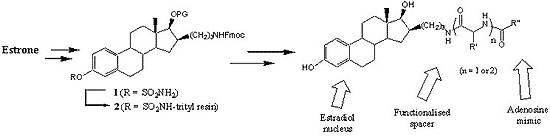
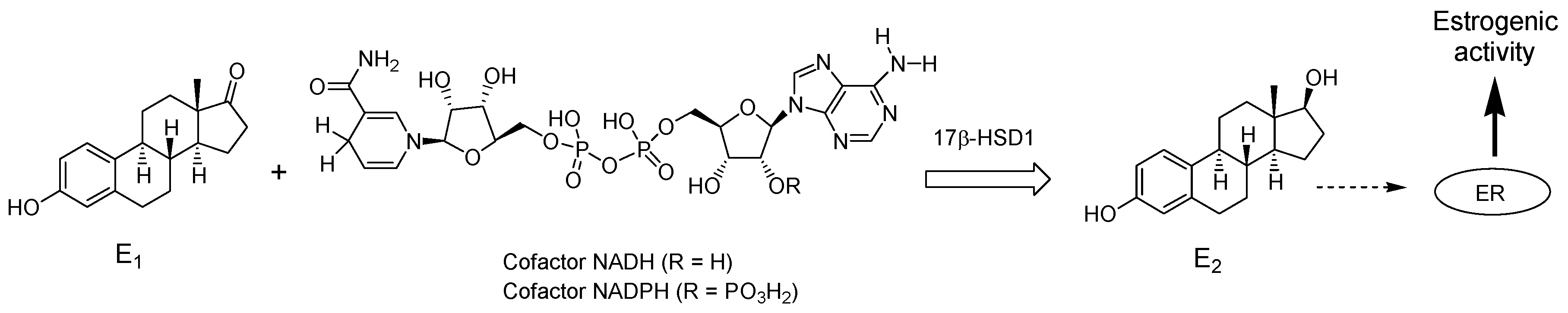
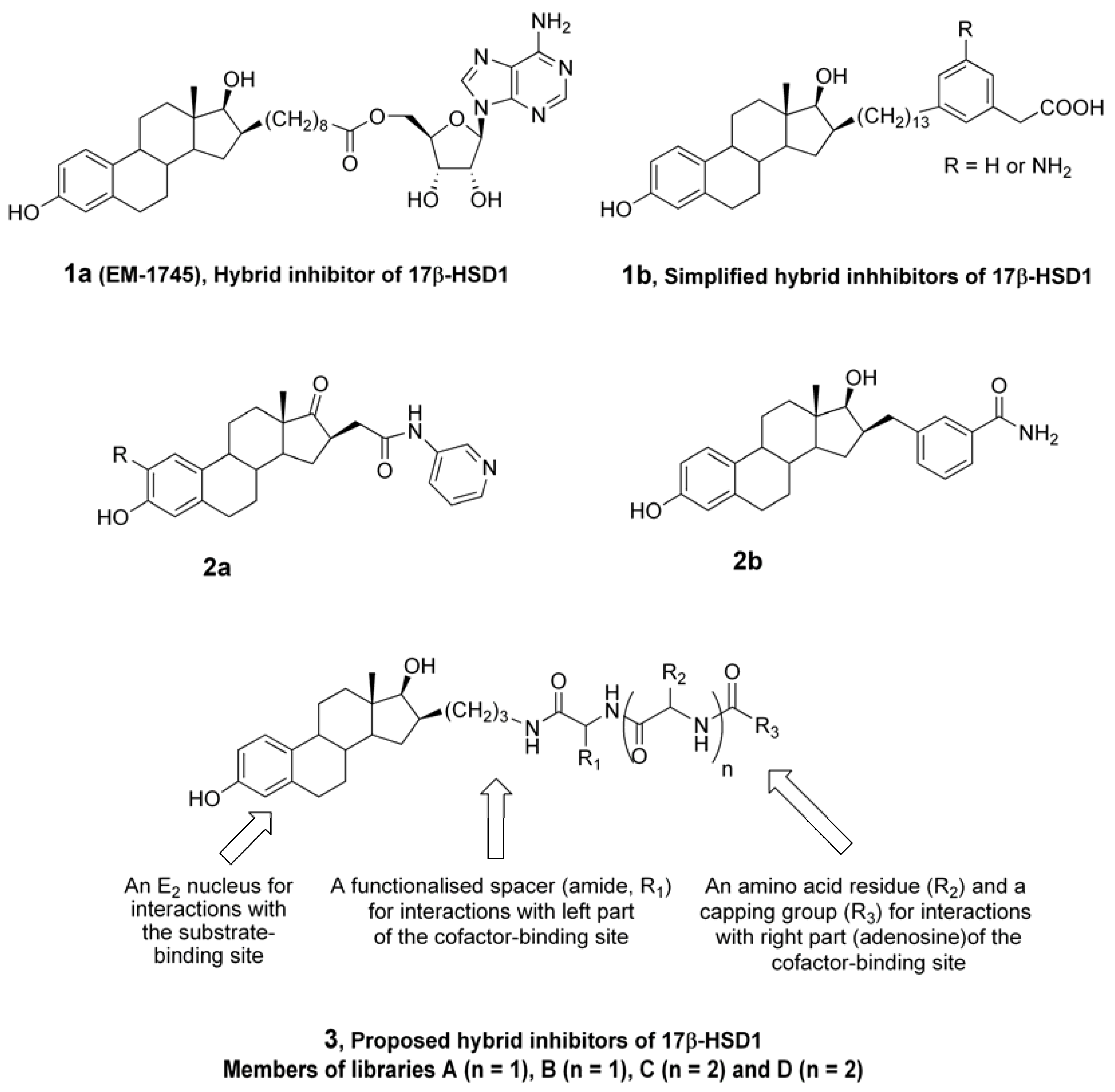
4.2. Chemical synthesis
4.2.1. 3-Sulfamoyloxy-16β-[3-(9H-fluoren-9-ylmethoxycarbonylamino)-propyl]-17β-acetoxy-estra-1,3,5(10)-triene (6a)
The starting material 3-(3-
tert-butyldimethylsilyloxy-17β-acetoxy-estra-1,3,5(10)-trien-16β-yl)-azido-propane (
4a) was synthesized as previously reported [
33]. A suspension of
4a (680 mg, 1.32 mmol) and 5% Pd/C (136 mg) in EtOAc (15 mL) and MeOH (160 mL) was stirred under a hydrogen atmosphere at room temperature. After 2 h, the resulting suspension was filtered through Celite, washed with MeOH and evaporated to dryness to afford the amine (620 mg, 96%) in the form of a viscous colourless oil in good purities without purification. To a solution of the crude amine (620 mg) in anhydrous THF (40 mL) under an argon atmosphere at 0 °C was added a solution of TBAF (1M in THF, 1.53 mL, 1.53 mmol). After 30 min, the reaction was quenched by addition of a saturated aqueous solution of NaHCO
3. The crude product was extracted with EtOAc and the organic phase was washed with brine, dried over MgSO
4 and evaporated to dryness to afford the phenol (585 mg) in the form of a yellowish solid. The crude phenol (500 mg) was dissolved in THF (120 mL) and H
2O (40 mL). At 0 °C, NaHCO
3 (1M in H
2O, 1.84 mL, 1.84 mmol) and Fmoc-OSu (570 mg, 1.69 mmol) were added and the reaction was stirred for 1 h at 0 °C. Then, the reaction mixture was quenched by addition of H
2O and the crude product was extracted with EtOAc and DCM. Each organic phase was washed with brine, dried over MgSO
4, and evaporated to dryness to provide
5a (1.05 g) in the form of a white foam. To a solution of crude phenol
5a (1.05 g) in dry DCM (120 mL) under an argon atmosphere were added 2,6-di-
t-butyl-4-methylpyridine (933 mg, 4.55 mmol) and NH
2SO
2Cl [
34] (421 mg, 3.64 mmol). The reaction was stirred for 24 h at room temperature and quenched by the addition of ice water. The crude product was extracted with DCM and the organic phase was washed with brine, dried over MgSO
4, and evaporated to dryness. Purification by flash chromatography (hexanes/EtOAc, 6:4) gained precursor
6a (604 mg, 82% yield for 4 steps) in the form of a white foam. IR (film) 3393 (NH and NH
2), 1716 (C=O, ester and carbamate), 1374 and 1188 (S=O, sulfamate);
1H-NMR (300 MHz, CDCl
3) δ 0.80 (s, 18-CH
3), 1.00 to 2.35 (m, CH and CH
2 of steroid skeleton and alkyl chain), 2.07 (s, CH
3CO), 2.84 (m, 6-CH
2), 3.18 (m, C
H2NH), 4.20 (t,
J = 6.8 Hz, CH
2C
H of Fmoc), 4.38 (d,
J = 6.9 Hz, C
H2CH of Fmoc), 4.73 (d,
J = 10.0 Hz, 17α-CH), 4.76 (m, N
HFmoc), 5.01 (s
br, NH
2), 7.01 (s, 4-CH), 7.05 (d,
J1 = 8.6 Hz,
J2 = 2.3 Hz, 2-CH), 7.28 (m, 1-CH and 2 × CH of Fmoc), 7.38 (t,
J = 7.4 Hz, 2 × CH of Fmoc), 7.57 (d,
J = 7.4 Hz, 2 × CH of Fmoc), 7.75 (d,
J = 7.4 Hz, 2 × CH of Fmoc);
13C-NMR (75 MHz, CDCl
3) δ 13.3, 21.0, 25.9 (2×), 27.0, 28.8, 29.5, 32.1, 37.5, 37.6, 38.2, 41.1, 43.4, 43.9, 47.3, 48.7, 66.6, 83.2, 118.9, 120.0 (2×), 121.9, 125.0 (2×), 126.8, 127.0 (2×), 127.7 (2×), 138.8, 139.5, 141.3 (2×), 144.0 (2×), 147.9 156.4, 171.2; LRMS calculated for C
38H
43N
2O
7S [M-H]
- 671.3, found 671.1 m/z.
4.2.2. 3-Sulfamoyloxy-16β-[3-(9H-fluoren-9-ylmethoxycarbonylamino)-propyl]-17β-(tetrahydro-2H-pyran-2-yl-oxy)-estra-1,3,5(10)-triene (6b)
