Two-Photon Polarization Dependent Spectroscopy in Chirality: A Novel Experimental-Theoretical Approach to Study Optically Active Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
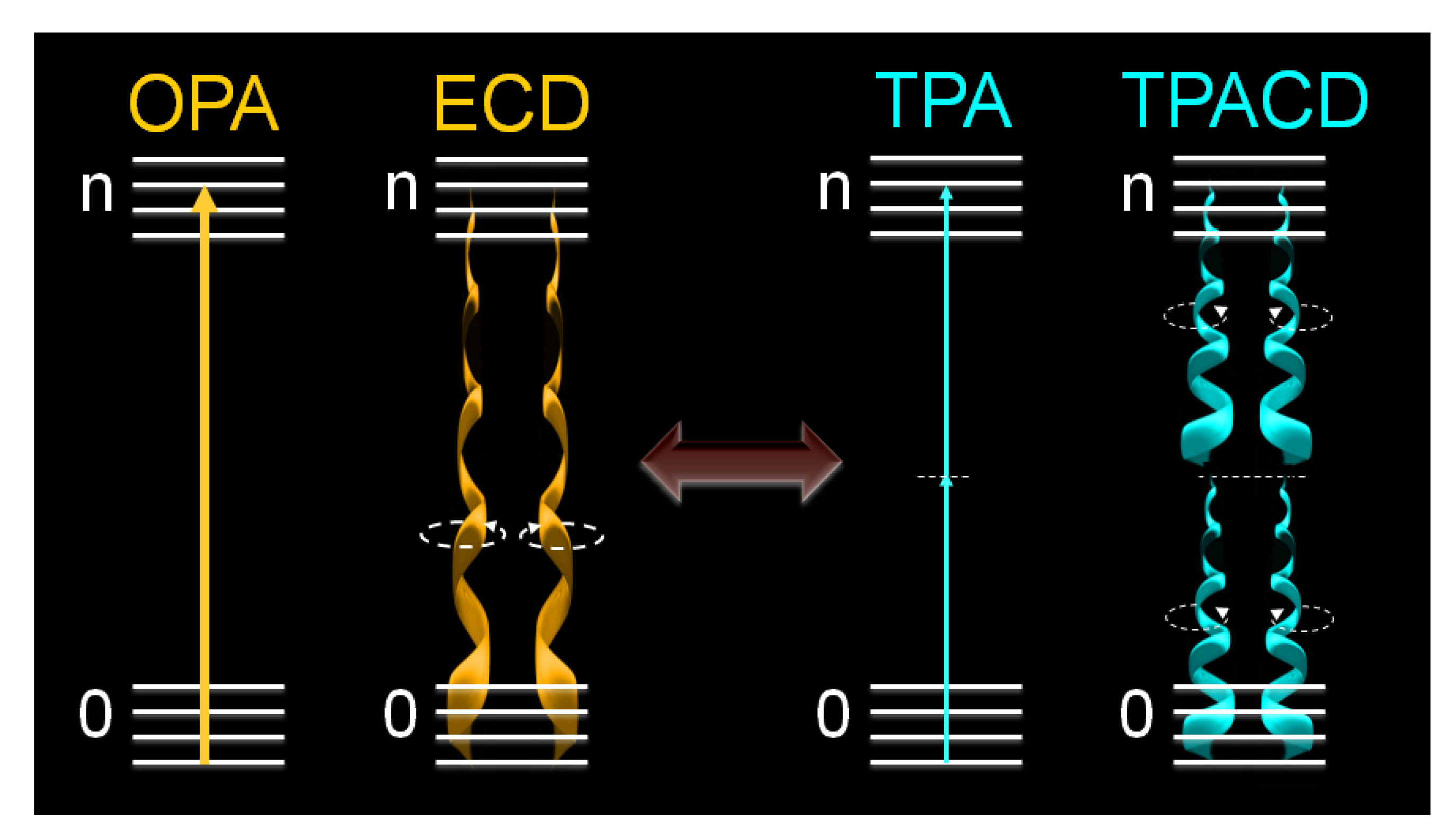
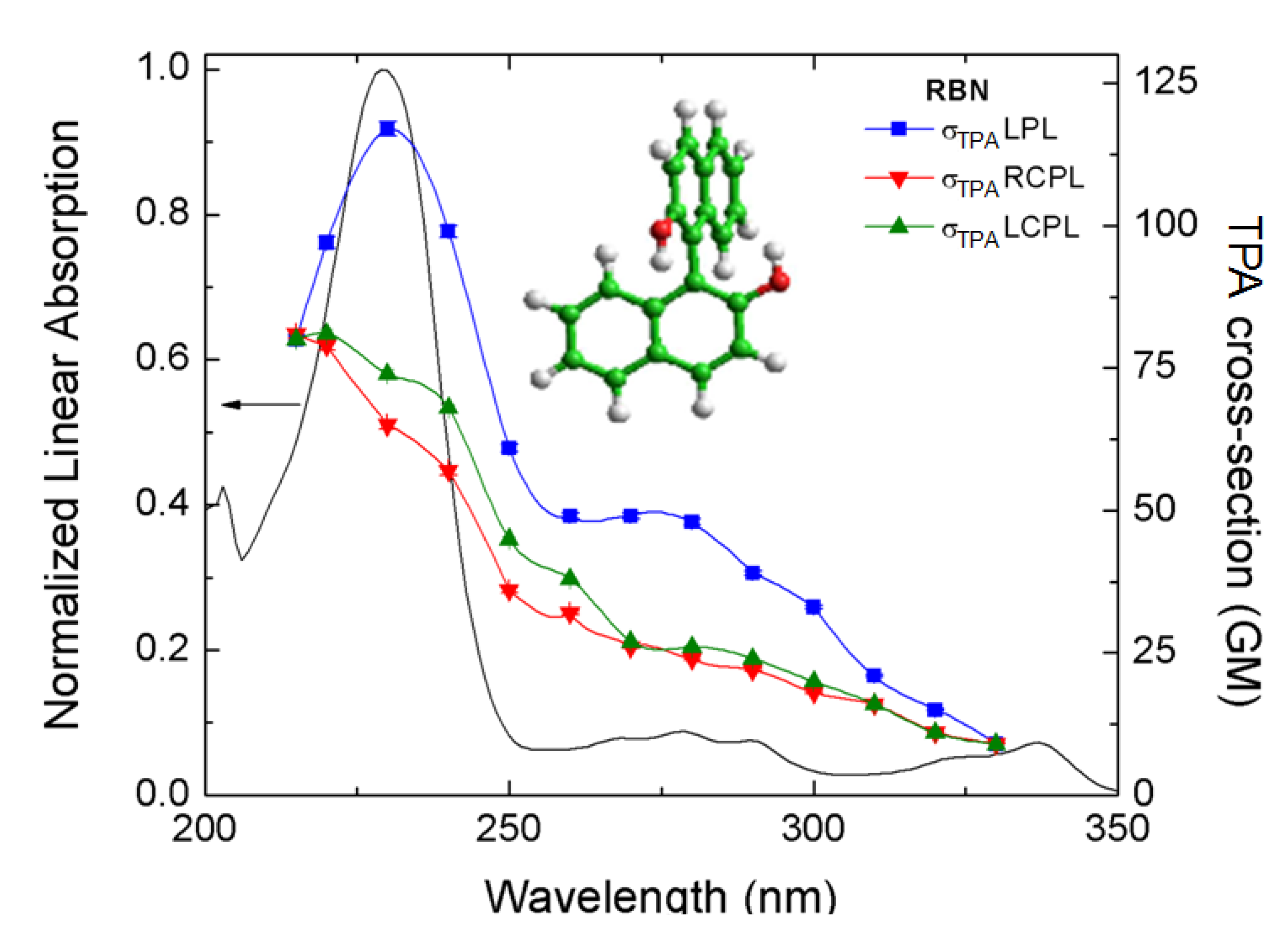
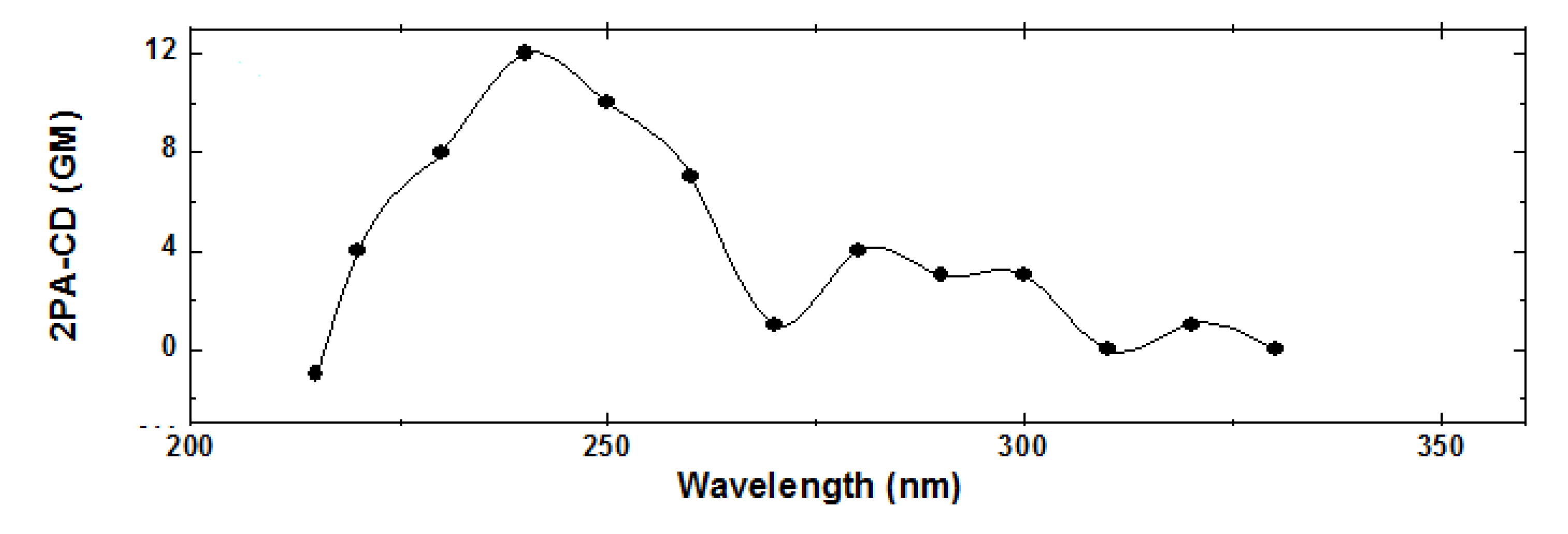
 , where δLTPA(λ) and δRTPA(λ) are the TPA cross-sections, δTPA(λ), for left and right circularly polarized light, respectively (Figure 1). Because at typical TPA excitation wavelengths the linear absorption is negligible and scattering minimized, the study of short wavelengths absorbing molecules becomes advantageous [38,39]. In addition, the fact that TPA transitions obey different selection rules than OPA (even-parity vs. odd-parity) leads to think that in chiral molecules ECD and TPACD should present different spectral features. Of course, these rules are very strict only in centrosymmetric molecules. As a result, one should expect greater differences in optically active compounds with a center of symmetry, which is contradictory. Nevertheless, it has already been predicted theoretically that in molecules that do not satisfy this condition, such as chiral molecules, significant differences between the ECD and TPACD spectra can be observed [31]. Therefore, being able to measure TPACD in chiral compounds whose excitation energies are in the UV, would provide structural and conformational information that is complementary to that obtained using ECD.
, where δLTPA(λ) and δRTPA(λ) are the TPA cross-sections, δTPA(λ), for left and right circularly polarized light, respectively (Figure 1). Because at typical TPA excitation wavelengths the linear absorption is negligible and scattering minimized, the study of short wavelengths absorbing molecules becomes advantageous [38,39]. In addition, the fact that TPA transitions obey different selection rules than OPA (even-parity vs. odd-parity) leads to think that in chiral molecules ECD and TPACD should present different spectral features. Of course, these rules are very strict only in centrosymmetric molecules. As a result, one should expect greater differences in optically active compounds with a center of symmetry, which is contradictory. Nevertheless, it has already been predicted theoretically that in molecules that do not satisfy this condition, such as chiral molecules, significant differences between the ECD and TPACD spectra can be observed [31]. Therefore, being able to measure TPACD in chiral compounds whose excitation energies are in the UV, would provide structural and conformational information that is complementary to that obtained using ECD. 
