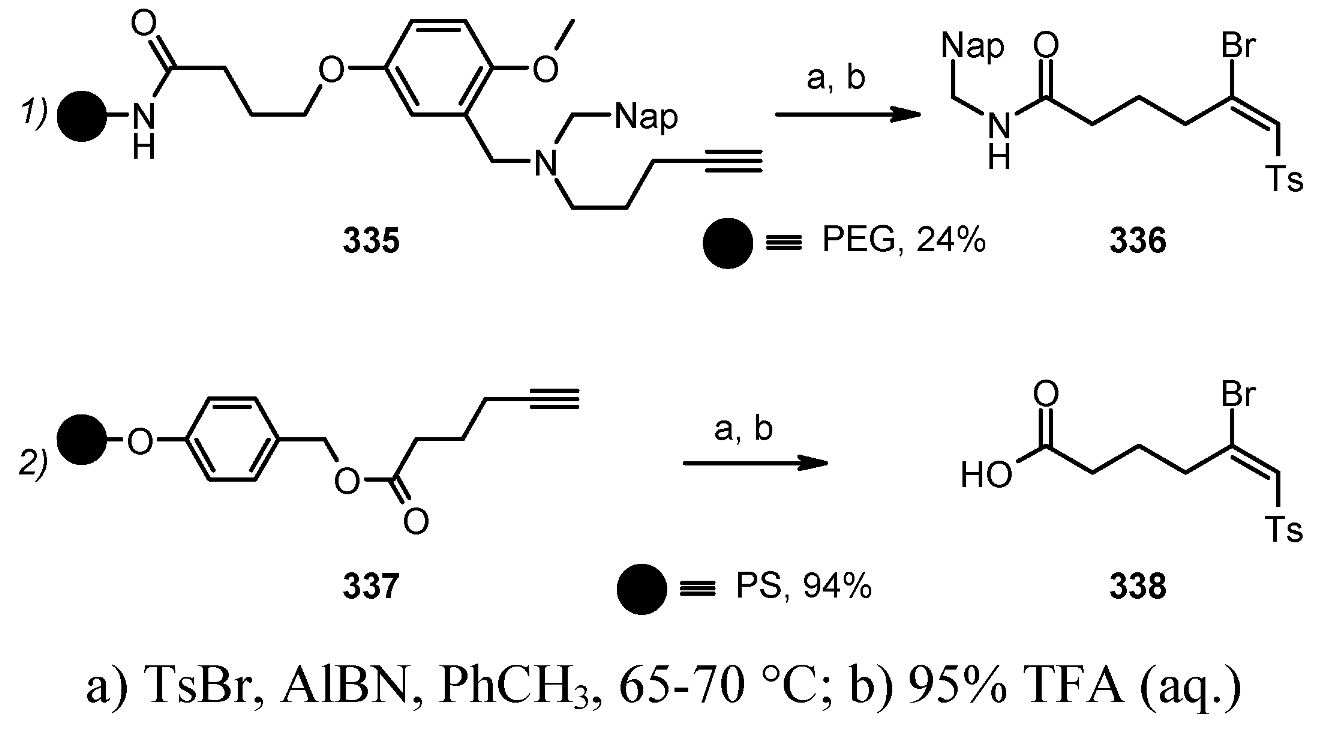{kind=link}
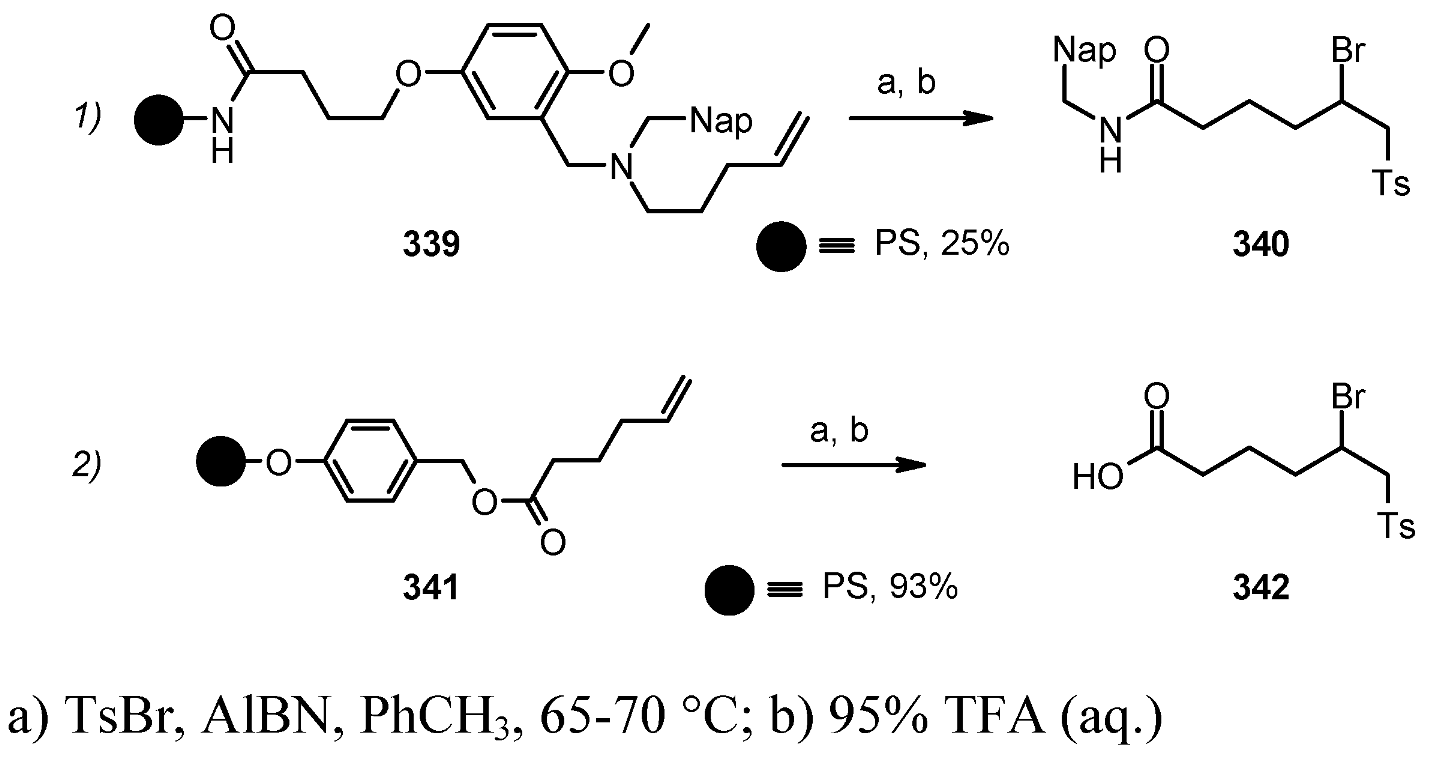{kind=link}
{kind=link}
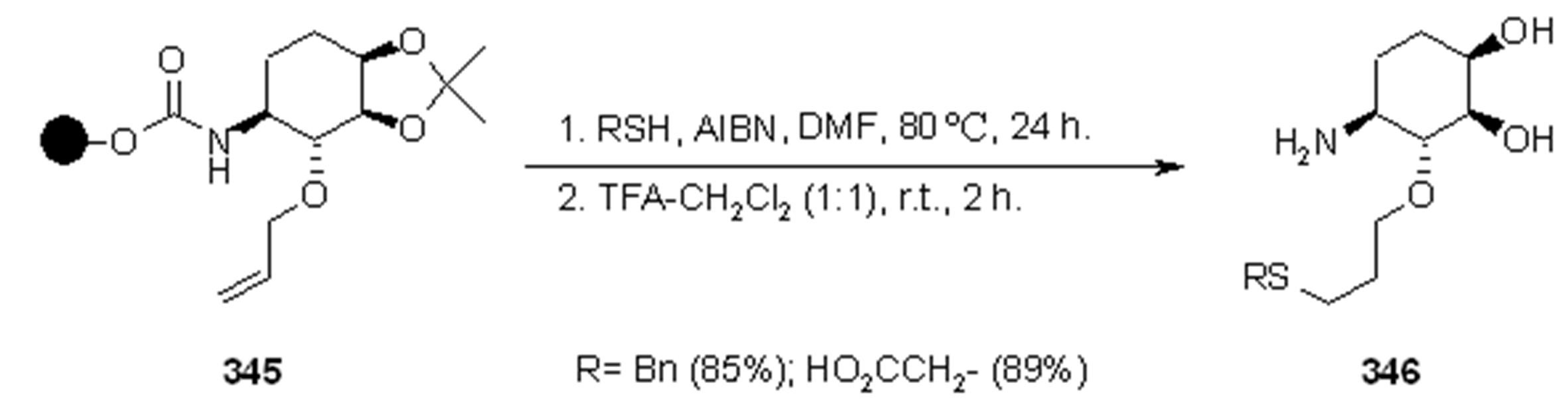{kind=link}
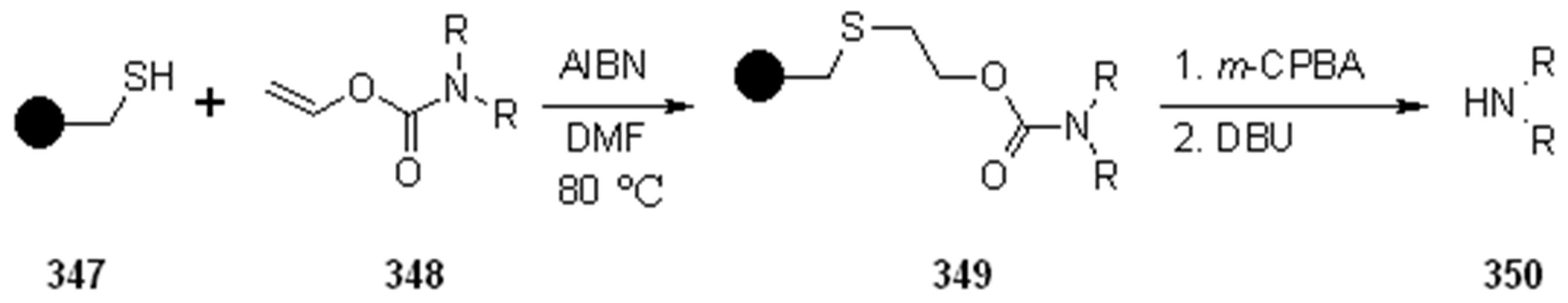{kind=link}
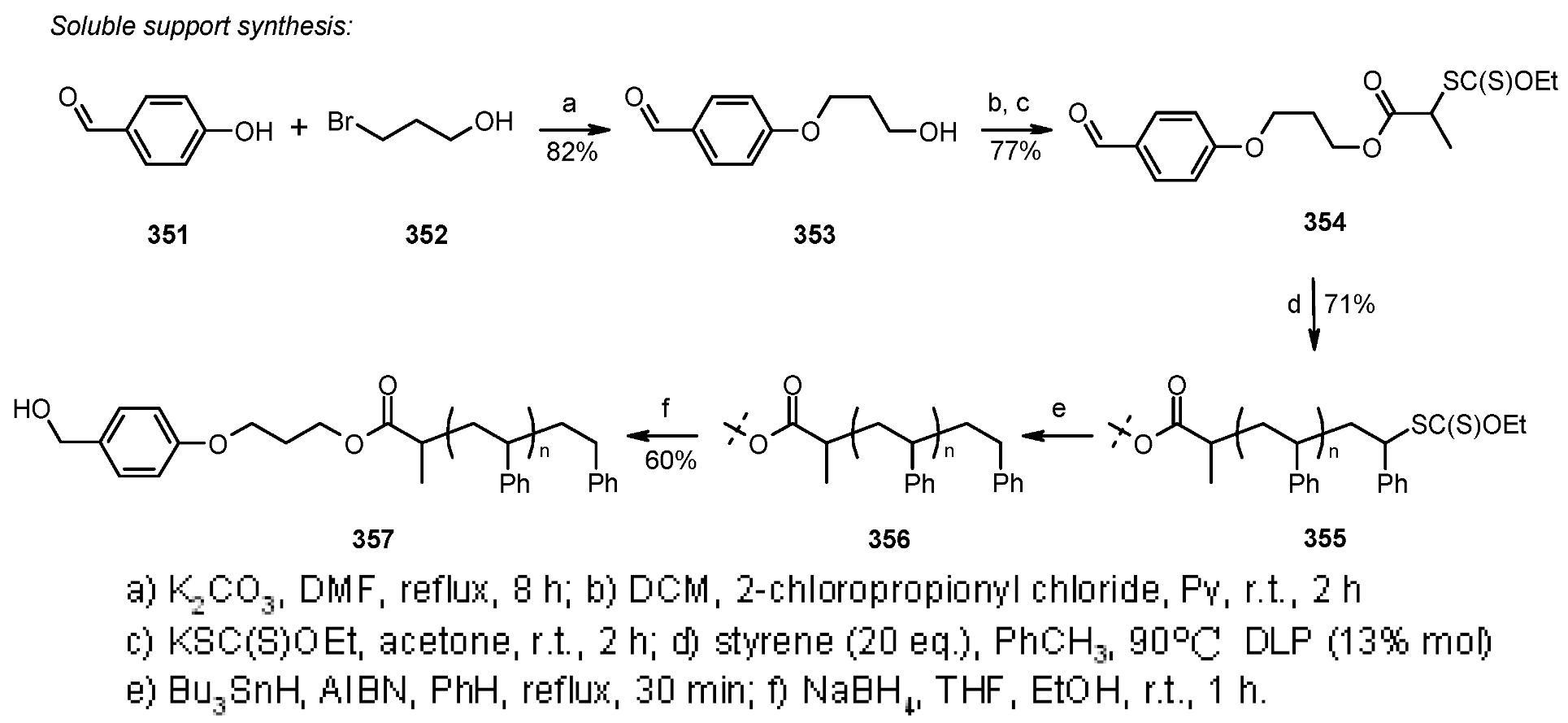{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| AIBN | Azo-bis-isobutyronitrile |
| AMBN | azobismethylisobutyronitrile |
| ATRC | Transition-metal-catalyzed atom transfer radical cyclization |
| 9-BBN | 9-Borabicyclo[3.3.1]nonane |
| CAN | Ceric ammonium nitrate |
| DAST | Diethylaminosulfur trifluoride |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DIC | Diisopropyl carbodiimide |
| DIPAD | Diisopropyl azadicarboxylate |
| DIPEA | diisopropylethylamine |
| DLP | lauroylperoxide |
| DMA | N,N-Dimethylacetamide |
| DMAP | 4-dimethylaminopyridine |
| DMF | N,N-Dimethylaminoformamide |
| EPHP | N-ethylpiperidine hypophosphite |
| FRET | Fluorescence Resonance Energy Transfer |
| HASC | α-Hetero-Atom Substituted Carbonyl |
| HBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HMPA | Hexamethylphosphoramide |
| IBX | 2-Iodoxybenzoic acid |
| LA | Lewis acid |
| NMM | N-methyl morpholine |
| NMP | N-methyl pyrrolidine |
| NMQ | N-methylquinolinium |
| PMHS | Polymethylhydrosiloxane |
| RCM | Ring-closing metathesis |
| SET | single electron transfer |
| SP | Solid-phase |
| SPS | Solid-phase synthesis |
| TBGH | Tributylgermanium hydride |
| TBTH | Tributyltin hydride |
| TEMPO | Tetramethyl pentahydropyridine oxide |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TON | Turnover number |
| TTF | Tetrathiofulvalene |
| TTMSS | Tris(trimethylsilyl)silane |
| Troc | 2,2,2-Trichloroethoxycarbonyl |
1. Introduction
2. Solid-Supported Reagents, Resins and Linkers
2.1. Linkers and Resins
2.1.1. Sulfur-Based Linkers













2.1.2. Selenium (and Tellurium)-based Linkers






















2.1.3. Oxygen-based Linkers
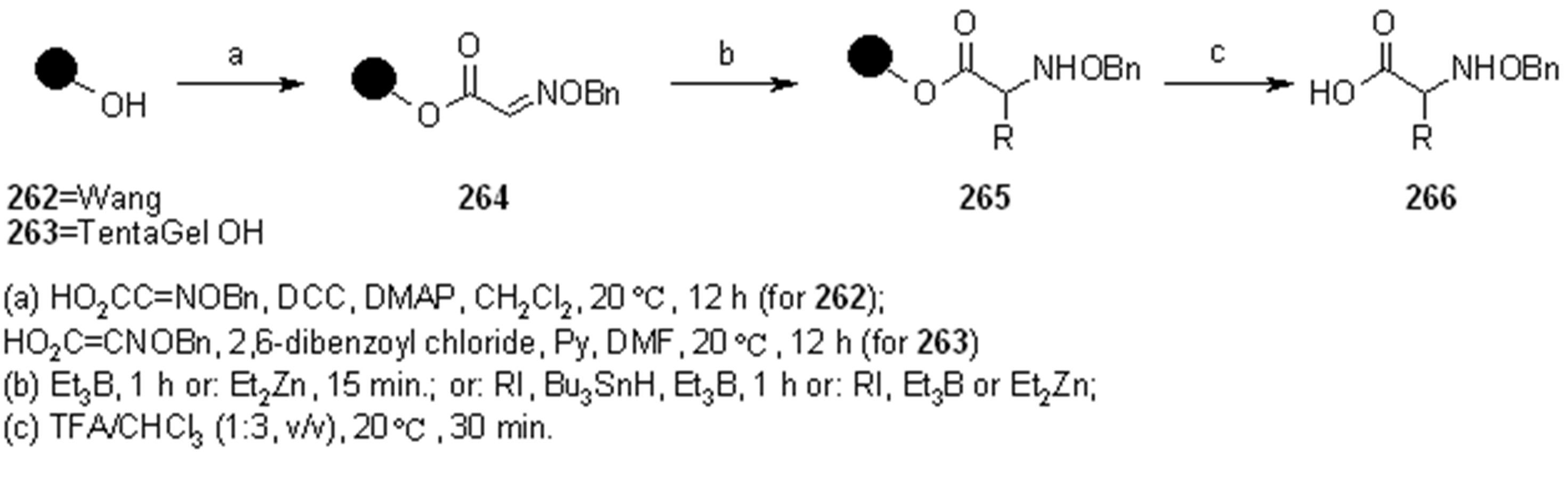
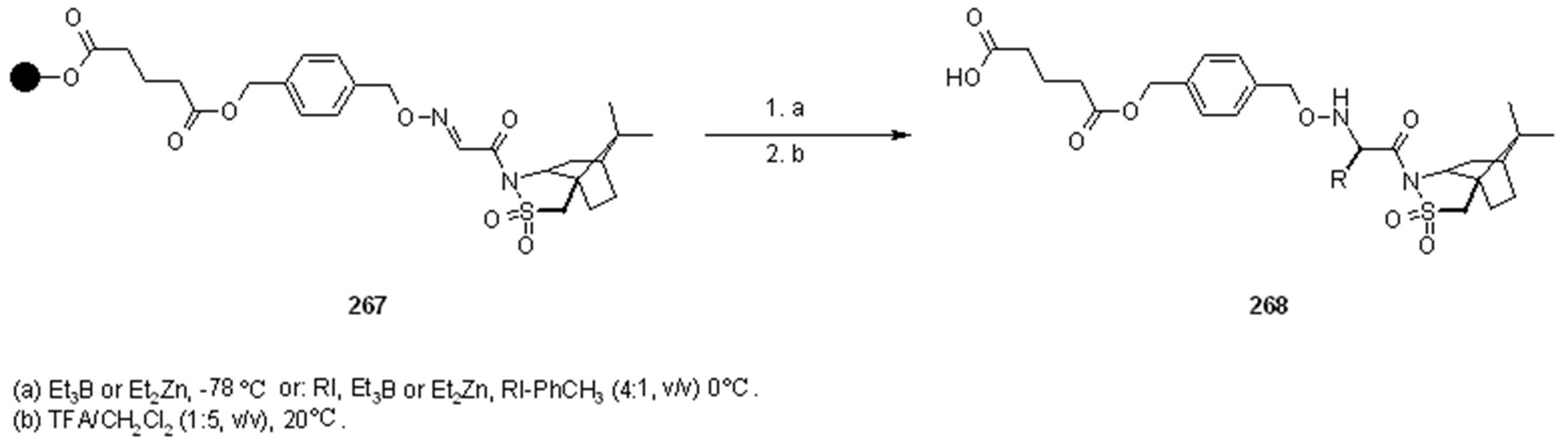
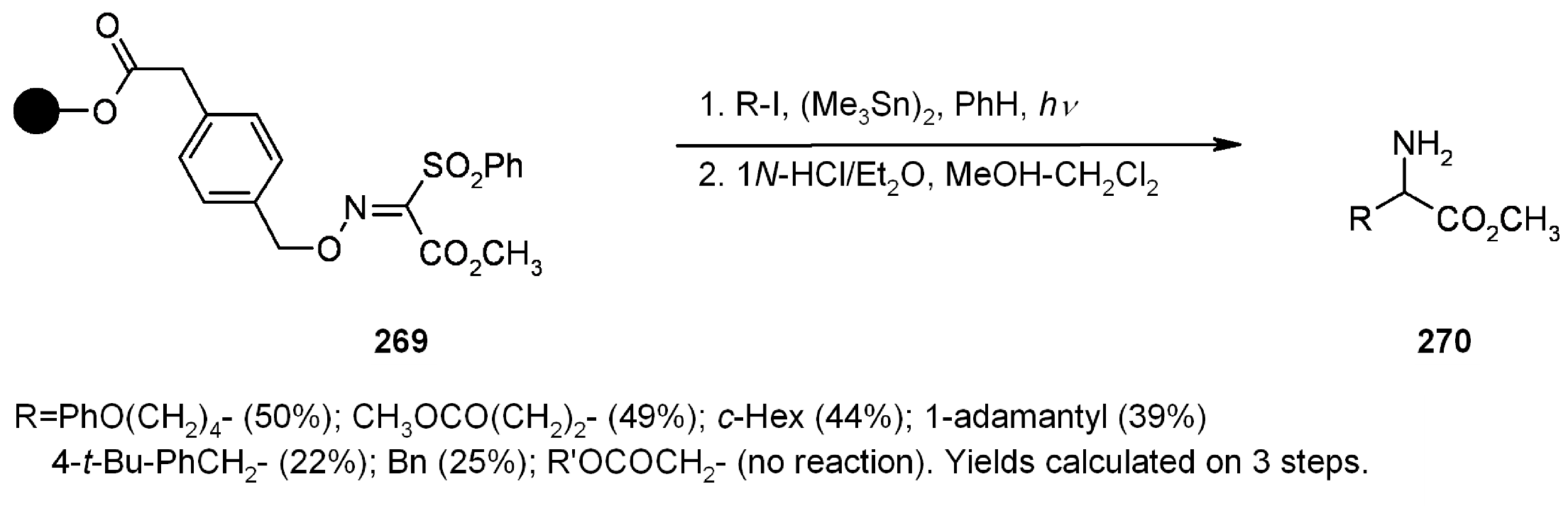
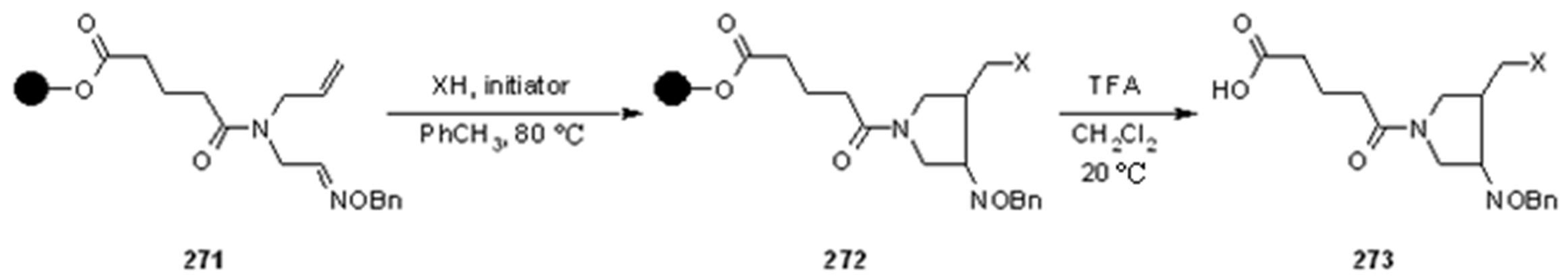
2.1.3.1. Cleavage of Hydroxylamine-based Linkers



2.1.3.2. Addition of Ketyl Radicals to α-β Unsaturated Esters



2.1.3.3. HASC Traceless Linker

2.1.4. “Carbon–Based” Linkers



2.2. Solid-Supported Reagents





























3. Exploitation of Radical Chemistry in SP Synthesis
3.1. Carbon-Carbon Bond Formation through Intermolecular Reactions
3.1.1. Substitutions Alpha to Carbonyl Groups







3.1.2. Radical Addition to Alkenes







3.1.3. Radical Addition to Oxime



3.2. Carbon-Carbon Bond Formation through Intramolecular Reactions
3.2.1. Radical Addition to Oximes




3.2.2. Radical Addition to Alkenes, Alkynes and Arenes





















3.3. Carbon-Heteroatom Formation












3.4. Other


4. Conclusions
Acknowledgements
References and Notes
- Gomberg, M. An Instance of Trivalent Carbon: Triphenylmethyl. J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar] [CrossRef]
- Geng, Y.; Discher, D.E.; Justynska, J.; Schlaad, H. Grafting Short Peptides onto Polybutadiene-block-poly(ethylene oxide): A Platform for Self-Assembling Hybrid Amphiphiles. Angew. Chem. Int. Ed. Engl. 2006, 45, 7578–7581. [Google Scholar] [CrossRef]
- Siegenthaler, K.O.; Schäfer, A.; Studer, A. Chemical Surface Modification via Radical C-C Bond-Forming Reactions. J. Am. Chem. Soc. 2007, 129, 5826–5827. [Google Scholar] [CrossRef]
- Hensarling, R.M.; Doughty, V.A.; Chan, J.W.; Patton, D.L. Clicking Polymer Brushes with Thiol-yne Chemistry: Indoors and Out. J. Am. Chem. Soc. 2009, 131, 14673–14675. [Google Scholar]
- Walling, C. Some properties of radical reactions important in synthesis. Tetrahedron 1985, 41, 3887–3900. [Google Scholar] [CrossRef]
- Newcomb, M. Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151–1176. [Google Scholar] [CrossRef]
- Gansäuer, A.; McGhee, A.; Procter, D. Radical Chemistry on Solid Support. In Radicals in Synthesis II; Springer: Berlin/Heidelberg, Germany, 2006; Volume 264, pp. 93–134. [Google Scholar]