3-(3-Hydroxy-17β-(tetrahydro-
2H-pyran-2-yl-oxy)-estra-1,3,5(10)-trien-16β-yl)-azidopropane (
4b) was synthesized as previously reported [
33]. A suspension of
4b (2.35 g, 5.35 mmol) and 5% Pd/C (470 mg) in MeOH (535 mL) was stirred under hydrogen atmosphere at room temperature. After 3 h, the resulting suspension was filtered through celite, washed with MeOH and evaporated to dryness to afford the amine (2.00 g) in good purities without purification. The crude amine (2.00 g) was dissolved in THF (360 mL) and H
2O (120 mL). At 0 °C, NaHCO
3 (1M in H
2O, 6.3 mL, 6.3 mmol) and Fmoc-OSu (1.96 g, 5.81 mmol) were added and the reaction was stirred for 1 h at 0 °C. The reaction mixture was then quenched by addition of H
2O and the crude product was extracted with EtOAc and DCM. Each organic phase was washed with brine, dried over MgSO
4, and evaporated to dryness to provide
5b (3.62 g) in the form of a white foam. To a solution of crude phenol
5b (3.62 g) in dry DCM (280 mL) under an argon atmosphere were added di-
tert-butyl-4-methylpyridine (2.92 g, 14.2 mmol) and NH
2SO
2Cl [
34] (1.31 g, 11.4 mmol). The reaction was stirred for 16 h at room temperature and quenched by addition of icewater. The crude product was extracted with DCM and the organic phase was washed with brine, dried over MgSO
4 and evaporated to dryness. Purification by flash chromatography (hexanes/EtOAc, 8:2 to 6:4) provided precursor
6b (2.00 g, 52% yield for 3 steps) as a white foam. The 17β-unprotected precursor (1.27 g) was also obtained. This product was reprotected with a THP using this procedure. The 17β-hydroxy precursor (1.27 g, 2.07 mmol) was dissolved in a minimum of dry THF (~2 mL) and DCM (40 mL) under an argon atmosphere at 0 °C. Then, 3,4-dihydro-2
H-pyran (187 µL, 2.07 mmol) and
p-TSA (39 mg, 0.21 mmol) were added and the reaction was stirred for 10 min. The reaction was quenched by addition of saturated aqueous solution of NaHCO
3 and the crude precursor was extracted with DCM. The organic phase was washed with brine, dried over MgSO
4 and evaporated under reduced pressure. Purification by flash chromatography (hexanes/EtOAc, 7:3 to 6:4) afforded precursor
6b (1.09 g, 74% yield). IR (film) 3392 (NH and NH
2), 1698 (C=O, carbamate), 1386 and 1187 (S=O, sulfamate);
1H-NMR (400 MHz, acetone-d
6) δ 0.83 and 0.87 (2s, 18-CH
3), 1.00 to 2.40 (m, CH and CH
2 of steroid skeleton and alkyl chain), 2.87 (m, 6-CH
2), 3.19 (m, C
H2NH), 3.47 and 3.92 (2m, OCH
2 of THP), 3.76 and 3.80 (2d,
J = 10.0 Hz, 17α-CH), 4.23 (t,
J = 7.1 Hz, CH
2C
H of Fmoc), 4.33 (d,
J = 7.2 Hz, C
H2CH of Fmoc), 4.64 and 4.71 (2m, CH of THP), 6.52 (NH), 7.08 (m, 2-CH, 4-CH and NH
2), 7.34 (m, 1-CH and 2 × CH of Fmoc), 7.42 (t,
J = 7.4 Hz, 2 × CH of Fmoc), 7.71 (d,
J = 7.5 Hz, 2 × CH of Fmoc), 7.88 (d,
J = 7.5 Hz, 2 × CH of Fmoc);
13C-NMR (100 MHz, acetone-d
6) δ 13.5 (14.3), 20.0 (20.5), 26.2 (26.3), 26.77, 26.84, 27.7, 29.1, 29.3 to 30.2 (1C under solvent peaks), 31.3 (31.6), 32.78 (32.82), 38.6, 38.70 (38.74), 39.0 (40.2), 41.5 (41.6), 44.2 (44.7), 44.6 (44.8), 48.0, 49.3 (49.5), 62.0 (63.0), 66.5, 86.1 (86.6), 98.2 (99.7), 120.0, 120.6 (2×), 122.8, 125.9 (2×), 127.2, 127.7 (2×), 128.5 (2×), 139.0, 139.6, 141.9 (2×), 145.0 (2×), 149.1, 156.9; LRMS calculated for C
41H
49N
2O
7S [M-H]
- 713.3, found 713.3 m/z.