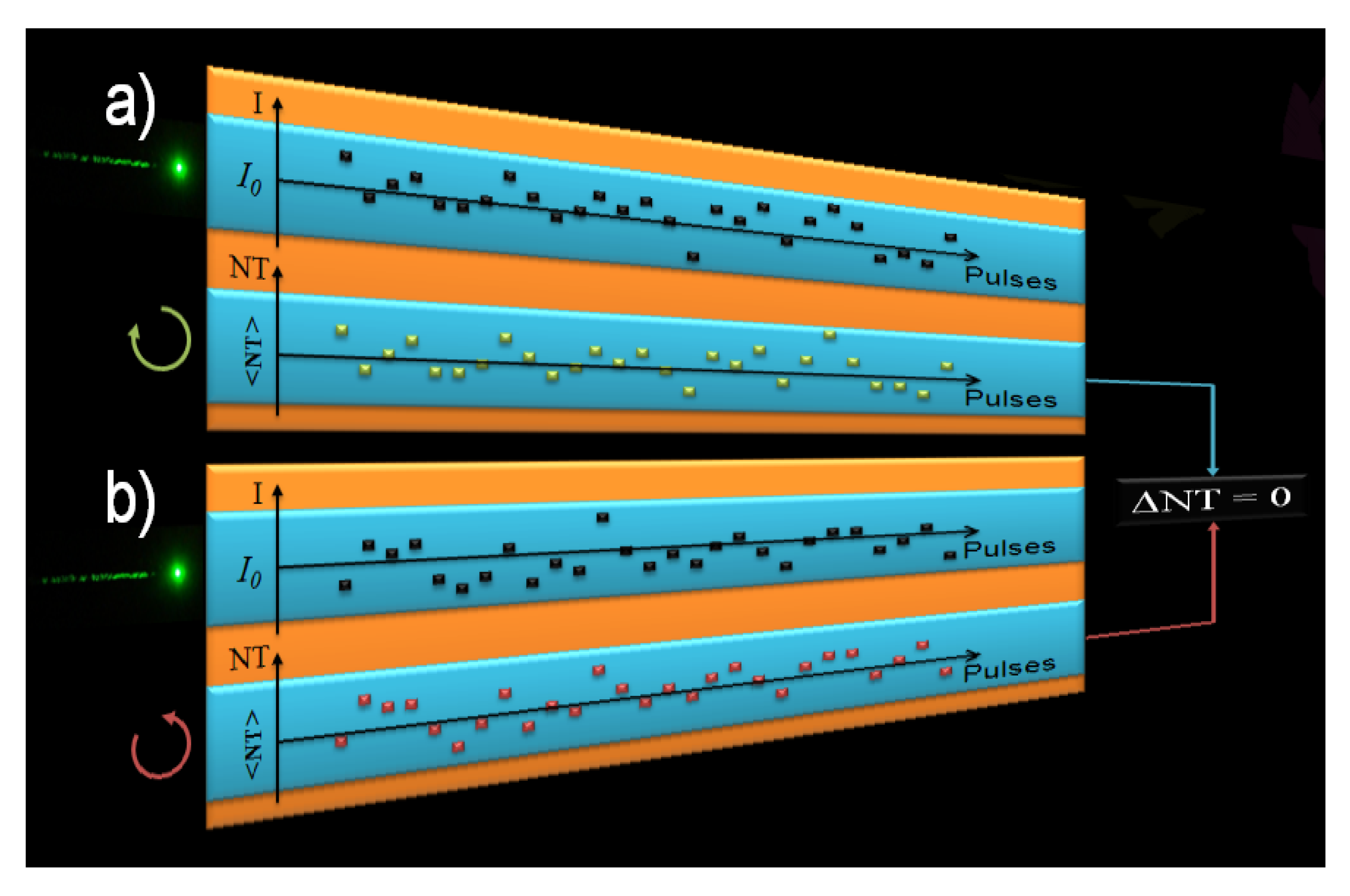
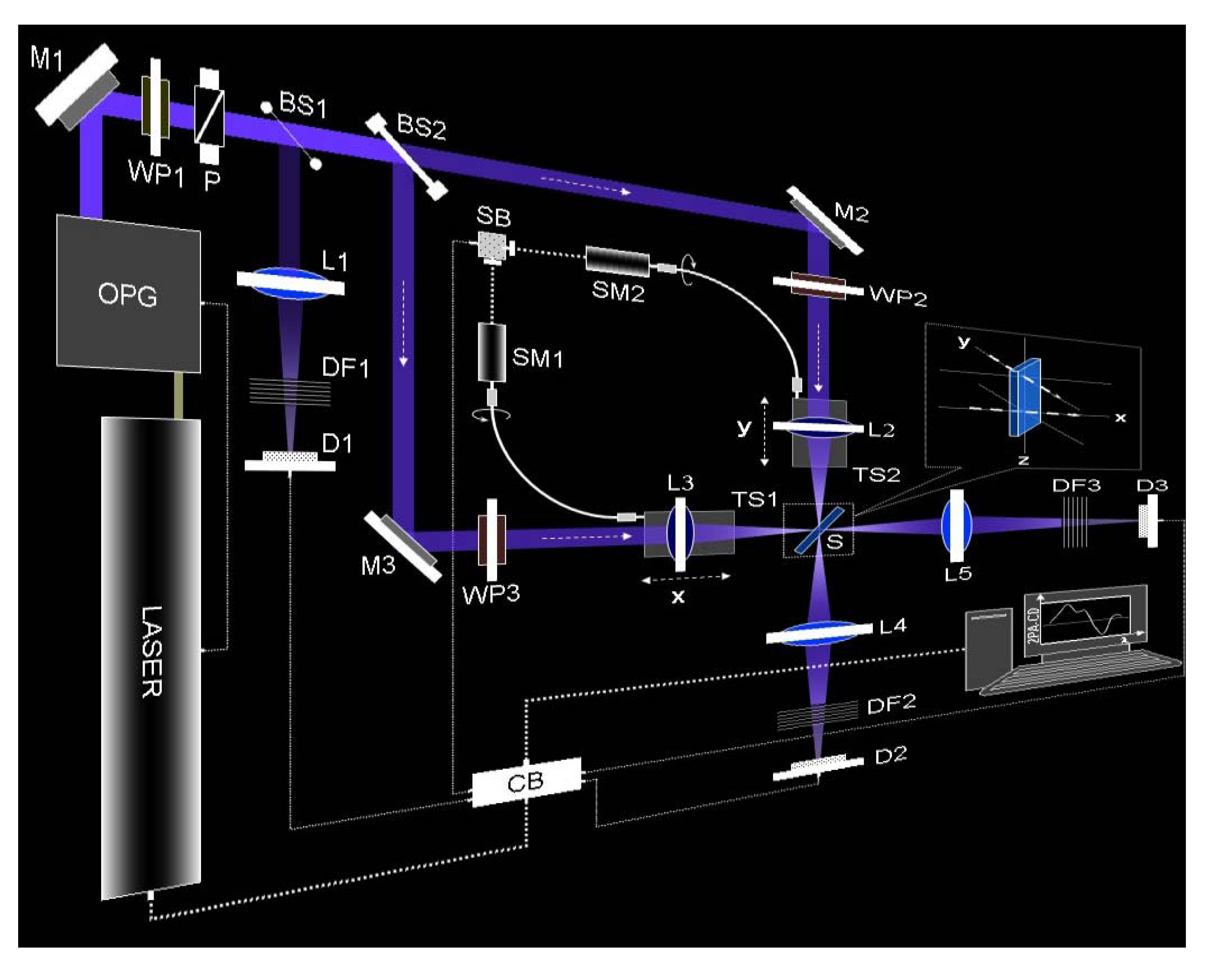
2. Experimental Approach for TPACD









3. Theory of TPACD
, was obtained by Tinoco in his 1975 paper as a semiclassical extension of the TPA formulae [27]. Quantum electrodynamical equivalent expressions were obtained by Power [28], by Andrews [46] and, in a series of papers, by Meath and Power [47,48,49,50], who were able to generalize the approach to the case of n photons [49], and considered also the modifications occurring in the formulae when elliptical polarization is assumed [50]. A nice discussion of the theoretical foundation of TPACD can be found in the book of Lin and co-workers [51]. In 1986 Szłucki and Stręk [52] predicted the possibility of detection of TPACD in the fluorescence of lanthanides (observed in 1995 by Gunde and Richardson [53]).