- Curran, D.P.; Yang, F.; Cheong, J.H. Relative Rates and Approximate Rate Constants for Inter- and Intramolecular Hydrogen Transfer Reactions of Polymer-Bound Radicals. J. Am. Chem. Soc. 2002, 124, 14993–15000. [Google Scholar] [CrossRef]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Convergent solid-phase peptide synthesis. Tetrahedron 1993, 49, 11065–11133. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile protecting groups and linkers. J. Chem. Soc. Perkin Trans. 1 2002, 125–142. [Google Scholar]
- Reitz, A.B. Recent advances in traceless linkers. Curr. Opin. Drug Discov. Dev. 1999, 2, 358–364. [Google Scholar]
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Scott, P.J.H.; Steel, P.G. Diversity Linker Units for Solid-Phase Organic Synthesis. Eur. J. Org. Chem. 2006, 2006, 2251–2268. [Google Scholar] [CrossRef]
- Jung, K.W.; Zhao, X.Y.; Janda, K.D. A linker that allows efficient formation of aliphatic C-H bonds on polymeric supports. Tetrahedron Lett. 1996, 37, 6491–6494. [Google Scholar] [CrossRef]
- Barluenga, S.; Dakas, P.Y.; Ferandin, Y.; Meijer, L.; Winssinger, N. Modular Asymmetric Synthesis of Aigialomycin D, a Kinase-Inhibitory Scaffold. Angew. Chem. Int. Ed. Engl. 2006, 45, 3951–3954. [Google Scholar] [CrossRef]
- Parsons, A.F.; Pettifer, R.M. A radical approach to N-desulfonylation. Tetrahedron Lett. 1996, 37, 1667–1670. [Google Scholar] [CrossRef]
- Luo, J.; Huang, W. A new strategy for solid phase synthesis of a secondary amide library using sulfonamide linker via radical traceless cleavage. Mol. Divers. 2003, 6, 33–41. [Google Scholar] [CrossRef]
- D'Herde, J.N.P.; J. de Clercq, P. Carbon-carbon bond formation on solid support. Application of the classical Julia-Lythgoe olefination. Tetrahedron Lett. 2003, 44, 6657–6659. [Google Scholar]
- Allin, S.M.; Bowman, W.R.; Karim, R.; Rahman, S.S. Aromatic homolytic substitution using solid phase synthesis. Tetrahedron 2006, 62, 4306–4316. [Google Scholar] [CrossRef]
- Roberts, B.P. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev. 1999, 28, 25–35. [Google Scholar] [CrossRef]
- McAllister, L.A.; Brand, S.; de Gentile, R.; Procter, D.J. The first Pummerer cyclisations on solid phase. Convenient construction of oxindoles enabled by a sulfur-link to resin. Chem. Commun. (Camb.) 2003, 2380–2381. [Google Scholar]
- Turner, K.L.; Baker, T.M.; Islam, S.; Procter, D.J.; Stefaniak, M. Solid-Phase Approach to Tetrahydroquinolones Using a Sulfur Linker Cleaved by SmI2. Org. Lett. 2006, 8, 329–332. [Google Scholar] [CrossRef]
- McAllister, L.A.; Turner, K.L.; Brand, S.; Stefaniak, M.; Procter, D.J. Solid Phase Approaches to N-Heterocycles Using a Sulfur Linker Cleaved by SmI2. J. Org. Chem. 2006, 71, 6497–6507. [Google Scholar] [CrossRef]
- McAllister, L.A.; McCormick, R.A.; Procter, D.J. Sulfide- and selenide-based linkers in phase tag-assisted synthesis. Tetrahedron 2005, 61, 11527–11576. [Google Scholar] [CrossRef]
- Ruhland, T.; Andersen, K.; Pedersen, H. Selenium-Linking Strategy for Traceless Solid-Phase Synthesis: Direct Loading, Aliphatic C-H Bond Formation upon Cleavage and Reaction Monitoring by Gradient MAS NMR Spectroscopy. J. Org. Chem. 1998, 63, 9204–9211. [Google Scholar]
- Ruhland, T.; Torang, J.; Pedersen, H.; Madsen, J.C.; Bang, K.S. Traceless Solid Phase Synthesis with Polystyrene-Bound Tellurium and in Comparison with Polystyrene-Bound Selenium. Synthesis 2004, 2323–2328. [Google Scholar]
- Ruhland, T.; Torang, J.; Pedersen, H.; Madsen, J.C.; Bang, K.S. Green Traceless Cleavage from Resin-Bound Selenium and Tellurium and Analysis by 2D 29Si/1H HR-MAS NMR Spectroscopy. Synthesis 2005, 1635–1640. [Google Scholar]
- Nicolaou, K.C.; Pastor, J.; Barluenga, S.; Winssinger, N. Polymer-supported selenium reagents for organic synthesis. Chem. Commun. (Camb.) 1998, 1947–1948. [Google Scholar]