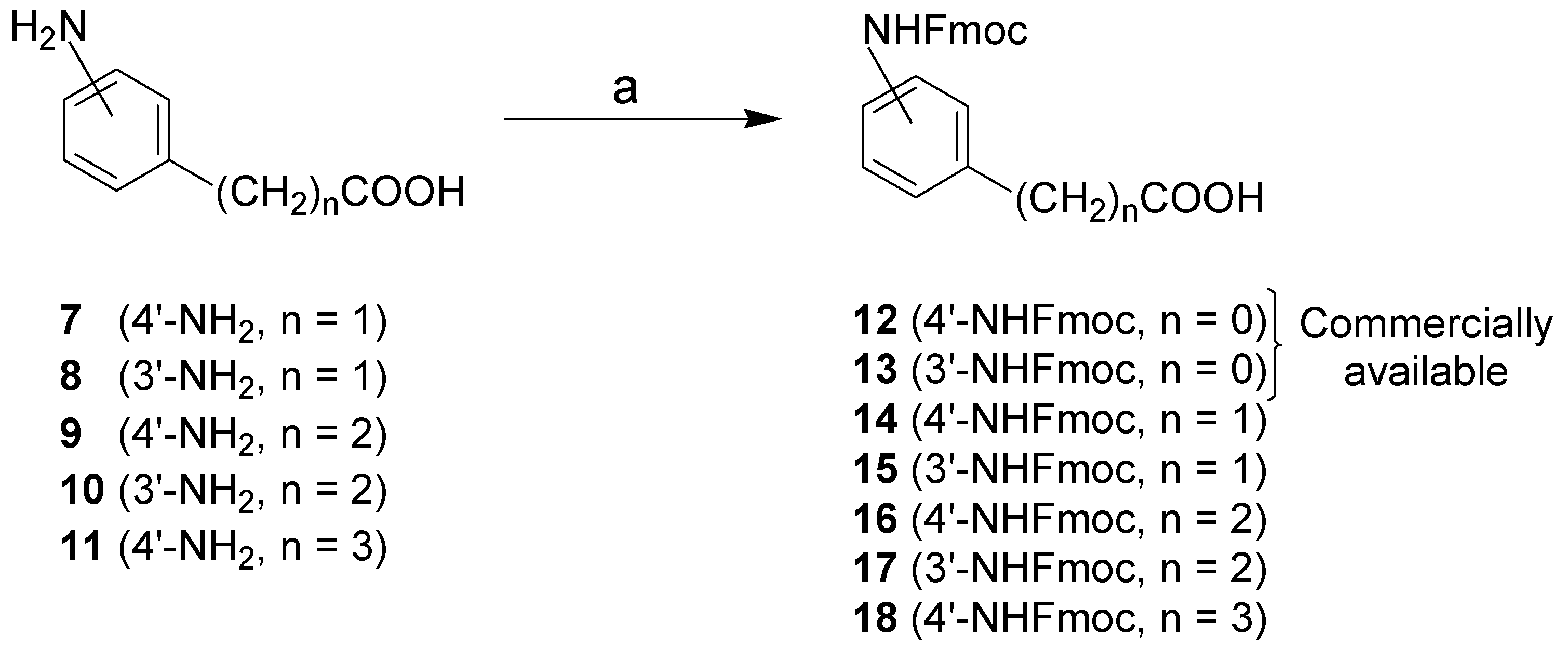
4.2.3. General procedure to synthesize Fmoc-NH protected carboxylic acid building blocks 14–18
Amines
9 and
10 were prepared as previously reported [
35]. Amines
7–11 (0.90–1.15 g, 5.45–6.93 mmol) were dissolved in THF (50 mL) and H
2O (10 mL). Then, NaHCO
3 (1M in H
2O, 13.6–17.3 mL, 13.6–17.3 mmol) and Fmoc-OSu (2.76–3.51 g, 8.17–10.4 mmol) were added. The reaction was stirred for 16 h at room temperature. The reaction mixture was then preabsorbed on silica gel and purified by flash chromatography (DCM/MeOH, 95:5 to 85:15) to afford
14–18 (1.35–2.00 g, 51–93% yield).
2-[4′-(9H-Fluoren-9-ylmethoxycarbonylamino)-phenyl]-acetic acid (14). White solid (1.72 g, 70% yield); IR (KBr) 3700–2200 (OH, COOH), 3336 (NH), 1708 (C=O, acid and carbamate); 1H-NMR (400 MHz, DMSO-d6) δ 3.47 (s, CH2COOH), 4.31 (t, J = 6.5 Hz, CH2CH of Fmoc), 4.49 (d, J = 6.2 Hz, CH2CH of Fmoc), 7.16 (d, J = 7.8 Hz, 2′-CH and 6′-CH), 7.39 (m, 3′-CH, 5′-CH and 4 × CH of Fmoc), 7.76 (d, J = 7.3 Hz, 2 × CH of Fmoc), 7.91 (d, J = 7.6 Hz, 2 × CH of Fmoc), 9.69 (sbr, NH); 13C-NMR (75 MHz, DMSO-d6) δ 40.3, 46.7, 65.5, 118.2 (2×), 120.2 (2×), 125.2 (2×), 127.1 (2×), 127.7 (2×), 129.5, 129.7 (2×), 137.5, 140.8 (2×), 143.8 (2×), 153.5, 173.2; LRMS calculated for C23H18NO4 [M-H]- 372.1, found 372.2 m/z.
2-[3′-(9H-Fluoren-9-ylmethoxycarbonylamino)-phenyl]-acetic acid (15). White solid (2.00 g, 81% yield); IR (KBr) 3600–2200 (OH, COOH), 3287 (NH), 1702 (C=O, acid and carbamate); 1H-NMR (400 MHz, DMSO-d6) δ 3.51 (s, CH2COOH), 4.32 (t, J = 6.7 Hz, CH2CH of Fmoc), 4.47 (d, J = 6.6 Hz, CH2CH of Fmoc), 6.90 (d, J = 7.5 Hz, 6′-CH), 7.21 (t, J = 7.7 Hz, 5′-CH), 7.40 (m, 2′-CH, 4′-CH and 4 × CH of Fmoc), 7.77 (d, J = 7.4 Hz, 2 × CH of Fmoc), 7.92 (d, J = 7.4 Hz, 2 × CH of Fmoc), 9.74 (sbr, NH), 12.37 (sbr,COOH); 13C-NMR (75 MHz, DMSO-d6) δ 41.0, 46.6, 65.6, 116.7, 119.2, 120.2 (2×), 123.6, 125.2 (2×), 127.2 (2×), 127.7 (2×), 128.6, 135.6, 139.0, 140.8 (2×), 143.8 (2×), 153.4, 172.6; LRMS calculated for C23H20NO4 [M+H]+ 374.1, found 374.1 m/z.
3-[4′-(9H-Fluoren-9-ylmethoxycarbonylamino)-phenyl]-propionic acid (16). White solid (1.35 g, 51% yield); IR (KBr) 3500–2000 (OH, COOH), 3331 (NH), 1702 (C=O, acid and carbamate); 1H-NMR (400 MHz, DMSO-d6) δ 2.50 (t, J = 7.5 Hz, CH2COOH), 2.76 (t, J = 7.5 Hz, CH2CH2COOH), 4.31 (t, J = 6.5 Hz, CH2CH of Fmoc), 4.48 (d, J = 6.3 Hz, CH2CH of Fmoc), 7.13 (d, J = 7.9 Hz, 2′-CH and 6′-CH), 7.39 (m, 3′-CH, 5′-CH and 4 × CH of Fmoc), 7.76 (d, J = 7.4 Hz, 2 × CH of Fmoc), 7.91 (d, J = 7.4 Hz, 2 × CH of Fmoc), 9.65 (sbr, NH), 12.13 (sbr, COOH); 13C-NMR (75 MHz, DMSO-d6) δ 29.7, 35.4, 46.6, 65.5, 118.4 (2×), 120.2 (2×), 125.1 (2×), 127.1 (2×), 127.7 (2×), 128.5 (2×), 134.9, 137.0, 140.8 (2×), 143.8 (2×), 153.4, 173.8; LRMS calculated for C24H22NO4 [M+H]+ 388.1, found 388.0 m/z.