 is an appropriate lineshape function, depending on the wavelength λ, the transition wavelength
is an appropriate lineshape function, depending on the wavelength λ, the transition wavelength  and the parameter γ discussed just below (E0n denotes the excitation energy). In practice, in our calculations the lineshape function used is either a Lorentzian:
and the parameter γ discussed just below (E0n denotes the excitation energy). In practice, in our calculations the lineshape function used is either a Lorentzian:



 for a Gaussian again expressed as a function of the ω (both functions are therefore normalized to unity on the ω axis). With the definition of Eqs. (3) and (4) each lineshape function is not normalized to unity in the wavelength domain, and it is not symmetric with respect to the λ =λ0n coordinate, where the function has its maximum. Quite often one approximates
for a Gaussian again expressed as a function of the ω (both functions are therefore normalized to unity on the ω axis). With the definition of Eqs. (3) and (4) each lineshape function is not normalized to unity in the wavelength domain, and it is not symmetric with respect to the λ =λ0n coordinate, where the function has its maximum. Quite often one approximates  , which restores both symmetry and normalization in the wavelength domain.
, which restores both symmetry and normalization in the wavelength domain. 

 indicates the matrix element between electronic states |i〉and |j〉of the α-component of the operator
indicates the matrix element between electronic states |i〉and |j〉of the α-component of the operator  , which is either the velocity operator
, which is either the velocity operator  :
:

 :
:

 :
:

 , and both the rotatory and the transition strengths are assumed to be given in a.u. (where they are usually of comparable size).
, and both the rotatory and the transition strengths are assumed to be given in a.u. (where they are usually of comparable size).4. The Computational Approach to TPACD

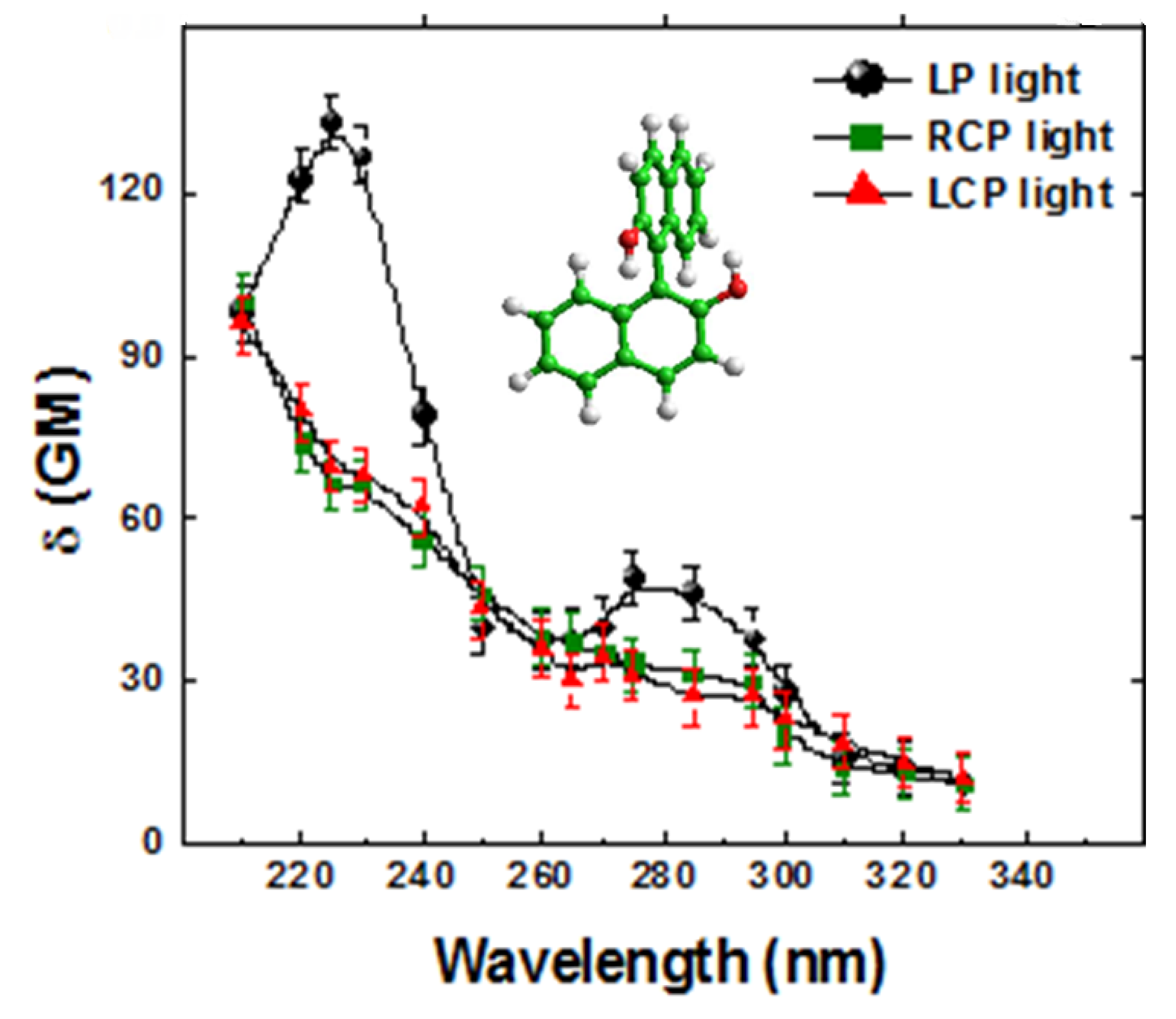
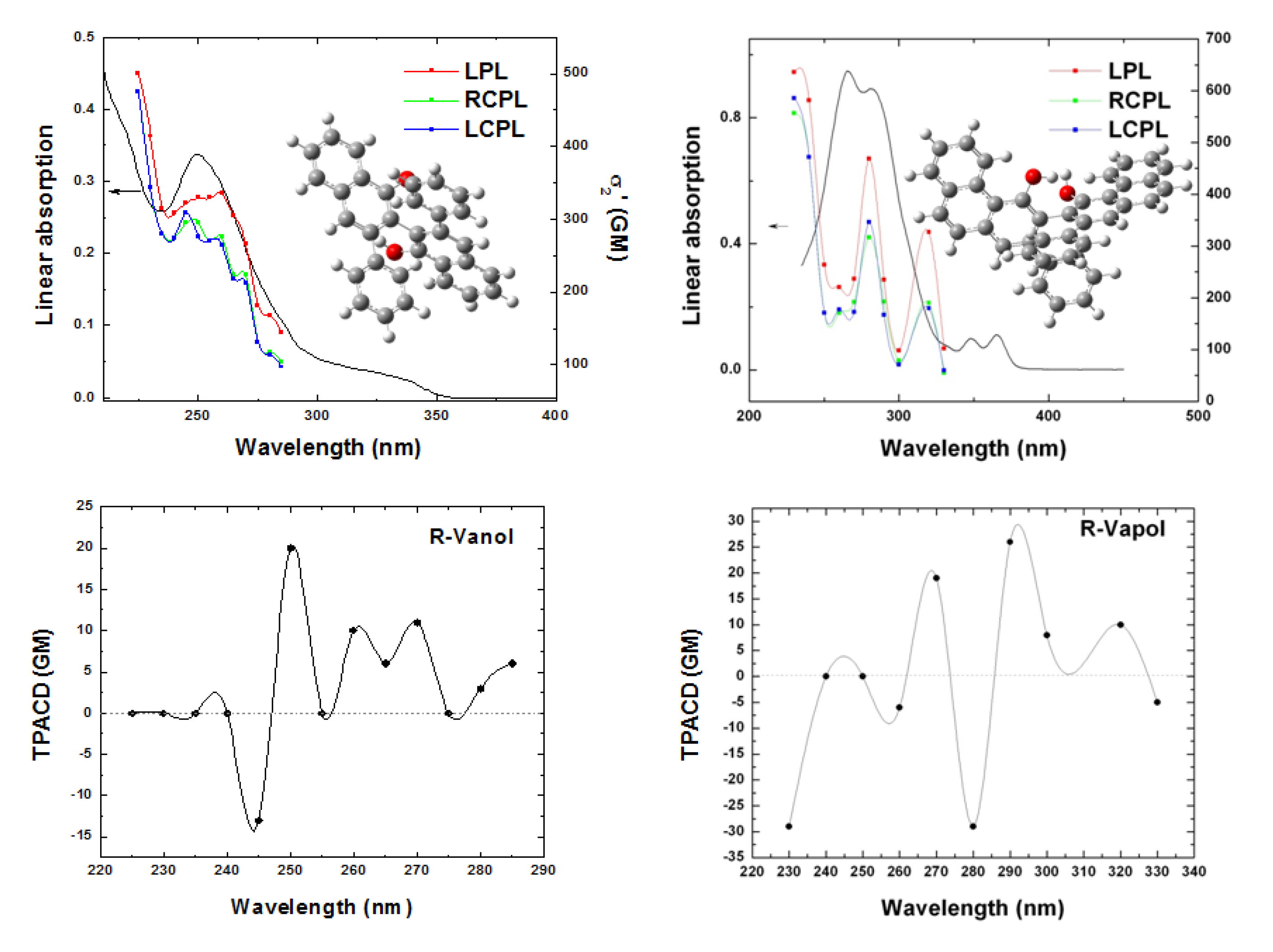
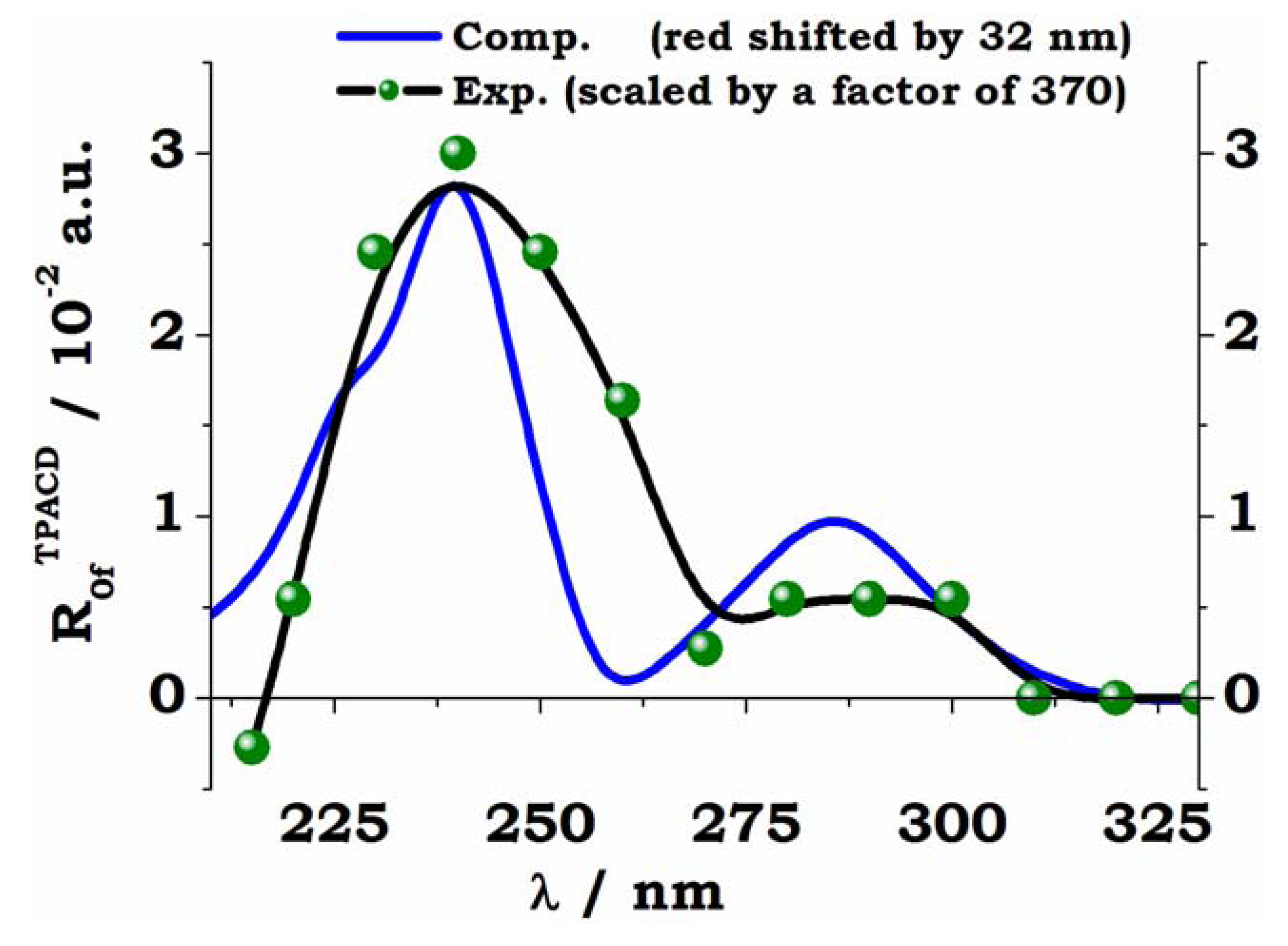
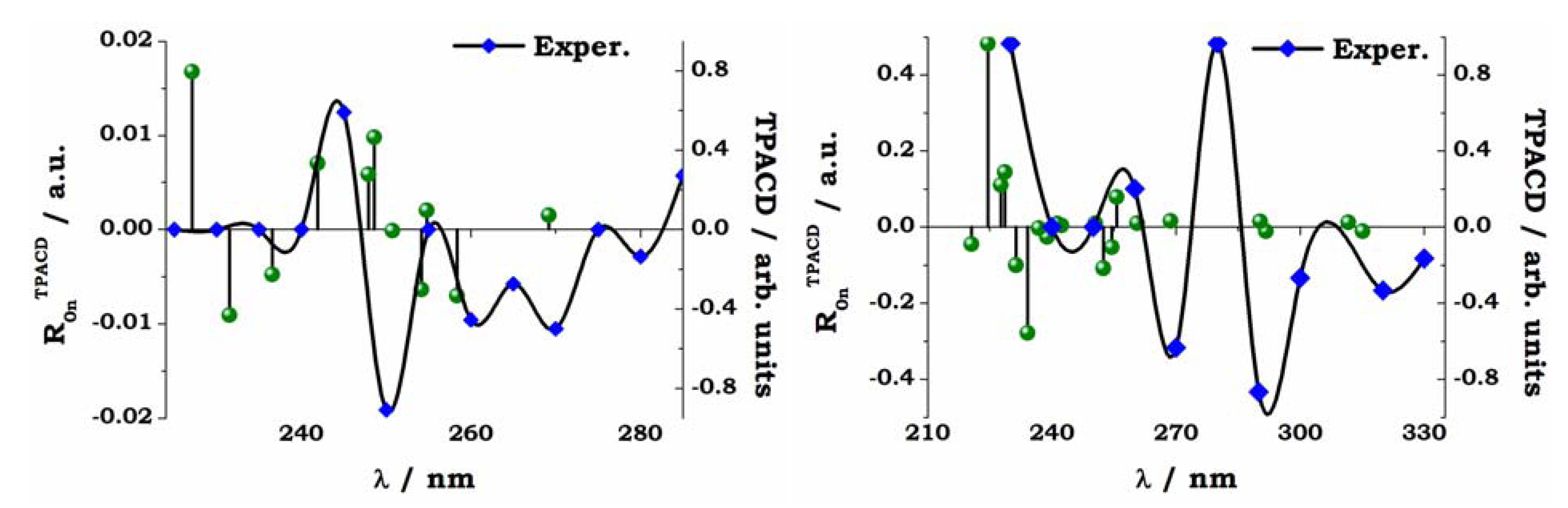
5. Early Computational Studies and Comparison of Experiment and Theory: BINOL, VANOL, VAPOL
 = 0.3 eV employed for the simulation, fee Ref. [66].