- Nicolaou, K.C.; Mitchell, H.J.; Fylaktakidou, K.C.; Suzuki, H.; Rodríguez, R.M. 1,2-Seleno Migrations in Carbohydrate Chemistry: Solution and Solid-Phase Synthesis of 2-Deoxy Glycosides, Orthoesters, and Allyl Orthoester. Angew. Chem. Int. Ed. Engl. 2000, 39, 1089–1093. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Fylaktakidou, K.C.; Mitchell, H.J.; van Delft, F.L.; Rodríguez, R.M.; Conley, S.R.; Jin, Z. Total Synthesis of Everninomicin 13,384-1—Part 4: Explorations of Methodology; Stereocontrolled Synthesis of 1,1′-Disaccharides, 1,2-Seleno Migrations in Carbohydrates, and Solution- and Solid-Phase Synthesis of 2-Deoxy Glycosides and Orthoesters. Chem. Eur. J. 2000, 6, 3166–3185. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Roecker, A.J.; Pfefferkorn, J.A.; Cao, G.Q. A Novel Strategy for the Solid-Phase Synthesis of Substituted Indolines. J. Am. Chem. Soc. 2000, 122, 2966–2967. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Roecker, A.J.; Hughes, R.; van Summeren, R.; Pfefferkorn, J.A.; Winssinger, N. Novel strategies for the solid phase synthesis of substituted indolines and indoles. Bioorg. Med. Chem. 2003, 11, 465–476. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Cao, G.Q.; Kim, S.; Kessabi, J. A Facile Method for the Solution and Solid-Phase Synthesis of Substituted [3.3.1] Bicycles. Org. Lett. 1999, 1, 807–810. [Google Scholar] [CrossRef]
- Berlin, S.; Ericsson, C.; Engman, L. Radical Carbonylation/Reductive Cyclization for the Construction of Tetrahydrofuran-3-ones and Pyrrolidin-3-ones. J. Org. Chem. 2003, 68, 8386–8396. [Google Scholar] [CrossRef]
- Fujita, K.I.; Watanabe, K.; Oishi, A.; Ikeda, Y.; Taguchi, Y. Preparation of Polymer-Supported Selenocyanates and their Application to Solid-Phase Oxyselenenylation-Deselenylation. Synlett 1999, 1999, 1760–1762. [Google Scholar] [CrossRef]
- Qian, H.; Huang, X. Polystyrene-Supported Selenosulfonates: Efficient Reagents for Regio- and Stereocontrolled Synthesis of Vinyl Sulfones. Synlett 2001, 1913–1916. [Google Scholar] [CrossRef]
- Qian, H.; Huang, X. Polystyrene-supported selenosulfonates: efficient reagents for the synthesis of acetylenic sulfones. Tetrahedron Lett. 2002, 43, 1059–1061. [Google Scholar] [CrossRef]
- Qian, H.; Huang, X. Solid-Phase Synthesis of alpha-Keto Sulfones. Synthesis 2006, 1934–1936. [Google Scholar]
- Qian, H.; Huang, X. Radical Cyclization of 1,6-Diene Using Polystyrene-Supported Selenosulfones. J. Comb. Chem. 2003, 5, 569–576. [Google Scholar] [CrossRef]
- Myers, R.M.; Langston, S.P.; Conway, S.P.; Abell, C. Reductive Cleavage of N-O Bonds Using Samarium(II) Iodide in a Traceless Release Strategy for Solid-Phase Synthesis. Org. Lett. 2000, 2, 1349–1352. [Google Scholar] [CrossRef]
- Gustafsson, M.; Olsson, R.; Andersson, C.M. General combinatorial synthesis of tertiary amines on solid support. A novel conditional release strategy based on traceless linking at nitrogen. Tetrahedron Lett. 2001, 42, 133–136. [Google Scholar] [CrossRef]
- Meloni, M.M.; Taddei, M. Solid-Phase Synthesis of beta-Lactams via the Miller Hydroxamate Approach. Org. Lett. 2001, 3, 337–340. [Google Scholar] [CrossRef]
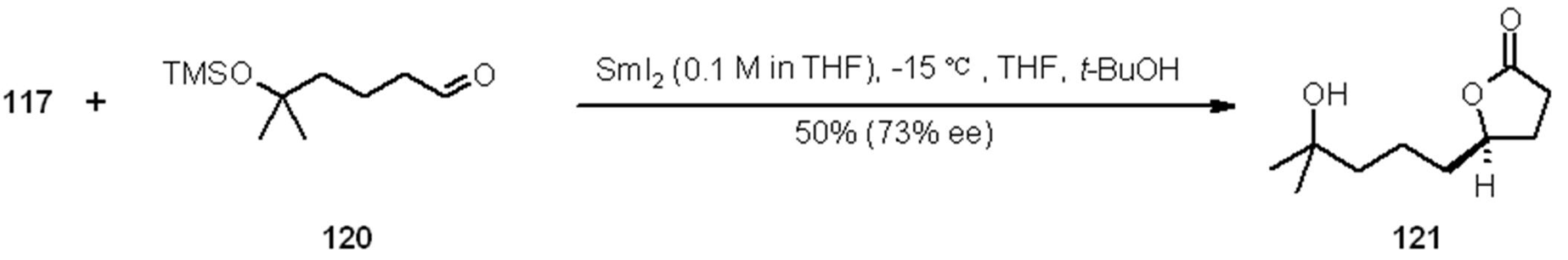
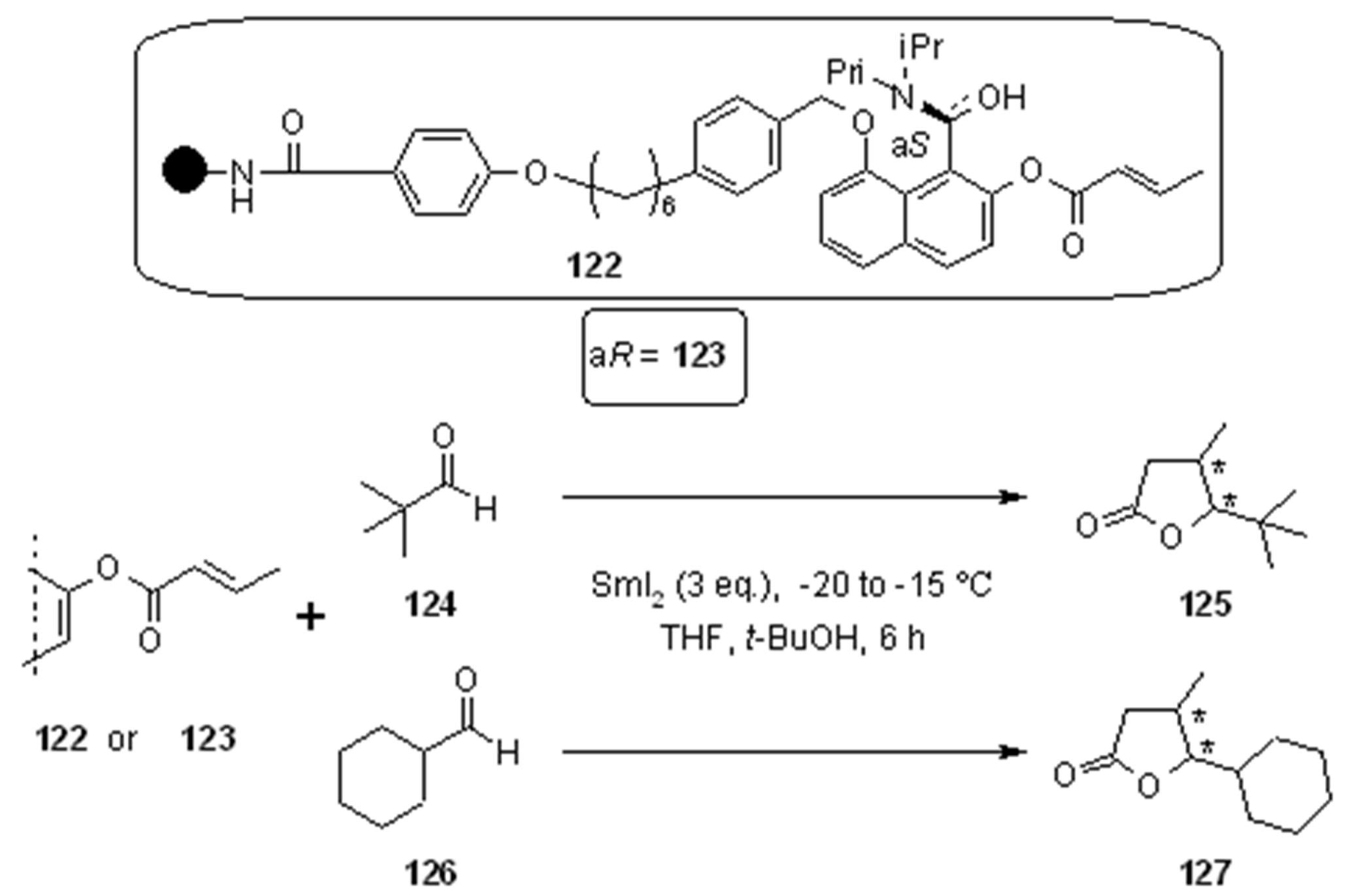
- Kerrigan, N.J.; Hutchison, P.C.; Heightman, T.D.; Procter, D.J. Application of an ephedrine chiral linker in a solid-phase, 'asymmetric catch-release' approach to gamma-butyrolactones. Chem. Commun. (Camb.) 2003, 21, 1402–1403. [Google Scholar]
- Kerrigan, N.J.; Hutchison, P.C.; Heightman, T.D.; Procter, D.J. Development of a solid-phase 'asymmetric resin-capture-release' process: application of an ephedrine chiral resin in an approach to gamma-butyrolactones. Org. Biomol. Chem. 2004, 2, 2476–2482. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Dai, W.M. Efficient Remote Axial-to-Central Chirality Transfer in Enantioselective SmI2-Mediated Reductive Coupling of Aldehydes with Crotonates of Atropisomeric 1-Naphthamides. J. Org. Chem. 2006, 71, 2445–2455. [Google Scholar] [CrossRef]
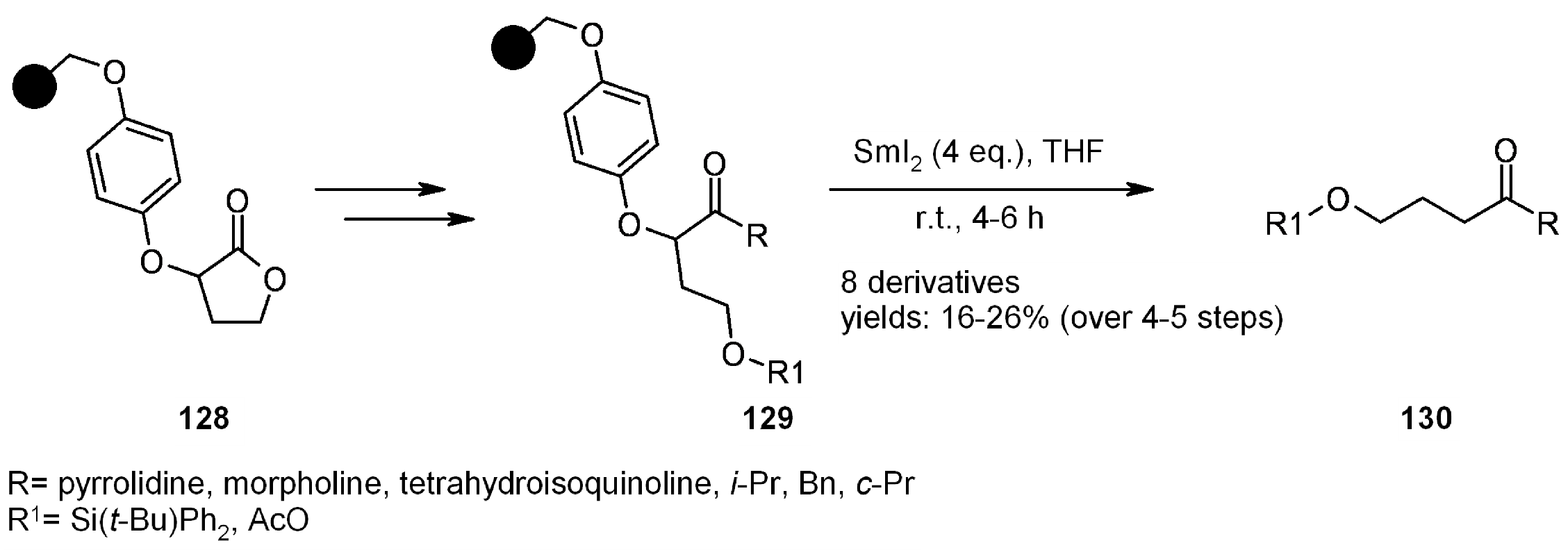
- McKerlie, F.; Procter, D.J.; Wynne, G. Reduction of alpha-aryloxy carbonyl compounds with samarium(II) iodide. A new traceless linker for the solid phase synthesis of carbonyl compounds. Chem. Commun. (Camb.) 2002, 21, 584–585. [Google Scholar]