3-[3′-(9H-Fluoren-9-ylmethoxycarbonylamino)-phenyl]-propionic acid (17). White solid (1.96 g, 93% yield); IR (KBr) 3500–2000 (OH, COOH), 3307 (NH), 1696 (C=O, acid and carbamate); 1H-NMR (400 MHz, DMSO-d6) δ 2.52 (t, J = 7.5 Hz, CH2COOH), 2.79 (t, J = 7.4 Hz, CH2CH2COOH), 4.32 (t, J = 6.6 Hz, CH2CH of Fmoc), 4.48 (d, J = 6.3 Hz, CH2CH of Fmoc), 6.87 (d, J = 7.5 Hz, 6′-CH), 7.18 (t, J = 7.7 Hz, 5′-CH), 7.40 (m, 2′-CH, 4′-CH and 4 × CH of Fmoc), 7.77 (d, J = 7.4 Hz, 2 × CH of Fmoc), 7.91 (d, J = 7.5 Hz, 2 × CH of Fmoc), 9.70 (sbr, NH), 12.18 (sbr, COOH); 13C-NMR (75 MHz, DMSO-d6) δ 30.5, 35.2, 46.7, 65.6, 116.2, 118.2, 120.2 (2×), 122.4, 125.2 (2×), 127.2 (2×), 127.7 (2×), 128.7, 139.1, 140.8 (2×), 141.5, 143.8 (2×), 153.4, 173.7; LRMS calculated for C24H22NO4 [M+H]+ 388.1, found 388.0 m/z.
4-[4′-(9H-Fluoren-9-ylmethoxycarbonylamino)-phenyl]-butanoic acid (18). White solid (1.59 g, 68% yield); IR (KBr) 3500–2000 (OH, COOH), 3345 (NH), 1708 (C=O, acid and carbamate); 1H-NMR (400 MHz, DMSO-d6) δ 1.77 (quintuplet, J = 7.5 Hz, CH2CH2COOH), 2.20 (t, J = 7.4 Hz, CH2COOH), 2.52 (m, CH2CH2CH2COOH), 4.31 (t, J = 6.5 Hz, CH2CH of Fmoc), 4.48 (d, J = 6.2 Hz, CH2CH of Fmoc), 7.09 (d, J = 7.9 Hz, 2′-CH and 6′-CH), 7.39 (m, 3′-CH, 5′-CH and 4 × CH of Fmoc), 7.76 (d, J = 7.4 Hz, 2 × CH of Fmoc), 7.91 (d, J = 7.5 Hz, 2 × CH of Fmoc), 9.65 (sbr, NH), 12.07 (sbr, COOH); 13C-NMR (75 MHz, DMSO-d6) δ 26.4, 33.0, 33.7, 46.7, 65.5, 118.4 (2×), 120.2 (2×), 125.2 (2×), 127.1 (2×), 127.7 (2×), 128.6 (2×), 135.6, 136.9, 140.8 (2×), 143.8 (2×), 153.5, 174.3; LRMS calculated for C25H24NO4 [M+H]+ 402.2, found 402.0 m/z.
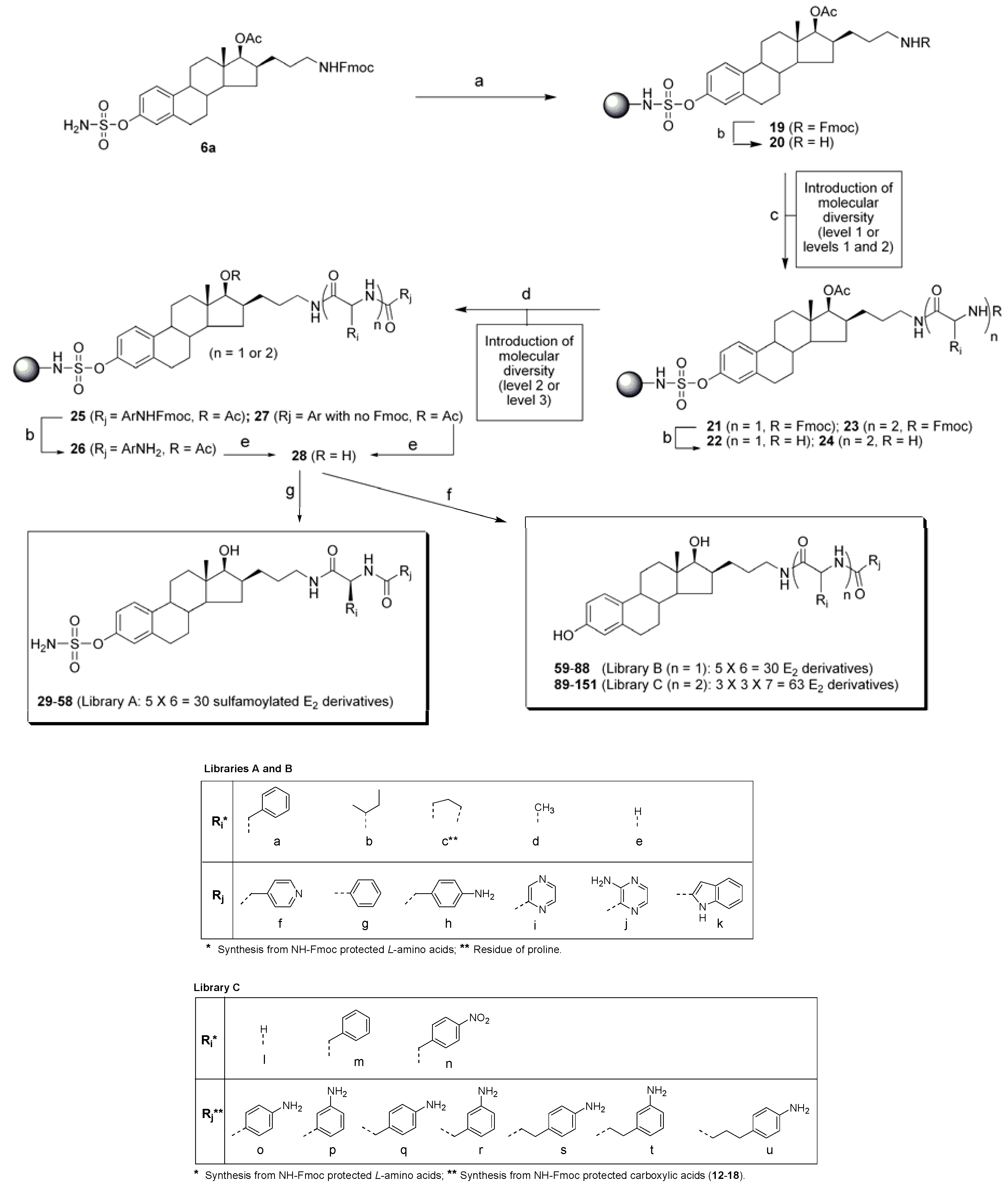
4.2.4. Synthesis of resin 20 for preparation of libraries A and B
The loading reaction was run in two 25 mL peptide flasks using the same amount of resin. In each peptide flask, a solution of precursor 6a (1.07 g, 1.62 mmol) in dry DCM (10 mL) was added to trityl chloride resin (1.09 g, 1.24 mmol/g theoretical loading) under an argon atmosphere. After 5 min, diisopropylethylamine (DIPEA, 2.16 mL) was added and the mixture was shaken for 12 h at room temperature. It is noteworthy to mention that to avoid total evaporation of DCM during the loading step, the argon inlet was removed after 30 min of reaction. The resin was filtered and washed with DCM (3×), MeOH (3×), and again with DCM, then dried overnight under a vacuum to afford 3.34 g of resin 19 globally. The loading yield, calculated by the mass increase, was 70%. The deprotection step was also run in two 25 mL peptide flasks using the same amount of resin. Resin 19 (1.67 g) was swollen in 15 mL of a solution of piperidine (20%) in DCM and shaken for 90 min at room temperature. Then, the resin was filtered and washed with DCM (3×), MeOH (3×), and again with DCM, and dried under a vacuum for 16 h. Acidic mini-cleavage (5% TFA/DCM, 10 min) of a sample of resin 20 and TLC analysis confirmed the complete deprotection of the secondary amine. The two batches of resin 20 prepared as described above were mixed before the next step. Then, 30 bottom fritted reaction vessels of the 96 solid-phase reaction block of ACT LabTech manual synthesizer were loaded with 80 mg of resin 20, which correspond to 0.052 mmol of compound 6a (without the Fmoc protective group).