= 0.3 eV employed for the simulation, fee Ref. [66].
= 0.3 eV employed for the simulation, fee Ref. [66].
= 0.3 eV employed for the simulation, fee Ref. [66].

6. Conclusions
Acknowledgements
References
- Biot, J.B. Phénomènes de polarisation successive, observés dans des fluides homogénes. Bull. Soc. Philomatique 1815, 190–192. [Google Scholar]
- van't Hoff, J.H. Sur les formules de structure dans l’espace. Bull. Soc. Chim. France 1875, 23, 295–301. [Google Scholar]
- Le Bel, J.A. Sur des relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull. Soc. Chim. France 1874, 22, 337–347. [Google Scholar]
- Pasteur, L. Recherches sur la dissymétrie moléculaire des produits organiques naturels. In Two Lectures Delivered to the Société Chimique de Paris, Paris, France, 20 January & 3 Feburary 1860.
- Schweitzer-Stenner, R. Secondary Structure Analysis of Polypeptides Based on an Excitonic Coupling Model to Describe the Band Profile of Amide I‘ of IR, Raman, and Vibrational Circular Dichroism Spectra. J. Phys. Chem. B 2004, 108, 16965–16975. [Google Scholar] [CrossRef]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to Study Proteins by Circular Dichroism. Biochim .Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Nafie, L.A. Vibrational optical activity: Comparison of theoretical and experimental results for (+)-(3R)-methylcyclohexanone. J. Chem. Phys. 1980, 73, 1567–1575. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Stephens, P.J. Ab initio calculation of atomic axial tensors and vibrational rotational strengths using density functional theory. Chem. Phys. Lett. 1996, 252, 211–220. [Google Scholar] [CrossRef]
- Barron, L.D. Raman Optical Activity: A New Probe of Stereochemistry and Magnetic Structure. Acc. Chem. Res. 1980, 13, 90–96. [Google Scholar] [CrossRef]
- Barron, L.D.; Hecht, L.; McColl, I.H.; Blanch, E.W. Raman Optical Activity Comes of Age. Mol. Phys. 2004, 102, 731–744. [Google Scholar]
- Prasad, P.L.; Nafie, L.A. Experimental Observations and Theoretical Predictions of Raman Optical Activity. In Proceedings of the VIIth International Conference on Raman Spectroscopy, Ottawa, Canada, 4-9 August 1980; Murphy, W.F., Ed.; North-Holland: New York, NY, USA, 1980; pp. 252–253. [Google Scholar]
- Diem, M.; Polavarapu, P.L.; Oboodi, M.; Nafie, L.A. Vibrational circular dichroism in amino acids and peptides. 4. Vibrational analysis, assignments, and solution-phase Raman spectra of deuterated isotopomers of alanine. J. Am. Chem. Soc. 1982, 104, 3329–3336. [Google Scholar] [CrossRef]
- Nafie, L.A.; Yu, G.-S.; Freedman, T.B. Raman Optical Activity of Biological Molecules. Vib.Spectrosc. 1995, 8, 231–239. [Google Scholar] [CrossRef]
- Yu, G.-S.; Freedman, T. B.; Nafie, L.A.; Deng, Z.; Polavarapu, P.L. Experimental Measurement and Ab Initio Calculation of Raman Optical Activity of L-Alanine and Its Deuterated Isotopomers. J. Phys. Chem. 1995, 99, 835–843. [Google Scholar]
- Fischer, P.; Hache, F. Nonlinear Optical Spectroscopy of Chiral Molecules. Chirality 2005, 17, 421–437. [Google Scholar] [CrossRef]
- Ji, N.; Ostroverkhov, V.; Belkin, M.; Shiu, Y.-J.; Shen, Y.-R. Toward Chiral Sum-Frequency Spectroscopy. J. Am. Chem. Soc. 2006, 128, 8845–8848. [Google Scholar] [CrossRef]
- Burke, B.J.; Moad, A.J.; Polizzi, M.A.; Simpson, G.J. Experimental Confirmation of the Importance of Orientation in the Anomalous Chiral Sensitivity of Second Harmonic Generation. J. Am. Chem. Soc. 2003, 125, 9111–9115. [Google Scholar]
- Mesnil, H.; Hache, F. Experimental evidence of third-order nonlinear dichroism in a liquid of chiral molecules. Phys. Rev. Lett. 2000, 85, 4257–4260. [Google Scholar] [CrossRef]
- Baranowska, A.; Rizzo, A.; Jansìk, B.; Coriani, S. Non linear effects in the interaction of time-dependent fields and chiral systems. A computational investigation. J. Chem. Phys. 2006, 125, 054107-1-1. [Google Scholar] [CrossRef]
- Power, E.A. Developments in the theory of multiphoton absorption by molecules (bound-bound): Applications of a chirooptic character. In New Frontiers in Quantum Electrodynamics and Quantum Optics; Barut, E.A., Ed.; Plenum Press: New York, NY, USA, 1990; pp. 253–266. [Google Scholar]
- Gedanken, A.; Tamir, M. Multiphoton optical rotator dispersion. Rev. Sci. Instrum. 1987, 58, 950–952. [Google Scholar]
- Cameron, R.; Tabisz, G.C. Observation of two-photon optical rotation by molecules. Mol. Phys. 1997, 90, 159–164. [Google Scholar]
- Qu, W.; Tabisz, G.C. Ab initio calculations of nonlinear optical rotation by several small chiral molecules and by uridine stereoisomers. J. Chem. Phys. 2006, 124, 184305-1-9. [Google Scholar] [CrossRef]
- Cameron, R.; Tabisz, G.C. Characterization of intensity-dependent optical rotation phenomena in chiral molecules in solution. J. Chem. Phys. 2007, 126, 224507-1-10. [Google Scholar] [CrossRef]
- De Boni, L.; Toro, C.; Hernández, F.E. Synchronized double L-scan technique for the simultaneous measurement of polarization dependence two-photon absorption in chiral molecules. Optics Lett. 2008, 33, 2958–2960. [Google Scholar] [CrossRef]
- Tinoco, I. Two-photon circular dichroism. J. Chem. Phys. 1975, 62, 1006–1009. [Google Scholar]
- Power, E.A. Two-photon circular dichroism. J. Chem. Phys. 1975, 63, 1348–1350. [Google Scholar] [CrossRef]
- Jansík, B.; Rizzo, A.; Ågren, H. Response theory calculations of two-photon circular dichroism. Chem. Phys. Lett. 2005, 414, 461–467. [Google Scholar] [CrossRef]
- Jansík, B.; Rizzo, A.; Bondo Pedersen, T.; Ågren, H. Origin invariant approaches to the calculation of two-photon circular dichroism. J. Chem. Phys. 2006, 125, 064113-1-11. [Google Scholar] [CrossRef]
- Jansík, B.; Rizzo, A.; Ågren, H. Ab initio study of the two-photon circular dichroism in chiral natural amino acids. J. Phys. Chem. B 2007, 111, 446–460, Erratum, ibid., 2409-2414.. [Google Scholar] [CrossRef]