- McKerlie, F.; Rudkin, I.M.; Wynne, G.; Procter, D.J. Evaluation of a new linker system cleaved using samarium(II) iodide. Application in the solid phase synthesis of carbonyl compounds. Org. Biomol. Chem. 2005, 3, 2805–2816. [Google Scholar] [CrossRef]
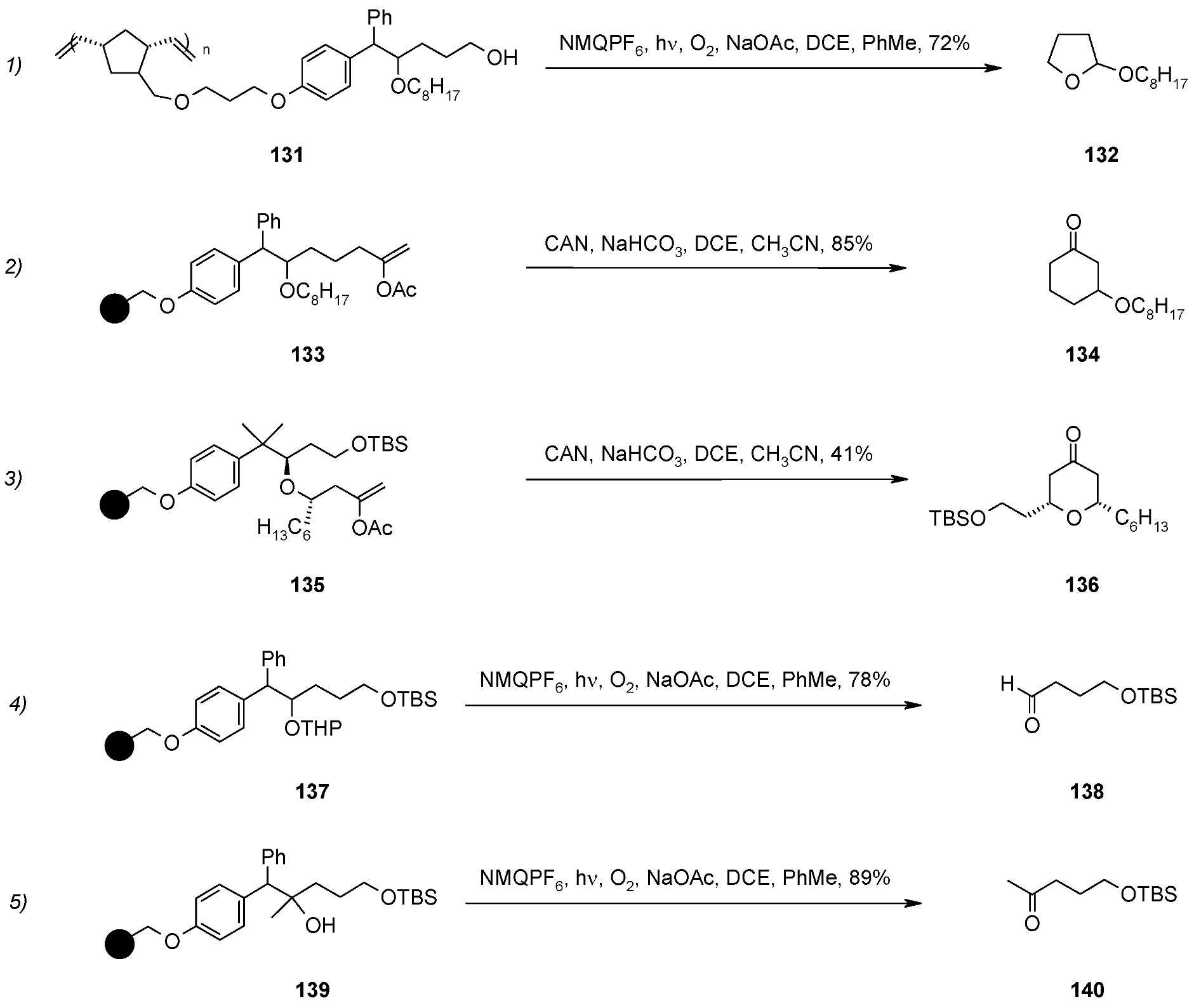
- Liu, H.; Wan, S.; Floreancig, P.E. Oxidative Cyclorelease from Soluble Polymeric Supports. J. Org. Chem. 2005, 70, 3814–3818. [Google Scholar] [CrossRef]
- Floreancig, P.E. Development and Applications of Electron-Transfer-Initiated Cyclization Reactions. Synlett 2007, 38, 191–203. [Google Scholar] [CrossRef]
- Whitehead, D.M.; Helliwell, P.A.; McKeown, S.C.; Routledge, A. Synthesis, chemical stability and application of a thiohydroxamic acid linker (THA) for solid-phase organic synthesis. React. Funct. Polym. 2009, 69, 884–890. [Google Scholar] [CrossRef]
- Horton, J.R.; Stamp, L.M.; Routledge, A. A photolabile 'traceless' linker for solid-phase organic synthesis. Tetrahedron Lett. 2000, 41, 9181–9184. [Google Scholar] [CrossRef]
- Whitehead, D.M.; Jackson, T.; McKeown, S.C.; Wilson, K.; Routledge, A. Application of X-ray Photoelectron Spectroscopy in Determining the Structure of Solid-Phase Bound Substrates. J. Comb. Chem. 2002, 4, 255–257. [Google Scholar] [CrossRef]
- Gerigk, U.; Gerlach, M.; Neumann, W.P.; Vieler, R.; Weintritt, V. Polymer-Supported Organotin Hydrides as Immobilized Reagents for Free Radical Synthesis. Synthesis 1990, 1990, 448–452. [Google Scholar] [CrossRef]
- Gerlach, M.; Joerdens, F.; Kuhn, H.; Neumann, W.P.; Peterseim, M. A polymer-supported organotin hydride and its multipurpose application in radical organic synthesis. J. Org. Chem. 1991, 56, 5971–5972. [Google Scholar] [CrossRef]
- Barton, D.H.R.; McCombie, S.W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc., Perkin Trans. 1 1975, 1574–1585. [Google Scholar]
- Neumann, W.P.; Peterseim, M. Elegant Improvement of the Deoxygenation of Alcohols Using a Polystyrene-Supported Organotin Hydride. Synlett 1992, 1992, 801–802. [Google Scholar]
- Ruel, G.; Ngo, K.T.; Dumartin, G.; Delmond, B.; Pereyre, M. Un nouvel hydrure organostannique greffé sur support insoluble. J. Organomet. Chem. 1993, 444, C18–C20. [Google Scholar]
- Dumartin, G.; Ruel, G.; Kharboutli, J.; Delmond, B.; Connil, M.F.; Jousseaume, B.; Pereyre, M. Straightforward Synthesis and Reactivity of Polymer-supported Organotin Hydrides. Synlett 1994, 1994, 952–954. [Google Scholar] [CrossRef]
- Dumartin, G.; Pourcel, M.; Delmond, B.; Donard, O.; Pereyre, M. In situ generated, polymer-supported organotin hydrides as clean reducing agents. Tetrahedron Lett. 1998, 39, 4663–4666. [Google Scholar] [CrossRef]
- Chemin, A.; Deleuze, H.; Maillard, B. Preparation and reactivity of a macroporous polymer-supported organotin hydride catalyst. Eur. Polym. J. 1998, 34, 1395–1404. [Google Scholar] [CrossRef]
- Boussaguet, P.; Delmond, B.; Dumartin, G.; Pereyre, M. Catalytic and supported Barton-McCombie deoxygenation of secondary alcohols: a clean reaction. Tetrahedron Lett. 2000, 41, 3377–3380. [Google Scholar] [CrossRef]
- Lopez, R.M.; Hays, D.S.; Fu, G.C. Bu3SnH-Catalyzed Barton-McCombie Deoxygenation of Alcohols. J. Am. Chem. Soc. 1997, 119, 6949–6950. [Google Scholar]
- Harendza, M.; Lessmann, K.; Neumann, W.P. A Polymer-Supported Distannane as Photochemical, Regenerable Source of Stannyl Radicals for Organic Synthesis. Synlett 1993, 1993, 283–285. [Google Scholar] [CrossRef]
- Junggebauer, J.; Neumann, W.P. An improved synthesis of a polymer-supported distannane and its application to radical formation. Tetrahedron 1997, 53, 1301–1310. [Google Scholar] [CrossRef]
- Enholm, E.J.; Gallagher, M.E.; Moran, K.M.; Lombardi, J.S.; Schulte, J.P. An Allylstannane Reagent on Non-Cross-Linked Polystyrene Support. Org. Lett. 1999, 1, 689–691. [Google Scholar] [CrossRef]
- Enholm, E.J.; Schulte, J.P. Convenient Catalytic Free Radical Reductions of Alkyl Halides Using an Organotin Reagent on Non-Cross-Linked Polystyrene Support. Org. Lett. 1999, 1, 1275–1277. [Google Scholar] [CrossRef]