4.2.5. Introduction of two levels of molecular diversity (libraries A and B)
Five stock solutions, each containing 2 equivalents (6×) of Fmoc-protected amino acid [Fmoc-L-Phe-OH (259 mg, 0.672 mmol), Fmoc-L-Ile-OH (236 mg, 0.672 mmol), Fmoc-L-Pro-OH (226 mg, 0.672 mmol), Fmoc-L-Ala-OH (208 mg, 0.672 mmol) or Fmoc-Gly-OH (199 mg, 0.672 mmol)], 2 equivalents (6×) of benzotriazole-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBOP) (348 mg, 0.672 mmol) and 2 equivalents (6×) of hydroxybenzotriazole (HOBt) (91 mg, 0.672 mmol), were prepared in dry DMF (5.0 mL). Prior to addition to the resin, 4 equivalents (6×) of DIPEA (230 µL, 1.34 mmol) were added to each stock solution. To each of the 30 resins 20 was added 0.8 mL of the appropriate stock solution. The resins were shaken under argon for 3 h at room temperature, then filtered, washed with DMF (2×), MeOH (3×) and DCM (3×) and dried under a vacuum. TLC analysis after a mini-cleavage test (5% TFA/DCM, 10 min) with samples of resins 21 revealed that the coupling reactions were not completed. Thus, the coupling step was repeated using the same procedure as described above. This time, TLC analysis after a mini-cleavage with sample of resins 21 indicated the completion of the coupling reaction. Thus, the resins 21 were reacted 1 h with 1.0 mL of a solution of piperidine (20%) in DCM to remove the Fmoc protective group. The resins were then filtered, washed with DCM (3×), MeOH (3×) and DCM (1×), and dried under a vacuum to give 5 groups of resin of general structure 22. The second level of molecular diversity was introduced by a similar coupling procedure described for the introduction of the first level. Thus, 6 stock solutions, each containing 2 equivalents (5x) of carboxylic acid building blocks [4-pyridyl-acetic acid (89 mg, 0.515 mmol), benzoic acid (63 mg, 0.515 mmol), 14 (192 mg, 0.515 mmol), 2-pyrazinecarboxylic acid (64 mg, 0.515 mmol), 3-aminopyrazine-2-carboxylic acid (72 mg, 0.515 mmol) or indole-2-carboxylic acid (83 mg, 0.515 mmol)], 2 equivalents (5x) of bromo-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBrOP) (240 mg, 0.515 mmol) and 2 equivalents (5x) of HOBt (70 mg, 0.515 mmol), were prepared in 4 mL of dry DMF and 4 equivalents (5x) of DIPEA (180 µL, 1.03 mmol) was added. To each of the 30 resins 22 was added 0.8 mL of the appropriate stock solution. The resins were shaken under an argon atmosphere for 3 h at room temperature, then filtered, washed with MeOH (1×), DMF (2×), MeOH (3×) and DCM (2×) and dried under a vacuum. The completion of the reaction was monitored by TLC analysis of acidic mini-cleavage of samples of resins 25 (when Rj contains a Fmoc protecting group) and 27 (when Rj contains any Fmoc protecting group). As for the introduction of the first level of molecular diversity, the acylation step was repeated (2 times) as described above to afford completion.
4.2.6. Removal of protective groups (libraries A and B)
Resins 25, containing the carboxylic acid 14 as building block were treated with 1 mL of a solution of piperidine (20%) in DCM to remove the Fmoc protective group. The mixtures were shaken for 1 h at room temperature and then filtered, washed with DCM (2×) and MeOH (2×), and dried under a vacuum. Completion of the reactions was confirmed by TLC analysis after a sampling mini-cleavage (5% TFA/DCM, 10 min) of resins 26. The acetate protective group of resins 26 or 27 was removed with 1 mL of a solution of 1 M MeONa in MeOH (25%) in THF. The mixtures were shaken for 48 h at room temperature and then, filtered, washed with MeOH (3×) and DCM (2×), and dried under a vacuum. TLC analysis of acidic mini-cleavage of sampling of resins 28 confirmed the complete deprotection of C17β-alcohol.
4.2.7. Generation of E2 derivatives (library B) by nucleophilic cleavage
To each of the 30 resins
28 was added a solution of diethylamine (DEA, 30% in THF, 1.5 mL). The mixtures were shaken for 24 h at room temperature, then filtered and washed with acetone (3×). The filtrates were collected in preweighed tubes and evaporated in a Speedvac apparatus. Each product was dissolved in EtOAc, evaporated twice, and dried under a vacuum pump in order to obtain the DEA-free product. The phenol derivatives
59–88 (2.6–18.8 mg, 10–63% overall yield from
19,
Table 2) were obtained in high average HPLC purity (90%) according to a random sampling of 6 library members, compounds
61,
69,
74,
78,
82 and
83.
16β-(N-4-Amino-phenylacetyl-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (61). Yellowish solid (8.2 mg, 26% yield); IR (film) 3292 (OH, NH and NH2), 1641 (C=O, amide); 1H- NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.80 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.77 (m, 6-CH2), 2.91 and 3.05 (2m, CHCH2Ph), 3.13 (t, J = 6.5 Hz, CH2NH), 3.37 (s, CH2PhNH2), 3.70 (d, J = 9.9 Hz, 17α-CH), 4.55 (m, COCHNH), 6.49 (d, J = 2.5 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 6.65 (d, J = 8.4 Hz, 2 × CH of aminophenyl), 6.90 (d, J = 8.4 Hz, 2 × CH of aminophenyl), 7.08 (d, J = 8.4 Hz, 1-CH), 7.20 (m, 5 × CH of phenyl); LRMS calculated for C38H48N3O4 [M+H]+ 610.4, found 610.4 m/z; HPLC purity = 85%.
16β-(N-3-Amino-2-pyrazinoyl-L-isoleucine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (69). Light yellow solid (6.4 mg, 22% yield); IR (film) 3312 (OH, NH and NH2), 1649 (C=O, amide); 1H-NMR (400 MHz, methanol-d4) δ 0.74 (s, 18-CH3), 0.90 to 2.30 (m, CH and CH2 of steroid skeleton and alkyl chain), 0.96 (t, J = 7.4 Hz, CH2CH3), 1.00 (d, J = 6.8 Hz, CHCH3), 2.77 (m, 6-CH2), 3.24 (m, CH2NH and CHCH3), 3.69 (d, J = 9.9 Hz, 17α-CH), 4.39 (d, J = 7.6 Hz, COCHNH), 6.49 (d, J = 2.6 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 7.08 (d, J = 8.5 Hz, 1-CH), 7.84 (d, J = 2.4 Hz, 1 × CH of pyrazine), 8.10 (d, J = 2.4 Hz, 1 × CH of pyrazine); LRMS calculated for C32H46N5O4 [M+H]+ 564.4, found 564.5 m/z; HPLC purity = 95%.