- Jansík, B.; Rizzo, A.; Ågren, H.; Champagne, B. Strong two-photon circular dichroism in helicenes: a theoretical investigation. J. Chem. Theory Comput. 2008, 4, 457–467. [Google Scholar] [CrossRef]
- Rizzo, A. Recent progress in the computation of non linear optical properties of chiral systems. In Proceedings of AIP Conference, Computational Methods in Science and Engineering: Theory and Computation: Old Problems and New Challenges. Lectures Presented at the International Conference on Computational Methods in Science and Engineering 2007 (ICCMSE 2007), Corfù, Greece, 25-30 September, 2007; Maroulis, G., Simos, T.E., Eds.; American Institute of Physics: College Park, MD, USA, 2007; 963, pp. 379–388. [Google Scholar]
- Rizzo, A.; Lin, N.; Ruud, K. Ab initio study of the one- and two-photon circular dichroism of R-(+)-3-methyl-cyclopentanone. J. Chem. Phys. 2008, 128, 164312-1-17. [Google Scholar] [CrossRef]
- Lin, N.; Santoro, F.; Rizzo, A.; Luo, Y.; Zhao, X.; Barone, V. Theory for vibronically resolved two-photon circular dichroism spectra. Application to (R)-(+)-3-methyl-cyclopentanone. J. Phys. Chem. A 2009, 113, 4198–4207. [Google Scholar]
- Guillaume, M.; Ruud, K.; Rizzo, A.; Monti, S.; Lin, Z.; Xu, X. Ab initio study of the one- and two-photon circular dichroism of (L)-Tryptophan. J. Phys. Chem. B 2010, 114, 6500–6512. [Google Scholar]
- Goppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 9, 273–295. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar]
- Cumpston, B.H.; Ananthavel, S.P.; Barlow, S.; Dyer, D.L.; Ehrlich, J.E.; Erskine, L.L.; Heikal, A.A.; Kuebler, S.M.; Lee, I.-Y.S.; McCord-Maughon, D.; Qin, J.; Röckel, H.; Rumi, M.; Wu, X.-L.; Marder, S.R.; Perry, J.W. Two-photon polymerization initiators for three-dimensional optical data storage and microfabrication. Nature 1999, 348, 51–54. [Google Scholar]
- Li, R.; Sullivan, R.; Al-Basheer, W.; Pagni, R.M.; Compton, R.N. Linear and nonlinear circular dichroism of R-(+)-3-methylcyclopentanone. J. Chem. Phys. 2006, 125, 144304-1-8. [Google Scholar]
- Bornschlegl, A.; Logé, C.; Boesl, U. Investigation of CD effects in the multi photon ionisation of R-(+)-3-methylcyclopentanone. Chem. Phys. Lett. 2007, 447, 187–191. [Google Scholar]
- Markowicz, P.; Samoc, M.; Cerne, J.; Prasad, P.; Pucci, A.; Ruggeri, G. Modified Z-scan techniques for investigations of nonlinear chiroptical effects. Opt. Expr. 2004, 12, 5209–5214. [Google Scholar] [CrossRef]
- Sheik-Bahae, M.; Said, A.A.; Wei, T.-H.; Hagan, D.J.; Van Stryland, E.W. Sensitive measurement of optical nonlinearities using a single beam. IEEE J. Quantum Electr. 1990, 26, 760–769. [Google Scholar]
- Toro, C.; De Boni, L.; Lin, N.; Santoro, F.; Rizzo, A.; Hernández, F.E. Two-Photon Absorption Circular Dichroism: “A new twist of nonlinear spectroscopy”. Chem. Eur. J. 2010, 16, 3504–3509. [Google Scholar] [CrossRef]
- Diaz, C.; Lin, N.; Hernandez, F.E.; Rizzo, A. University of Central Florida, Orlando, FL, USA. Unpublished work, 2011.
- Andrews, D.L. A two-chromophore model for two-photon circular dichroism. Chem. Phys. 1976, 16, 419–424. [Google Scholar] [Green Version]
- Meath, W.J.; Power, E.A. On the importance of permanent moments in multiphoton absorption using perturbation theory. J. Phys. B. At. Mol. Phys. 1984, 17, 763–782. [Google Scholar] [CrossRef]
- Meath, W.J.; Power, E.A. On the effects of diagonal dipole matrix elements in multi-photon resonance profiles using two-level systems as models. Mol. Phys. 1984, 51, 585–600. [Google Scholar] [CrossRef]
- Meath, W.J.; Power, E.A. Differential multiphoton absorption by chiral molecules and the effect of permanent moments. J. Phys. B.: At. Mol. Phys. 1987, 20, 1945–1964. [Google Scholar] [CrossRef]
- Meath, W.J.; Power, E.A. On the Interaction of Elliptically Polarized Light with Molecules; the Effects of Both Permanent and Transition Multipole Moments on Multiphoton Absorption and Chiroptical Effects. J. Mod. Optics 1989, 36, 977–1002. [Google Scholar] [CrossRef]
- Lin, S.H.; Fujimura, Y.; Neusser, H.J.; Schlag, E.W. Multiphoton Spectroscopy of Molecules; Academic Press: New York, NY, USA, 1984. [Google Scholar]
- Szłucki, J.; Stręk, W. Two photon circular dichroism in lanthanide (III) complexes. J. Chem. Phys. 1986, 85, 5547–5550. [Google Scholar]
- Gunde, K.E.; Richardson, F.S. Fluorescence-detected two-photon circular dichroism of Gd3+ in trigonal Na3[Gd(C4H4O5)3]·2NaClO4·6H2O. Chem. Phys. 1995, 194, 195–200. [Google Scholar] [CrossRef]
- Schellman, J.A. Circular dichroism and optical rotation. Chem. Rev. 1975, 75, 323–331. [Google Scholar]
- Barron, L.D. Molecular Light Scattering and Optical Activity; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism. Principles and Applications, 2nd ed; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Craig, D.P.; Thirunamachandran, T. Molecular Quantum Electrodynamics. An Introduction to Radiation Molecule Interaction; Dover Publications Inc.: New York, NY, USA, 1984. [Google Scholar]
- Olsen, J.; Jørgensen, P. Linear and non linear response functions for an exact state and for an MCSCF state. J. Chem. Phys. 1985, 82, 3235–3264. [Google Scholar]
- Olsen, J.; Jørgensen, P. Time-dependent response theory with applications to self-consistent field and multiconfigurational self-consistent field wave functions. In Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific: Singapore, Singapore, 1995; pp. 857–990, Part II. [Google Scholar]
- Dalgaard, E. Quadratic response functions within the time-dependent Hartree-Fock approximation. Phys. Rev. A 1982, 26, 42–52. [Google Scholar]
- DALTON, a molecular electronic structure program, Release 2.0 (2005). Available online: http://www.kjemi.uio.no/software/dalton/dalton.html (accessed on 15 February 2011).