- Corey, E.J.; Suggs, J.W. Method for catalytic dehalogenations via trialkyltin hydrides. J. Org. Chem. 1975, 40, 2554–2555. [Google Scholar] [CrossRef]
- Ueno, Y.; Chino, K.; Watanabe, M.; Moriya, O.; Okawara, M. Homolytic carbocyclization by use of a heterogeneous supported organotin catalyst. A new synthetic route to 2-alkoxytetrahydrofurans and gamma-butyrolactones. J. Am. Chem. Soc. 1982, 104, 5564–5566. [Google Scholar] [CrossRef]
- Mochida, K.; Sugimoto, H.; Yasuo, Y. First polymer-supported organogermanium hydrides and their reduction of organic halides. Polyhedron 1997, 16, 1767–1770. [Google Scholar] [CrossRef]
- Bowman, W.R.; Krintel, S.L.; Schilling, M.B. New Solid Phase Triorganogermanium Hydrides for Radical Synthesis. Synlett 2004, 2004, 1215–1218. [Google Scholar]
- Patro, B.; Merrett, M.; Murphy, J.A.; Sherrington, D.C.; Morrison, M.G.J.T. Radical-polar crossover reactions with polymer-supported tetrathiafulvalene. Tetrahedron Lett. 1999, 40, 7857–7860. [Google Scholar]
- Baguley, P.A.; Walton, J.C. Flight from the Tyranny of Tin: The Quest for Practical Radical Sources Free from Metal Encumbrances. Angew. Chem. Int. Ed. Engl. 1998, 37, 3072–3082. [Google Scholar] [CrossRef]
- Fletcher, R.; Kizil, M.; Lampard, C.; Murphy, J.A.; Roome, S.J. Tetrathiafulvalene-mediated stereoselective synthesis of the tetracyclic core of Aspidosperma alkaloids. J. Chem. Soc., Perkin Trans. 1 1998, 2341–2352. [Google Scholar]
- Callaghan, O.; Lampard, C.; Kennedy, A.R.; Murphy, J.A. A novel total synthesis of (+/-)-aspidospermidine. J. Chem. Soc., Perkin Trans. 1 1999, 995–1002. [Google Scholar]
- de Luca, L.; Giacomelli, G.; Porcu, G.; Taddei, M. A New Supported Reagent for the Photochemical Generation of Radicals in Solution. Org. Lett. 2001, 3, 855–857. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Crich, D.; Motherwell, W.B. The invention of new radical chain reactions. Part VIII. Radical chemistry of thiohydroxamic esters; A new method for the generation of carbon radicals from carboxylic acids. Tetrahedron 1985, 41, 3901–3924. [Google Scholar] [CrossRef]
- Clark, A.J.; Filik, R.P.; Haddleton, D.M.; Radigue, A.; Sanders, C.J.; Thomas, G.H.; Smith, M.E. Solid-Supported Catalysts for Atom-Transfer Radical Cyclization of 2-Haloacetamides. J. Org. Chem. 1999, 64, 8954–8957. [Google Scholar]
- Clark, A.J.; Geden, J.V.; Thom, S. Solid-Supported Copper Catalysts for Atom-Transfer Radical Cyclizations: Assessment of Support Type and Ligand Structure on Catalyst Performance in the Synthesis of Nitrogen Heterocycles. J. Org. Chem. 2006, 71, 1471–1479. [Google Scholar]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Cutting, J.; Dupont, W.; Kruse, L.; Silberman, L.; Thomas, R.C. 5-Endo-trigonal reactions: a disfavoured ring closure. J. Chem. Soc. Chem. Commun. 1976, 736–738. [Google Scholar]
- Clark, A.J.; Dell, C.P.; Ellard, J.M.; Hunt, N.A.; McDonagh, J.P. Efficient room temperature copper(I) mediated 5-endo radical cyclisations. Tetrahedron Lett. 1999, 40, 8619–8623. [Google Scholar]
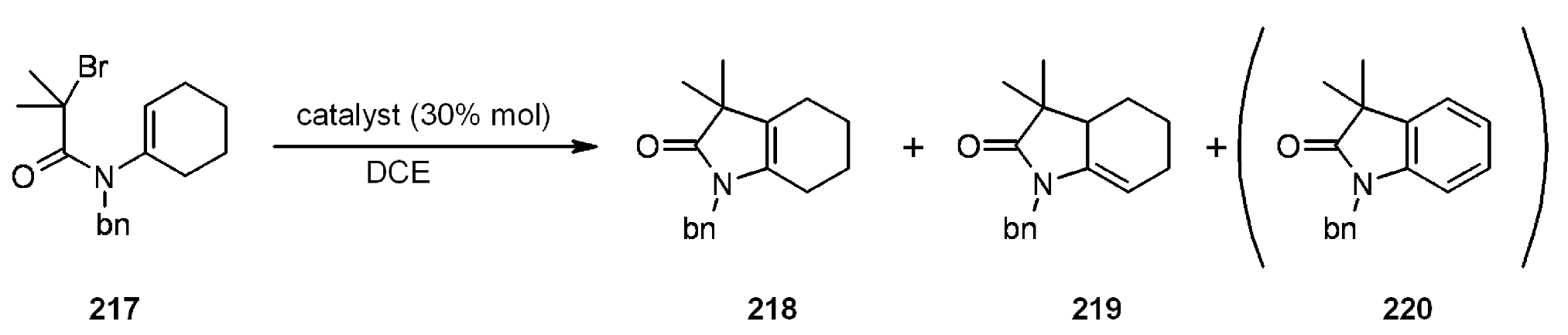
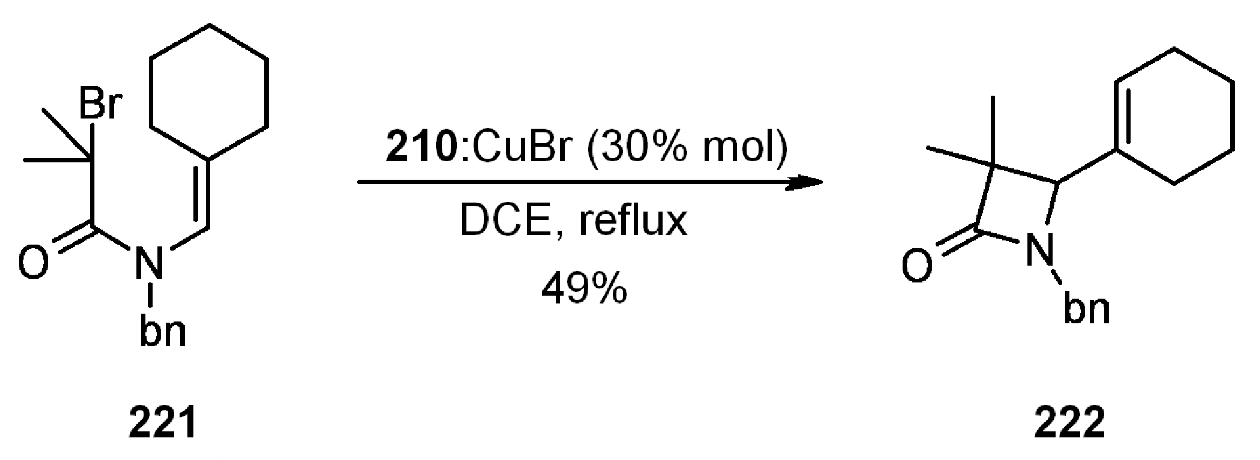
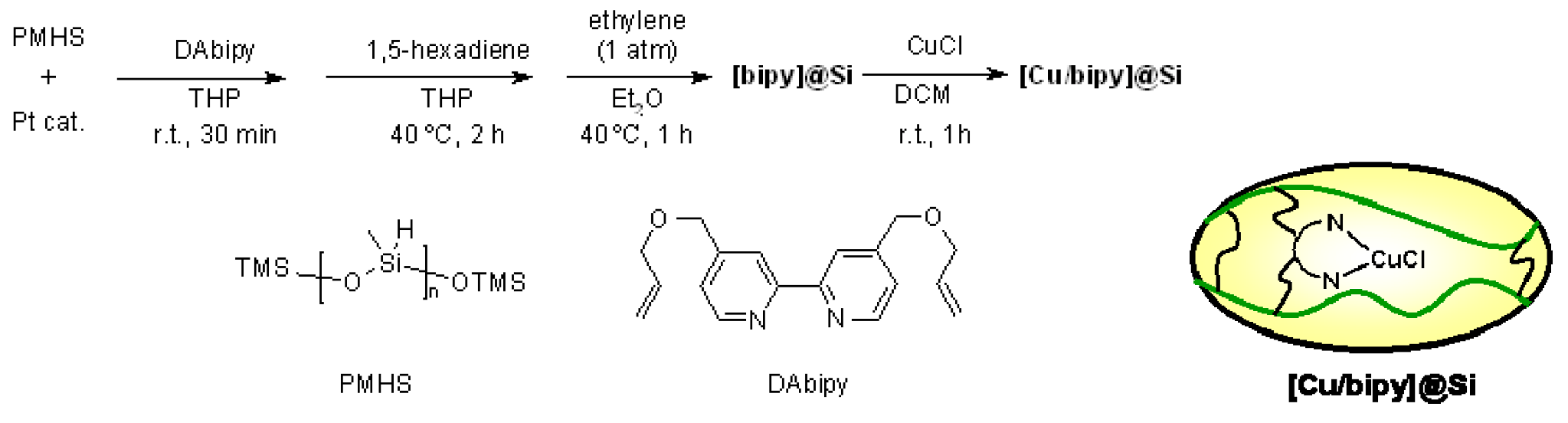
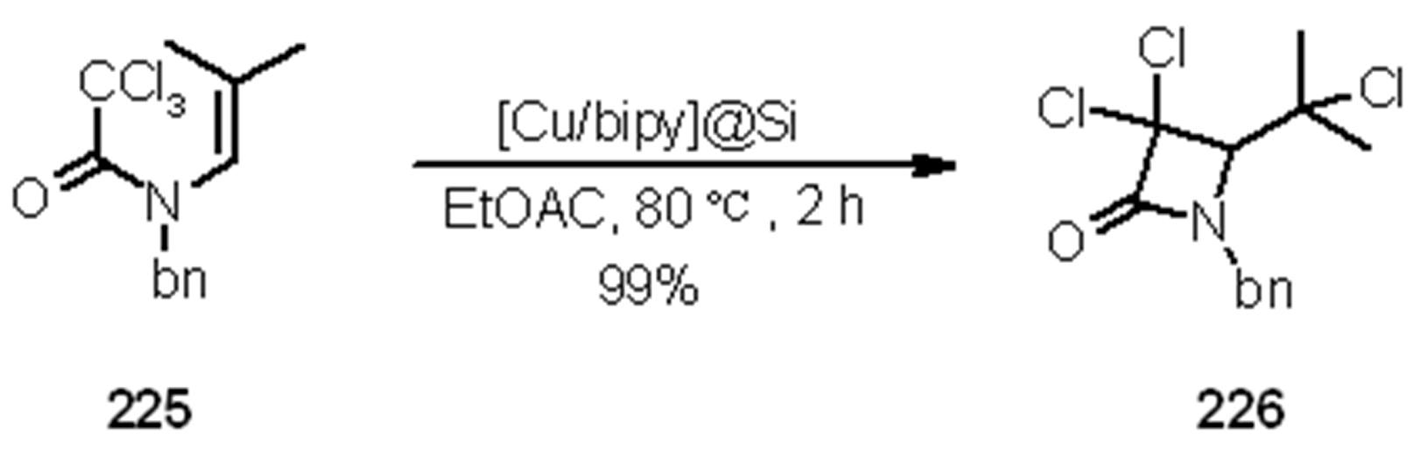
- Motoyama, Y.; Kamo, K.; Yuasa, A.; Nagashima, H. Catalytic atom-transfer radical cyclization by copper/bipyridine species encapsulated in polysiloxane. Chem. Commun. (Camb.) 2010, 46, 2256–2258. [Google Scholar] [CrossRef]
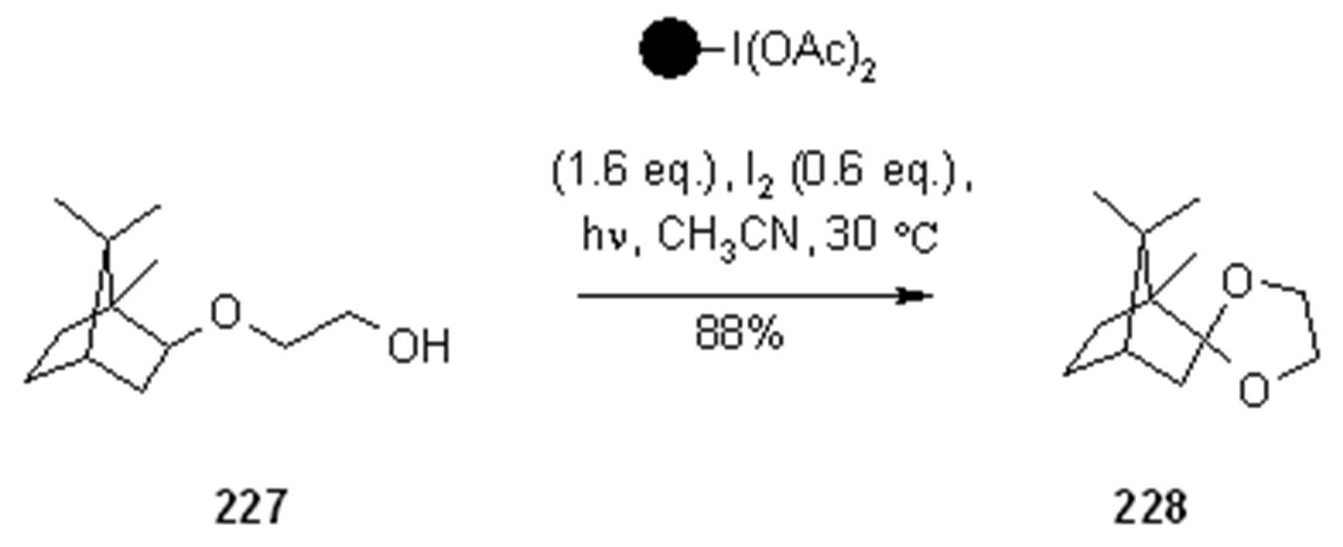
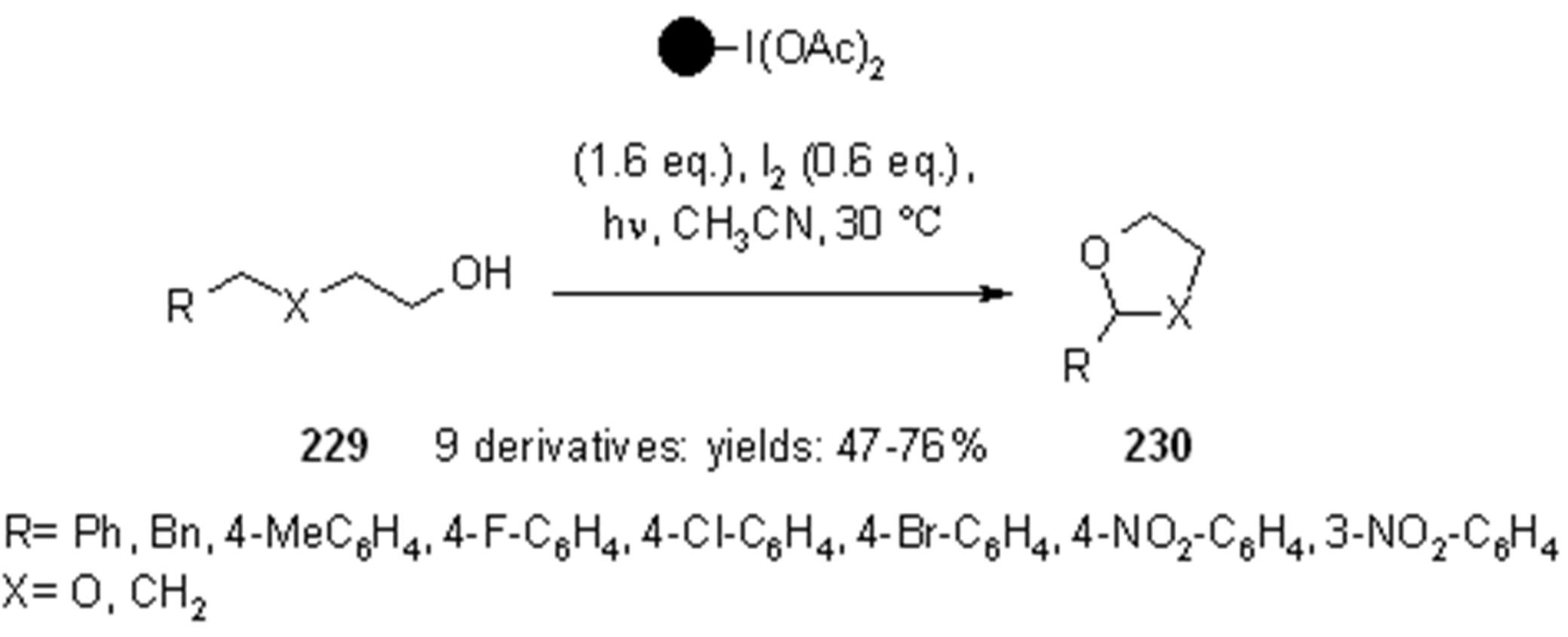
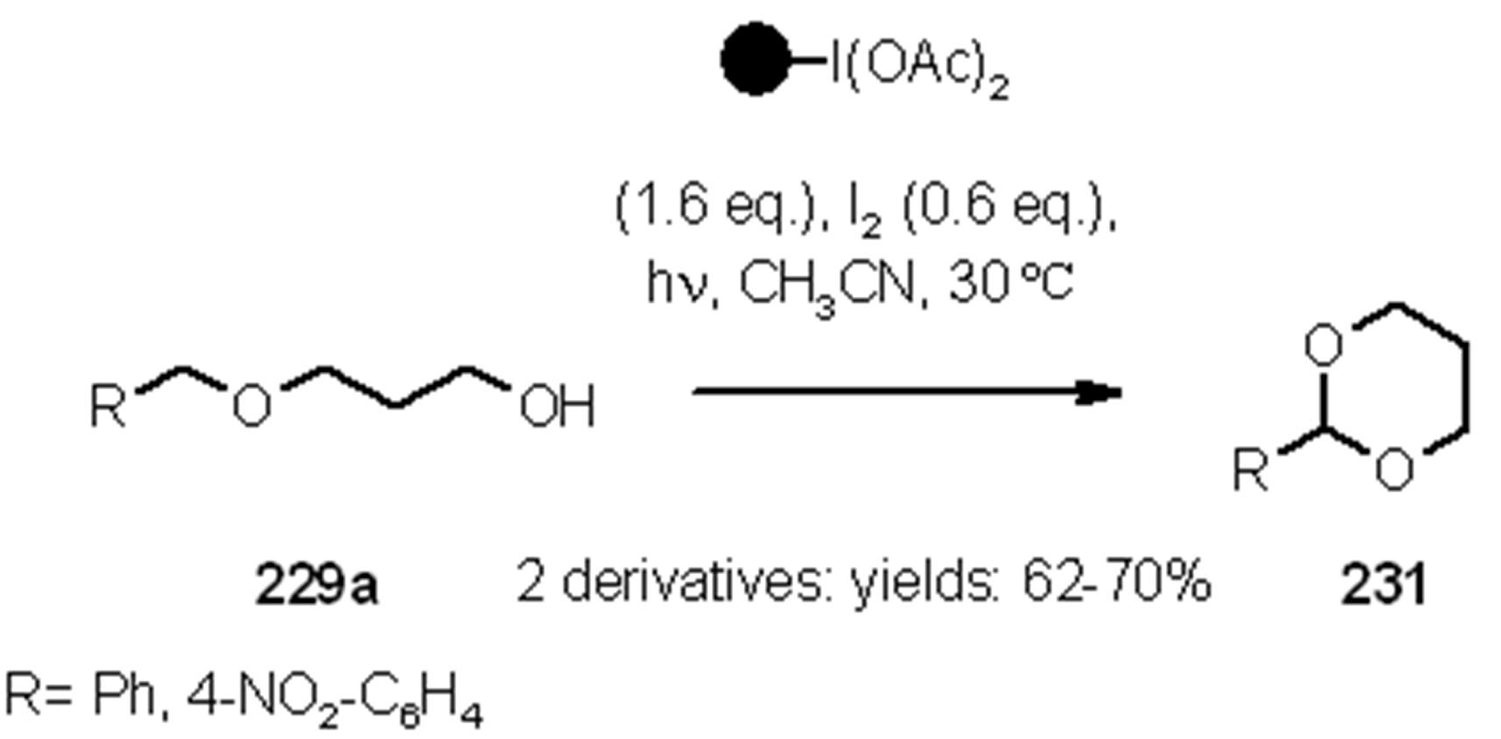
- Teduka, T.; Togo, H. I2-Mediated Photochemical Preparation of 2-Substituted 1,3-Dioxolanes and Tetrahydrofurans from Alcohols with Polymer-Supported Hypervalent Iodine Reagent, PSDIB. Synlett 2005, 2005, 923–926. [Google Scholar]
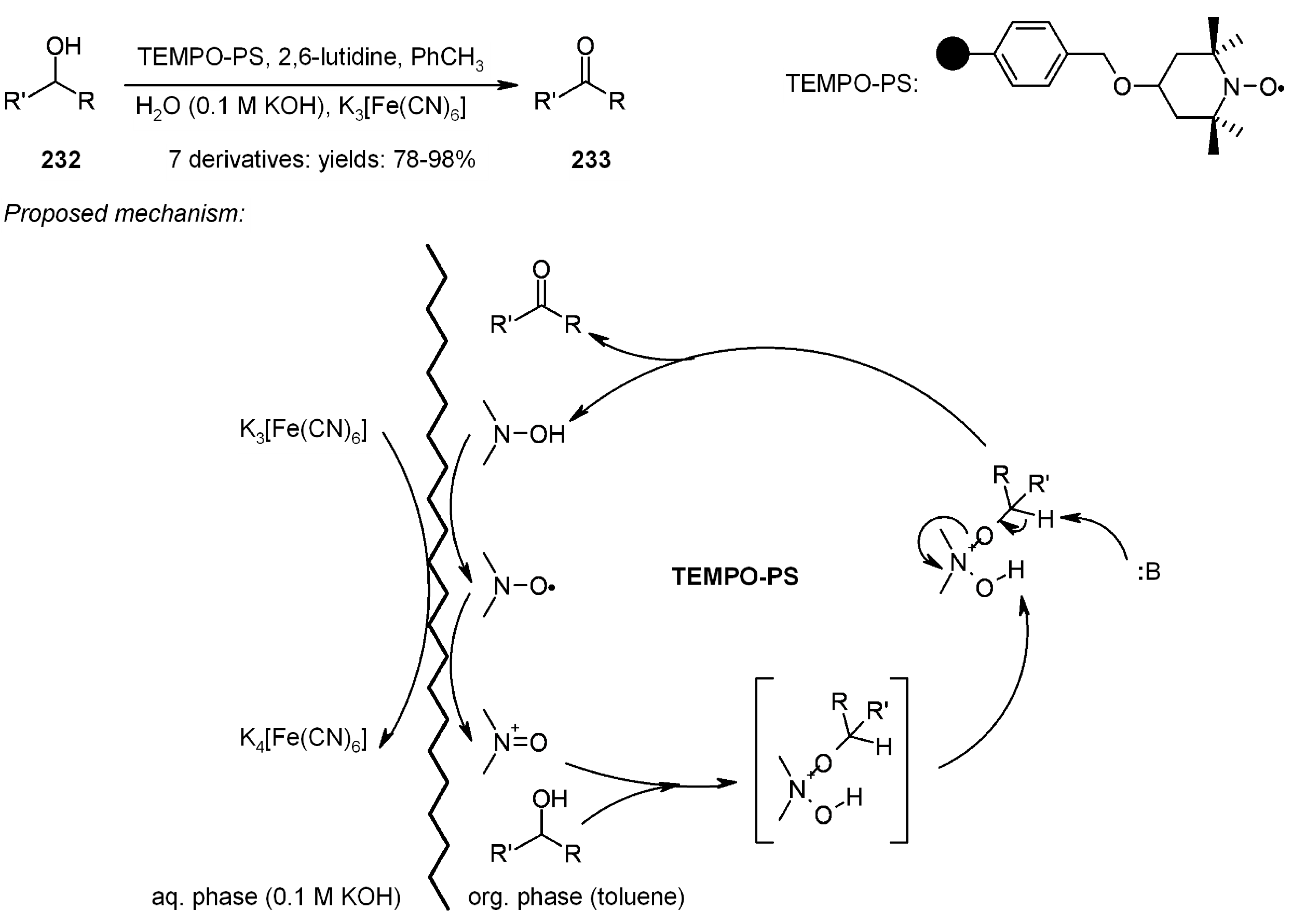
- Kashiwagi, Y.; Ikezoe, H.; Ono, T. Oxidation of Alcohols with Nitroxyl Radical Resins under Two-Phase Conditions. Synlett 2006, 2006, 69–72. [Google Scholar] [CrossRef]