16β-(N-2-Pyrazinoyl-L-proline-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (74). Yellowish solid (11.7 mg, 43% yield); IR (film) 3312 (OH and NH), 1631 (C=O, amide); 1H-NMR (400 MHz, methanol-d4) δ 0.70 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 0.78 and 0.79 (2s, 18-CH3), 2.79 (m, 6-CH2), 3.05 and 3.23 (2m, CH2NH), 3.71 to 3.95 (m, CH2N of proline and 17α-CH), 4.60 and 5.05 (2m, CHN of proline), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 7.08 (d, J = 8.5 Hz, 1-CH), 8.69 (m, 2 × CH of pyrazine), 9.05 (dd, J1 = 2.5 Hz, J2 = 1.5 Hz, 1 × CH of pyrazine); LRMS calculated for C31H41N4O4 [M+H]+ 533.3, found 533.4 m/z; HPLC purity = 91%.
16β-(N-Benzoyl-L-alanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (
78). Light yellow solid (5.4 mg, 21% yield). HPLC purity = 90% [50% of MeOH/H
2O (90:10) and 50% of H
2O, both containing 20 mM of NH
4OAc]. The characterization of compound
78 was reported earlier by us [
30].
16β-(N-1H-Indole-2-carbonyl-L-alanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (82). Light yellow solid (3.7 mg, 13% yield); IR (film) 3292 (OH and NH), 1650 (C=O, amide); 1H -NMR (400 MHz, methanol-d4) δ 0.72 (s, 18-CH3), 0.85 to 2.30 (m, CH and CH2 of steroid skeleton and alkyl chain), 1.50 (d, J = 7.2 Hz, CHCH3), 2.75 (m, 6-CH2), 3.22 (m, CH2NH), 3.69 (d, J = 9.9 Hz, 17α-CH), 4.59 (q, J = 7.2 Hz, CHCH3), 6.48 (d, J = 2.6 Hz, 4-CH), 6.55 (d, J = 8.4 Hz, 2-CH), 7.06 (m, 1-CH and 1 × CH of indole), 7.20 (m, 2 × CH of indole), 7.43 (dd, J1 = 8.3 Hz, J2 = 0.8 Hz, 1 × CH of indole), 7.61 (d, J = 8.1 Hz, 1 × CH of indole); LRMS calculated for C33H42N3O4 [M+H]+ 544.3, found 544.5 m/z; HPLC purity = 89%.
16β-(N-4-Pyridylacetyl-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (83). White solid (7.3 mg, 28% yield); IR (film) 3284 (OH and NH), 1649 (C=O, amide); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.80 to 2.30 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.79 (m, 6-CH2), 3.23 (m, CH2NH), 3.69 (s, CH2pyridine), 3.71 (d, J = 10.0 Hz, 17α-CH), 3.86 (s, COCH2NH), 6.49 (d, J = 2.5 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 7.09 (d, J = 8.6 Hz, 1-CH), 7.43 (dd, J1 = 4.6 Hz, J2 = 1.4 Hz, 2′-CH and 6′-CH of pyridine), 8.48 (dd, J1 = 4.6 Hz, J2 = 1.6 Hz, 3′-CH and 5′-CH of pyridine); LRMS calculated for C30H40N3O4 [M+H]+ 506.3, found 506.5 m/z; HPLC purity = 88%.
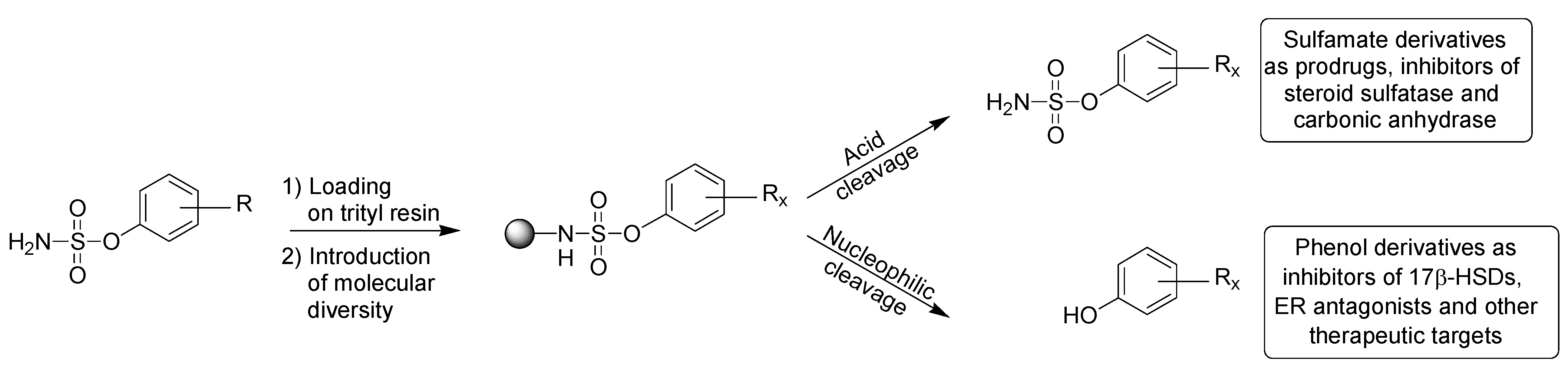
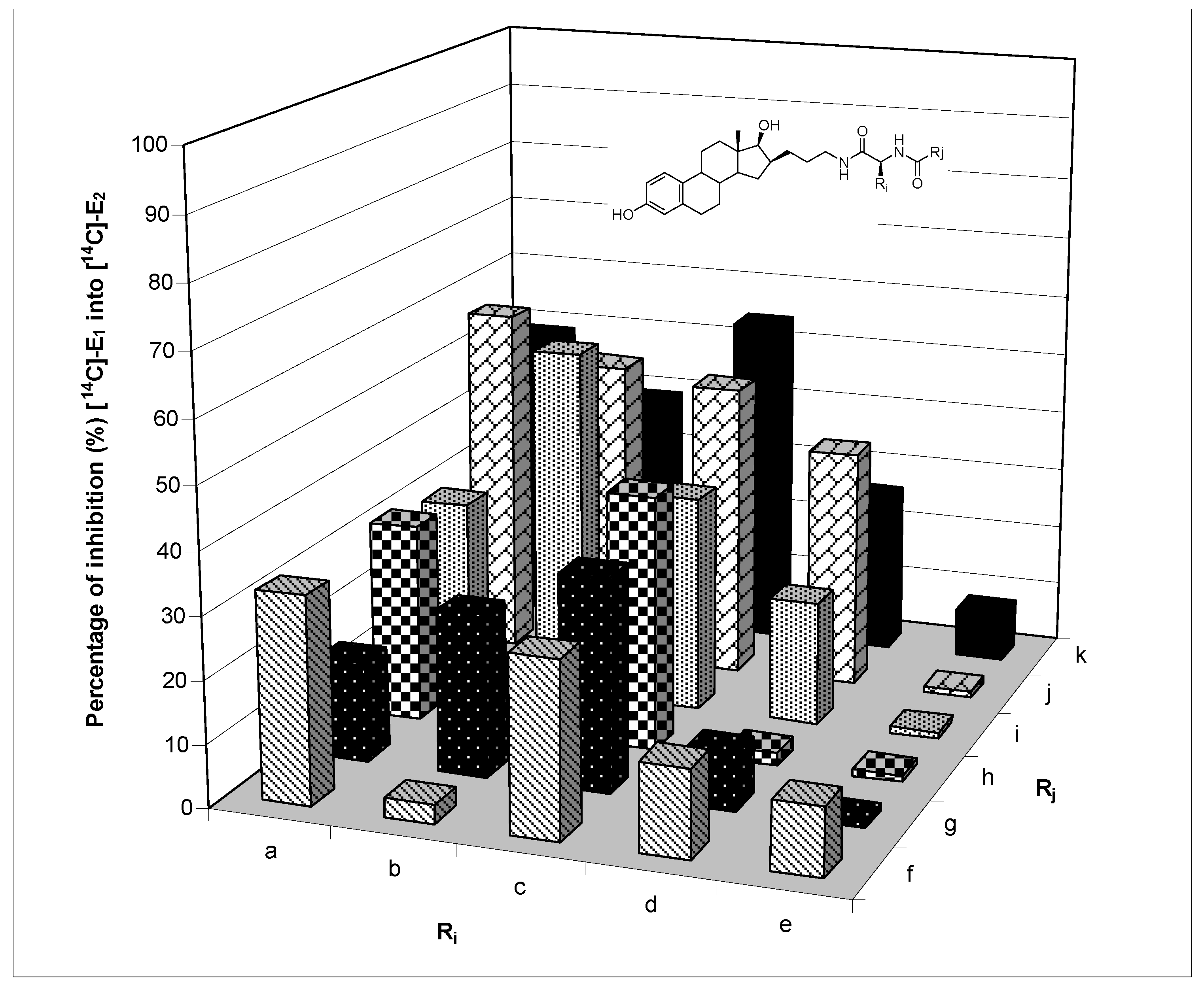
4.2.8. Generation of sulfamoylated E2 derivatives (library A) by acidic cleavage
To each of the 30 resins
28 which have already been treated by nucleophilic cleavage conditions, was added 1 mL of a solution of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (30%) in DCM. The mixtures were shaken for 6 h at room temperature, then filtered and washed with acetone (2×). The filtrates were collected in preweighed tubes, evaporated in a Speedvac apparatus and dried under a vacuum pump. The sulfamate derivatives
29–58 (10.5–18.2 mg, 35–53% overall yield from
19,
Table 1) were obtained in high average HPLC purity (90%) according to a random sampling of six library members, compounds
30,
38,
41,
49,
51 and
58.