- Hettema, H.; Jensen, H.J.Aa.; Jørgensen, P.; Olsen, J. Quadratic response functions from a multiconfigurational self-consistent field wave function. J. Chem. Phys. 1992, 97, 1174–1190. [Google Scholar]
- Hättig, C.; Christiansen, O.; Jørgensen, P. Multiphoton transition moments and absorption cross section in coupled cluster response theory employing variational transition moment functional. J. Chem. Phys. 1998, 108, 8331–8354. [Google Scholar] [CrossRef]
- Christiansen, O.; Jørgensen, P.; Hättig, C. Response Functions from Fourier Component Variational Perturbation Theory applied to a Time-Averaged Quasienergy. Int. J. Quantum. Chem. 1998, 68, 1–52. [Google Scholar] [CrossRef]
- Sałek, P.; Vahtras, O.; Helgaker, T.; Ågren, H. Density-functional theory of linear and nonlinear time-dependent molecular properties. J. Chem. Phys. 2002, 117, 9630–9645. [Google Scholar] [CrossRef]
- Lin, N.; Santoro, F.; Zhao, X.; Toro, C.; De Boni, L.; Hernández, F.E.; Rizzo, A. Computational challenges in simulating and analyzing experimental linear and nonlinear circular dichroism spectra. R-(+)-1,1'-bi(2-naphthol) as a prototype case. J. Phys. Chem. B 2011, 115, 811–824. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Yanai, Y.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Helgaker, T.; Sałek, P.; Keal, T.W.; Lutnæs, O.B.; Tozer, D.J.; Handy, N.C. Assessment of a Coulomb-attenuated exchange-correlation energy functional. Phys. Chem. Chem. Phys. 2006, 8, 558–562. [Google Scholar]
- Paterson, M.J.; Christiansen, O.; Pawłowski, F.; Jørgensen, P.; Hättig, C.; Helgaker, T.; Sałek, P. Benchmarking Two-Photon Absorption With CC3 Quadratic Response Theory, and Comparison With Density Functional Response Theory. J. Chem. Phys. 2006, 124, 054322-1-10. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3063. [Google Scholar]
- Li, R.; Sullivan, R.; Al-Basheer, W.; Pagni, R.M.; Compton, R.N. Linear and nonlinear circular dichroism of R-(+)-3-methylcyclopentanone. J. Chem. Phys. 2006, 125, 144304:1–144304:8. [Google Scholar]
- Boesl von Grafenstein, U.; Bornschlegl, A. Circular Dichroism Laser Mass Spectrometry: Differentiation of 3-Methylcyclopentanone Enantiomers. ChemPhysChem 2006, 7, 2085–2087. [Google Scholar]
- Jacquemin, D.; Wathelet, V.; Perpète, E.A.; Adamo, C. Extensive TD-DFT benchmark: singlet-excited states of organic molecules. J. Chem. Theor. Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef]
- Santoro, F.; Barone, V.; Improta, R. Can TD-DFT calculations accurately describe the excited states behavior of stacked nucleobases? The cytosine dimer as a test case. J. Comput. Chem. 2008, 29, 957–964. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation energies in density functional theory: An evaluation and a diagnostic test. J. Chem. Phys. 2008, 128, 044118-1-8. [Google Scholar]
- Kriech, M.A.; Conboy, J.C. Using the Intrinsic Chirality of a Molecule as a Label-Free Probe to Detect Molecular Adsorption to a Surface by Second Harmonic Generation. Appl. Spectr. 2005, 59, 746–753. [Google Scholar]
- Prasad, L.; Polavarapu, P.L.; Petrovic, A.G.; Vick, S.E.; Wulff, W.D.; Ren, H.; Ding, Z.; Staples, R.J. Absolute Configuration of 3,3’-Diphenyl-[2,2’-binaphthalene]-1,1’-diol Revisited. J. Org. Chem. 2009, 74, 5451–5457. [Google Scholar]
- Petrovic, A.G.; Vick, S.E.; Polavarapu, P.L. Determination of the Absolute Stereochemistry of Chiral Biphenanthryls in Solution Phase Using Chiroptical Spectroscopic Methods: 2,2’-Diphenyl-[3,3’-biphenanthrene]-4,4’-diol. Chirality 2008, 20, 501–510. [Google Scholar] [CrossRef]
- Wanapun, D.; Wampler, R.D.; Begue, N.J.; Simpson, G.J. Polarization-dependent two-photon absorption for the determination of protein secondary structure: A theoretical study. Chem. Phys. Lett. 2008, 455, 6–12. [Google Scholar] [CrossRef]
- Toro, C.; De Boni, L.; Lin, N.; Santoro, F.; Rizzo, A.; Hernández, F.E. Two-Photon Absorption Circular-Linear Dichroism of Axial Enantiomers. Chirality 2010, 22, E202–E210. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hernández, F.E.; Rizzo, A. Two-Photon Polarization Dependent Spectroscopy in Chirality: A Novel Experimental-Theoretical Approach to Study Optically Active Systems. Molecules 2011, 16, 3315-3337. https://doi.org/10.3390/molecules16043315
Hernández FE, Rizzo A. Two-Photon Polarization Dependent Spectroscopy in Chirality: A Novel Experimental-Theoretical Approach to Study Optically Active Systems. Molecules. 2011; 16(4):3315-3337. https://doi.org/10.3390/molecules16043315
Chicago/Turabian StyleHernández, Florencio E., and Antonio Rizzo. 2011. "Two-Photon Polarization Dependent Spectroscopy in Chirality: A Novel Experimental-Theoretical Approach to Study Optically Active Systems" Molecules 16, no. 4: 3315-3337. https://doi.org/10.3390/molecules16043315
APA StyleHernández, F. E., & Rizzo, A. (2011). Two-Photon Polarization Dependent Spectroscopy in Chirality: A Novel Experimental-Theoretical Approach to Study Optically Active Systems. Molecules, 16(4), 3315-3337. https://doi.org/10.3390/molecules16043315