- Sibi, M.P.; Chandramouli, S.V. Intermolecular free radical reactions on solid support. Allylation of esters. Tetrahedron Lett. 1997, 38, 8929–8932. [Google Scholar] [CrossRef]
- Enholm, E.J.; Gallagher, M.E.; Jiang, S.; Batson, W.A. Free Radical Allyl Transfers Utilizing Soluble Non-Cross-Linked Polystyrene and Carbohydrate Scaffold Supports. Org. Lett. 2000, 2, 3355–3357. [Google Scholar] [CrossRef]
- Enholm, E.J.; Gallagher, M.E. Free Radical Reactions on Soluble Supports from Ring-Opening Metathesis. Org. Lett. 2001, 3, 3397–3399. [Google Scholar] [CrossRef]
- Baytas, S.N.; Wang, Q.; Karst, N.A.; Dordick, J.S.; Linhardt, R.J. Solid-Phase Chemoenzymatic Synthesis of C-Sialosides. J. Org. Chem. 2004, 69, 6900–6903. [Google Scholar] [CrossRef]
- Zhu, X.; Ganesan, A. Intermolecular Conjugate Addition of Alkyl Radicals on Solid Phase. J. Comb. Chem. 1999, 1, 157–162. [Google Scholar] [CrossRef]
- Kumar, H.M.S.; Chakravarthy, P.P.; Rao, M.S.; Reddy, P.S.R.; Yadav, J.S. Free radical addition of haloalkanes to polymer bound olefins and its application to the solid-phase synthesis of pyrethroids. Tetrahedron Lett. 2002, 43, 7817–7819. [Google Scholar]
- Yim, A.M.; Vidal, Y.; Viallefont, P.; Martinez, J. Solid-phase synthesis of alpha-amino acids by radical addition to adehydroalanine derivative. Tetrahedron Lett. 1999, 40, 4535–4538. [Google Scholar]
- Caddick, S.; Hamza, D.; Wadman, S.N.; Wilden, J.D. Solid-Phase Intermolecular Radical Reactions 2: Synthesis of C-Glycopeptide Mimetics via a Novel Acrylate Acceptor. Org. Lett. 2002, 4, 1775–1777. [Google Scholar] [CrossRef]
- Miyabe, H.; Fujishima, Y.; Naito, T. Novel Synthesis of alpha-Amino Acid Derivatives through Triethylborane-Induced Solid-Phase Radical Reactions. J. Org. Chem. 1999, 64, 2174–2175. [Google Scholar] [CrossRef]
- Miyabe, H.; Nishimura, A.; Fujishima, Y.; Naito, T. Carbon-carbon bond construction on solid support: triethylborane-induced radical reactions of oxime ethers. Tetrahedron 2003, 59, 1901–1907. [Google Scholar] [CrossRef]
- Miyabe, H.; Ueda, M.; Naito, T. Carbon-Carbon Bond Construction Based on Radical Addition to C=N Bond. Synlett 2004, 2004, 1140–1157. [Google Scholar]
- Miyabe, H. Development of Solid-Phase Radical Reactions Using Oxime Ethers as a Radical Acceptor. Yakugaku Zasshi 2000, 120, 667–676. [Google Scholar]
- Miyabe, H. Development of Carbon Radical Addition to Imine Derivatives. Yakugaku Zasshi 2003, 123, 285–294. [Google Scholar] [CrossRef]
- Miyabe, H.; Konishi, C.; Naito, T. Stereocontrol in Solid-Phase Radical Reactions: Radical Addition to Oxime Ether Anchored to Polymer Support. Org. Lett. 2000, 2, 1443–1445. [Google Scholar] [CrossRef]
- Miyabe, H.; Konishi, C.; Naito, T. Diastereoselective Solid-Phase Radical Addition to Oxime Ether Anchored to Polymer Support. Chem. Pharm. Bull. 2003, 51, 540–544. [Google Scholar] [CrossRef]
- Jeon, G.H.; Yoon, J.Y.; Kim, S.; Kim, S.S. Radical Reaction of Phenylsulfonyl Oxime Ethers on Solid Support: Application to the Synthesis of alpha-Amino Esters. Synlett 2000, 2000, 128–130. [Google Scholar] [CrossRef]
- Miyabe, H.; Tanaka, H.; Naito, T. Triethylborane-induced solid-phase radical cyclization of oxime ethers. Tetrahedron Lett. 1999, 40, 8387–8390. [Google Scholar] [CrossRef]
- Miyabe, H.; Fujii, K.; Tanaka, H.; Naito, T. Solid-phase tandem radical addition-cyclisation reaction of oxime ethers. Chem. Commun. (Camb.) 2001, 831–832. [Google Scholar]
- Miyabe, H.; Tanaka, H.; Naito, T. Solid-Phase Tandem Radical Addition-Cyclization Reaction: Triethylborane-Induced Reaction of Oxime Ethers Anchored to Polymer Support. Chem. Pharm. Bull. 2004, 52, 842–847. [Google Scholar] [CrossRef]
- Akamatsu, H.; Fukase, K.; Kusumoto, S. Solid-Phase Synthesis of Indol-2-ones by Microwave-Assisted Radical Cyclization. Synlett 2004, 2004, 1049–1053. [Google Scholar]
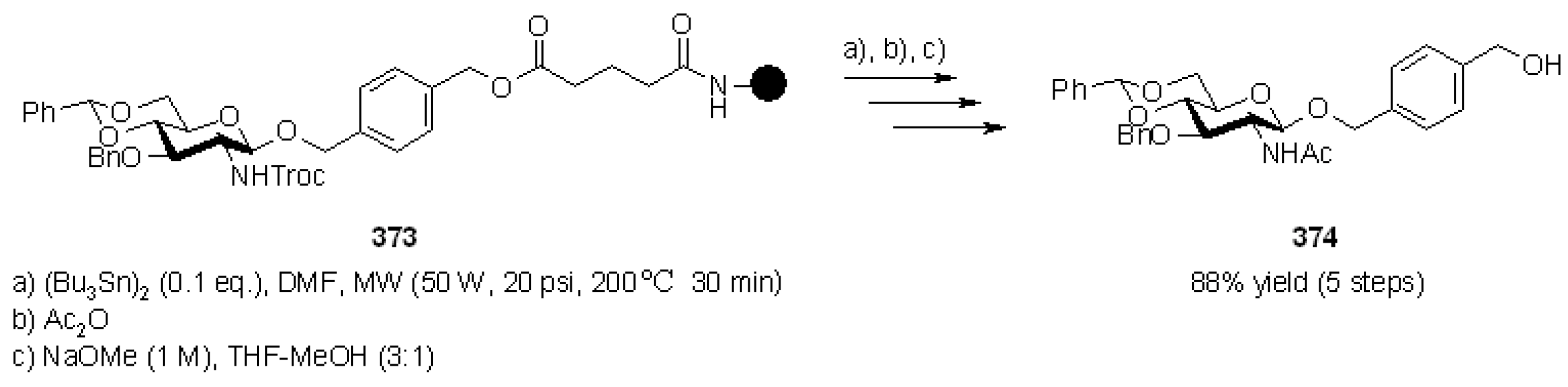
- Tokimoto, H.; Fukase, K. New deprotection method of the 2,2,2-trichloroethoxycarbonyl (Troc) group with (Bu3Sn)2. Tetrahedron Lett. 2005, 46, 6831–6832. [Google Scholar] [CrossRef]