3-O-Sulfamate-16β-(N-benzoyl-L-phenylalanine-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene (30). Light yellow solid (15.4 mg, 45% yield); IR (film) 3307 (OH, NH and NH2), 1636 (C=O, amide), 1374 and 1184 (S=O, sulfamate); 1H-NMR (400 MHz, methanol-d4) δ 0.77 (s, 18-CH3), 0.95 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.88 (m, 6-CH2), 3.05 and 3.19 (2m, CHCH2Ph and CH2NH), 3.72 (d, J = 9.8 Hz, 17α-CH), 4.80 (m, COCHNH), 7.02 (d, J = 2.4 Hz, 4-CH), 7.06 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 7.20 to 7.55 (m, 1-CH, 5 × CH of phenyl, and 3′-CH, 4′-CH and 5′-CH of benzoyl), 7.76 (dd, J1 = 8.5 Hz, J2 = 1.4 Hz, 2′-CH and 6′-CH of benzoyl); LRMS calculated for C37H46N3O6S [M+H]+ 660.3, found 660.4 m/z; HPLC purity = 89%.
3-O-Sulfamate-16β-(N-2-pyrazinoyl-L-isoleucine-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene (
38). Light yellow solid (16.2 mg, 50% yield). HPLC purity = 90%. The characterization of compound
38 was reported earlier by us [
30].
3-O-Sulfamate-16β-(N-4-pyridylacetyl-L-proline-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene (41). Light yellow solid (15.0 mg, 47% yield); IR (film) 3292 (OH, NH and NH2), 1641 (C=O, amide), 1374 and 1184 (S=O, sulfamate); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.80 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.89 (m, 6-CH2), 3.22 (t, J = 7.1 Hz, CH2NH), 3.33 (COCH2pyridine under solvent peaks), 3.70 (m, 17α-CH and CH2N of proline), 4.40 (m, CHN of proline), 7.03 (d, J = 2.4 Hz, 4-CH), 7.06 (dd, J1 = 8.5 Hz, J2 = 2.5 Hz, 2-CH), 7.34 (d, J = 6.6 Hz, 1-CH), 7.40 (dd, J1 = 4.6 Hz, J2 = 1.6 Hz, 2′-CH and 6′-CH of pyridine), 8.47 (dd, J1 = 4.5 Hz, J2 = 1.6 Hz, 3′-CH and 5′-CH of pyridine); LRMS calculated for C33H45N4O6S [M+H]+ 625.3, found 625.4 m/z; HPLC purity = 89%.
3-O-Sulfamate-16β-(N-4-amino-phenylacetyl-L-alanine-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene (49). Light yellow solid (12.1 mg, 38% yield); IR (film) 3308 (OH, NH and NH2), 1652 (C=O, amide), 1374 and 1184 (S=O, sulfamate); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.90 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 1.33 (d, J = 7.1 Hz, CHCH3), 2.89 (m, 6-CH2), 3.18 (t, J = 7.1 Hz, CH2NH), 3.43 (s, CH2PhNH2), 3.73 (d, J = 9.8 Hz, 17α-CH), 4.30 (q, J = 7.2 Hz, CHCH3), 6.70 (dd, J1 = 6.4 Hz, J2 = 2.0 Hz, 2 × CH of aminophenyl), 7.04 (m, 2-CH, 4-CH and 2 × CH of aminophenyl), 7.35 (d, J = 8.6 Hz, 1-CH); LRMS calculated for C32H45N4O6S [M+H]+ 613.3, found 613.3 m/z; HPLC purity = 91%.
3-O-Sulfamate-16β-(N-3-amino-pyrazinoyl-L-alanine-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene (51). Light yellow solid (14.5 mg, 47% yield); IR (film) 3346 (OH, NH and NH2), 1646 (C=O, amide), 1375 and 1186 (S=O, sulfamate); 1H-NMR (400 MHz, methanol-d4) δ 0.77 (s, 18-CH3), 0.95 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 1.47 (d, J = 7.1 Hz, CHCH3), 2.88 (m, 6-CH2), 3.23 (m, CH2NH), 3.72 (d, J = 9.8 Hz, 17α-CH), 4.54 (q, J = 7.1 Hz, CHCH3), 7.02 (d, J = 2.4 Hz, 4-CH), 7.06 (dd, J1 = 8.6 Hz, J2 = 2.5 Hz, 2-CH), 7.33 (d, J = 8.6 Hz, 1-CH), 7.86 (d, J = 2.4 Hz, 1 × CH of pyrazine), 8.13 (d, J = 2.4 Hz, 1 × CH of pyrazine); LRMS calculated for C29H41N6O6S [M+H]+ 601.3, found 601.3 m/z; HPLC purity = 90%.
3-O-Sulfamate-16β-(N-1H-indole-2-carbonyl-glycine-aminopropyl)-17β-hydroxy-estra-1,3,5(10)-triene 58). White solid (13.0 mg, 41% yield); IR (film) 3292 (OH, NH and NH2), 1636 (C=O, amide), 1374 and 1186 (S=O, sulfamate); 1H-NMR (400 MHz, methanol-d4) δ 0.75 (s, 18-CH3), 0.95 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.88 (m, 6-CH2), 3.26 (m, CH2NH), 3.71 (d, J = 9.9 Hz, 17α-CH), 4.05 (s, COCH2NH), 7.02 (d, J = 2.5 Hz, 4-CH), 7.06 (m, 2-CH and 1 × CH of indole), 7.14 (d, J = 0.8 Hz, 1 × CH of indole), 7.22 (dt, J1 = 8.0 Hz, J2 = 1.2 Hz, 1 × CH of indole), 7.33 (d, J = 8.4 Hz, 1-CH), 7.44 (dd, J1 = 8.3 Hz, J2 = 0.8 Hz, 1 × CH of indole), 7.62 (d, J = 8.1 Hz, 1 × CH of indole); LRMS calculated for C32H41N4O6S [M+H]+ 609.3, found 609.2 m/z; HPLC purity = 89%.