- Allin, S.M.; Bowman, W.R.; Elsegood, M.R.J.; McKee, V.; Karim, R.; Rahman, S.S. Synthetic applications of aryl radical building blocks for cyclisation onto azoles. Tetrahedron 2005, 61, 2689–2696. [Google Scholar]
- Berteina, S.; de Mesmaeker, A.; Wendeborn, S. Functionalization via Radical Cyclization of Cyclohexenediol Derivatives Bound to Polystyrene. Synlett 1999, 1999, 1121–1123. [Google Scholar] [CrossRef]
- Du, X.; Armstrong, R.W. Synthesis of Benzofuran Derivatives on Solid Support via SmI2-Mediated Radical Cyclization. J. Org. Chem. 1997, 62, 5678–5679. [Google Scholar] [CrossRef]
- Du, X.; Armstrong, R.W. SmI2-mediated sequential radical cyclization/anionic capture of aryl iodides on solid support. Tetrahedron Lett. 1998, 39, 2281–2284. [Google Scholar] [CrossRef]
- Berteina, S.; Mesmaeker, A.D. Application of radical chemistry to solid support synthesis. Tetrahedron Lett. 1998, 39, 5759–5762. [Google Scholar] [CrossRef]
- Berteina, S.; Wendeborn, S.; de Mesmaeker, A. Radical and Palladium-Mediated Cyclization of Ortho-Iodo Benzyl Enamines: Application to Solid Phase Synthesis. Synlett 1998, 1998, 1231–1233. [Google Scholar] [CrossRef]
- Chevet, C.; Jackson, T.; Santry, B.; Routledge, A. A Cleaner Approach to Solid-Supported Radical Chemistry: Application of Hypophosphite Salts to Intramolecular C-C Bond Formation on the Solid-Phase. Synlett 2005, 2005, 477–480. [Google Scholar]
- Helliwell, P.A.; Bailey, V.A.; Chevet, C.; Clarke, D.B.; Lloyd, A.; Macarthur, R.; Routledge, A. A mass spectrometric investigation into microenvironmental effects in solid-supported radical chemistry. React. Funct. Polym. 2010, 70, 110–115. [Google Scholar] [CrossRef]
- Routledge, A.; Abell, C.; Balasubramanian, S. An Investigation into Solid-Phase Radical Chemistry-Synthesis of Furan Rings. Synlett 1997, 1997, 61–62. [Google Scholar] [CrossRef]
- Jia, G.; Iida, H.; Lown, J.W. Solid-Phase Synthesis of 1-Chloromethyl-1,2-dihydro-3H-benz[e]indole (seco-CBI) and a Polyamide Conjugate. Synlett 2000, 2000, 603–606. [Google Scholar] [CrossRef]
- Watanabe, Y.; Ishikawa, S.; Takao, G.; Toru, T. Radical cyclization on solid support: Synthesis of gamma-butyrolactones. Tetrahedron Lett. 1999, 40, 3411–3414. [Google Scholar] [CrossRef]
- Enholm, E.J.; Cottone, J.S. Highly Diastereoselective Radical Cyclizations on Soluble Ring Opening Metathesis Supports. Org. Lett. 2001, 3, 3959–3962. [Google Scholar] [CrossRef]
- Renaud, P.; Gerster, M. Use of Lewis Acids in Free Radical Reactions. Angew. Chem. Int. Ed. Engl. 1998, 37, 2562–2579. [Google Scholar] [CrossRef]
- Murakata, M.; Jono, T.; Mizuno, Y.; Hoshino, O. Construction of Chiral Quaternary Carbon Centers by Catalytic Enantioselective Radical-Mediated Allylation of alpha-Iodolactones Using Allyltributyltin in the Presence of a Chiral Lewis Acid. J. Am. Chem. Soc. 1997, 119, 11713–11714. [Google Scholar] [CrossRef]
- Porter, N.A.; Giese, B.; Curran, D.P. Acyclic stereochemical control in free-radical reactions. Acc. Chem. Res. 1991, 24, 296–304. [Google Scholar] [CrossRef]
- Nishida, M.; Hayashi, H.; Yamaura, Y.; Yanaginuma, E.; Yonemitsu, O.; Nishida, A.; Kawahara, N. A simple preparation of (R)-(2-cyclopentenyl)acetic acid and (R)-(2-cyclohexenyl)acetic acid using beta-diastereoselective radical cyclization in the presence of Lewis acid. Tetrahedron Lett. 1995, 36, 269–272. [Google Scholar]
- Nishida, M.; Ueyama, E.; Hayashi, H.; Ohtake, Y.; Yamaura, Y.; Yanaginuma, E.; Yonemitsu, O.; Nishida, A.; Kawahara, N. Lewis Acid-Promoted Diastereoselective Radical Cyclization Using Chiral alpha,beta-Unsaturated Esters. J. Am. Chem. Soc. 1994, 116, 6455–6456. [Google Scholar]
- Badone, D.; Bernassau, J.M.; Cardamone, R.; Guzzi, U. Diastereoselective Chelation-Controlled Radical Cyclization of Chiral Oxazolidinone-Derived 2-Alkenamides and Modeling of the Transition State. Angew. Chem. Int. Ed. Engl. 1996, 35, 535–538. [Google Scholar] [CrossRef]
- Lamberto, M.; Corbett, D.F.; Kilburn, J.D. Thiol-mediated free radical cyclisations of isocyanides on solid support. Tetrahedron Lett. 2004, 45, 8541–8543. [Google Scholar] [CrossRef]
- Harrowven, D.C.; May, P.J.; Bradley, M. Sulfur-mediated radical cyclisation reactions on solid support. Tetrahedron Lett. 2003, 44, 503–506. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Schiesser, C.H. Regio- and stereo-selectivity of alkenyl radical ring closure: A theoretical study. Tetrahedron 1985, 41, 3925–3941. [Google Scholar] [CrossRef]
- Spellmeyer, D.C.; Houk, K.N. Force-field model for intramolecular radical additions. J. Org. Chem. 1987, 52, 959–974. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.L.; Choi, H.S.; Yoon, W.H.; He, Y.; Fong, K.C. Total Synthesis of the CP Molecules CP-263,114 and CP-225,917 − Part 1: Synthesis of Key Intermediates and Intelligence Gathering. Angew. Chem. Int. Ed. Engl. 1999, 38, 1669–1675. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.L.; Fong, K.C.; He, Y.; Yoon, W.H.; Choi, H.S. Total Synthesis of the CP Molecules CP-225,917 and CP-263,114 − Part 2: Evolution of the Final Strategy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1676–1678. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Zhong, Y.L.; Baran, P.S. New Synthetic Technology for the Rapid Construction of Novel Heterocycles − Part 1: The Reaction of Dess-Martin Periodinane with Anilides and Related Compounds. Angew. Chem. Int. Ed. Engl. 2000, 39, 622–625. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. Iodine(V) Reagents in Organic Synthesis. Part 3: New Routes to Heterocyclic Compounds via o-Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244. [Google Scholar]
- Caddick, S.; Hamza, D.; Wadman, S.N. Solid-phase intermolecular radical reactions 1. Sulfonyl radical addition to isolated alkenes and alkynes. Tetrahedron Lett. 1999, 40, 7285–7288. [Google Scholar] [CrossRef]
- Plourde Jr, R.; Johnson Jr, L.L.; Longo, R.K. Solid-Phase Free Radical Addition of Thiols to Allyl Ethers. Synlett 2001, 2001, 439–441. [Google Scholar]
- Timár, Z.; Gallagher, T. Constructing the 2-(thiobenzyl)ethyl carbamate linker via thiyl radical addition. Tetrahedron Lett. 2000, 41, 3173–3176. [Google Scholar] [CrossRef]
- Dublanchet, A.C.; Lusinchi, M.; Zard, S.Z. Xanthates and solid-phase chemistry. A new soluble polymer analogue of Wang resin. Tetrahedron 2002, 58, 5715–5721. [Google Scholar] [CrossRef]
- Back, T.G.; Zhai, H. Cyclizations and cycloadditions of acetylenic sulfones on solid supports. Chem. Commun. (Camb.) 2006, 21, 326–328. [Google Scholar] [CrossRef]
- Attardi, M.E.; Taddei, M. The Barton radical decarboxylation on solid phase. A versatile synthesis of peptides containing modified amino acids. Tetrahedron Lett. 2001, 42, 3519–3522. [Google Scholar] [CrossRef]
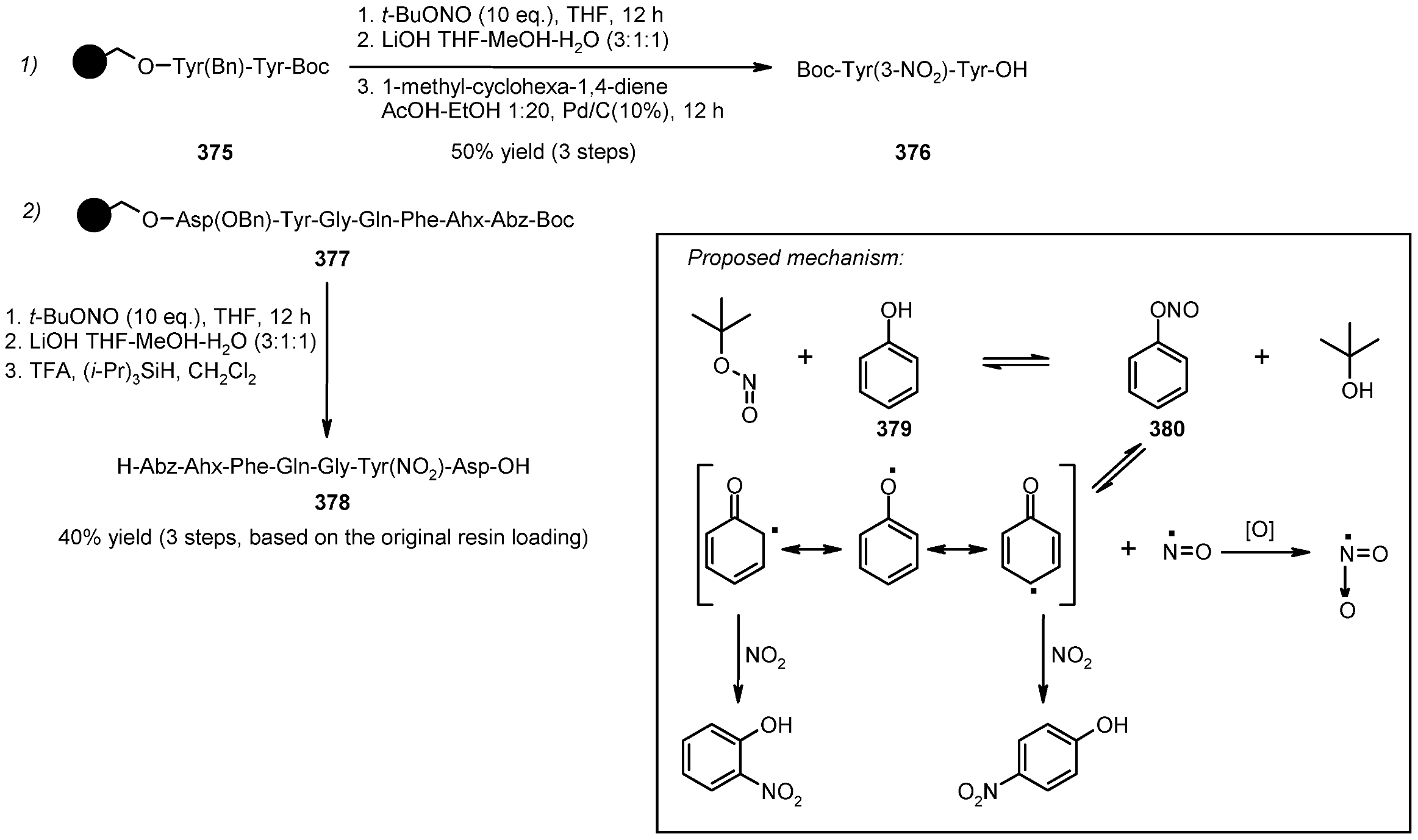
- Koley, D.; Colon, O.C.; Savinov, S.N. Chemoselective Nitration of Phenols with tert-Butyl Nitrite in Solution and on Solid Support. Org. Lett. 2009, 11, 4172–4175. [Google Scholar] [CrossRef]
- Sample Availability: not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mirizzi, D.; Pulici, M. From Polymer to Small Organic Molecules: A Tight Relationship between Radical Chemistry and Solid-Phase Organic Synthesis. Molecules 2011, 16, 3252-3314. https://doi.org/10.3390/molecules16043252
Mirizzi D, Pulici M. From Polymer to Small Organic Molecules: A Tight Relationship between Radical Chemistry and Solid-Phase Organic Synthesis. Molecules. 2011; 16(4):3252-3314. https://doi.org/10.3390/molecules16043252
Chicago/Turabian StyleMirizzi, Danilo, and Maurizio Pulici. 2011. "From Polymer to Small Organic Molecules: A Tight Relationship between Radical Chemistry and Solid-Phase Organic Synthesis" Molecules 16, no. 4: 3252-3314. https://doi.org/10.3390/molecules16043252
APA StyleMirizzi, D., & Pulici, M. (2011). From Polymer to Small Organic Molecules: A Tight Relationship between Radical Chemistry and Solid-Phase Organic Synthesis. Molecules, 16(4), 3252-3314. https://doi.org/10.3390/molecules16043252