4.2.9. Synthesis of resin 20 for preparation of library C
The loading procedure described above for the preparation of libraries A and B was used without any significant modifications. Briefly, trityl chloride resin (10.1 g, 1.50 mmol/g theoretical loading) and precursor 6a (5.11 g, 7.6 mmol) were put in a round 250 mL dry flask equipped with a septum and an argon inlet. DCM (100 mL) was added and the reaction mixture was stirred for 3 min. Then, DIPEA (24.3 mL) was added and the mixture was shaken at room temperature. After 30 min of reaction, the argon inlet was removed. After 16 h, the mixture was filtered, washed with DCM (3×) and MeOH (3×), and dried under a vacuum to afford 14.1 g of resin 19. The loading yield, calculated by the mass increase, was 42%. It is noteworthy to mention that the loading could not be higher than 50% because 2 equivalents of trityl chloride resin were used for 1 equivalent of precursor 6a. Then 63 fritted bottom reaction vessels of the 96 solid-phase reaction block of ACT LabTech manual synthesizer were loaded with 200 mg of resin 19. A solution of piperidine (20%) in DCM was added to resins 19 and the mixtures were shaken for 1 h at room temperature to remove the Fmoc protective group. Then, the resin was filtered and washed with DCM (2×), MeOH (3×), and dried under a vacuum for 16 h. The completion of the deprotection was proven by acidic mini-cleavage as described above. Thus, each reaction vessel contains resins loading 0.109 mmol of compound 6a (without the Fmoc protective group).
4.2.10. Introduction of three levels of molecular diversity (library C)
The levels of molecular diversity were introduced using a similar procedure as the one described above for the preparation of libraries A and B. To introduce the first level of molecular diversity, three stock solutions, each containing 2 equivalents (21×) of Fmoc-protected amino acid [Fmoc-Gly-OH (1.36 g, 4.56 mmol), Fmoc-L-Phe-OH (1.77 g, 4.56 mmol) or Fmoc-L-4-NO2-Phe-OH (1.98 g, 4.56 mmol)], 2 equivalents (21×) of PyBOP (2.37 g, 4.56 mmol) and 2 equivalents (21×) of HOBt (616 mg, 4.56 mmol), were prepared in dry DMF (42 mL). Prior to addition to the resin, 4 equivalents (21×) of DIPEA (1.59 mL, 9.12 mmol) were added to each stock solution. To each of the 63 resins 20 was added 2.0 mL of the appropriate stock solution. The resins were shaken under an argon atmosphere for 2 h at room temperature, then filtered, washed with DMF (2×), DCM (2×) and MeOH (3×) and dried under a vacuum. This procedure was repeated (3 times) until completion of the reaction was afforded and verified by acidic mini-cleavage as described above. The cleavage of Fmoc group of resins 21 was then performed by a 1 h treatment with 2.0 mL of a solution of piperidine (20%) in DCM. The resins were then filtered, washed with DCM (3×), MeOH (3×), and dried under a vacuum to give three groups of resins of general structure 22. The second level of molecular diversity was introduced using the same building blocks and the same procedure described above for the introduction of the first level of molecular diversity. This time, the coupling procedure was repeated twice to afford completion of the reaction by TLC analysis after acidic mini-cleavage. Resins of general structure 23 were then treated with a solution of piperidine (20%) in DCM for 1 h at room temperature. The resins were filtered, washed with DCM (2×) and MeOH (2×), and dried to afford resins of general structure 24. The third level of molecular diversity was introduced by coupling resins 24 with carboxylic acid building blocks 12–18. Seven stock solutions, each containing 2 equivalents (9x) of carboxylic acid building blocks 12–18 [12 (701 mg, 1.95 mmol), 13 (701 mg, 1.95 mmol), 14 (729 mg, 1.95 mmol), 15 (729 mg, 1.95 mmol), 16 (756 mg, 1.95 mmol), 17 (756 mg, 1.95 mmol) or 18 (784 mg, 1.95 mmol)], 2 equivalents (9x) of PyBrOP (911 mg, 1.95 mmol) and 2 equivalents of HOBt (264 mg, 1.95 mmol), were prepared in dry DMF (18 mL) and 4 equivalents (9x) of DIPEA (681 µL, 3.90 mmol) were added. To each of the 63 resins 24 was added 2.0 mL of the appropriate stock solution. The resins were shaken under an argon atmosphere for 2 h at room temperature, then filtered, washed with DMF (2×), DCM (2×) and MeOH (3×) and dried under a vacuum to afford resins of general structure 25 (n = 2). The completion of the coupling step was proven by acidic mini-cleavage and TLC analysis of samples of resins 25.
4.2.11. Removal of protective groups (library C)
The Fmoc protective group from carboxylic acid building blocks on resins 25 (n = 2) was removed with the same procedure described above for the preparation of libraries A and B. The acetate protective group of resins 26 (n = 2) was removed using a similar procedure described above for the preparation of libraries A and B. However, the mixtures were shaken for 100 h instead of 48 h in order to obtain complete deprotection of the steroidal C17β-hydroxy by TLC analysis after acidic mini-cleavage of samples of resins 28 (n = 2).
4.2.12. Generation of E2 derivatives (library C) by nucleophilic cleavage
To each of the 63 resins
28 (n = 2) was added 1.5 mL of a solution of DEA (30%) in THF. The mixtures were shaken for 50 h at room temperature. Then 1.0 mL of a solution of DEA (30%) in THF was added to each of the 63 resins and the mixtures were shaken for an additional 16 h. The mixtures were next filtered and washed with THF (1×) and MeOH (3×). The filtrates were collected in preweighed tubes and evaporated in a Speedvac apparatus. Each product was dissolved in THF, evaporated and dried under a vacuum pump in order to obtain the DEA-free product. The phenol derivatives
89–151 (5.3–33.5 mg, 6–41% overall yield from
19,
Table 3) were obtained in purity around 70 to 75% according to TLC analysis of all library members and
1H-NMR and LRMS analysis of a random sampling of 14 library members, compounds
89,
94,
99,
103,
109,
111,
114,
118,
121,
126,
133,
143,
144 and
148.
16β-(N-4-Aminobenzoyl-glycine-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (89). Yellowish solid (16.4 mg, 27% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 3.23 (m, CH2NH), 3.69 (d, J = 10.4 Hz, 17α-CH), 3.86 and 3.99 (2m, 2 × COCH2NH), 6.49 (d, J = 2.2 Hz, 4-CH), 6.55 (dd, J1 = 8.6 Hz, J2 = 2.5 Hz, 2-CH), 6.68 (d, J = 7.0 Hz, 3′-CH and 5′-CH of 4-aminobenzoyl), 7.09 (d, J = 8.4 Hz, 1-CH), 7.69 (dd, J1 = 8.5 Hz, J2 = 1.4 Hz, 2′-CH and 6′-CH of 4-aminobenzoyl); LRMS calculated for C32H43N4O5 [M+H]+ 563.3, found 563.3 m/z.
16β-(N-3-Aminophenyl-propanoyl-glycine-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (94). Yellowish solid (16.5 mg, 26% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.79 and 0.80 (2s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.58 (t, J = 7.6 Hz, COCH2CH2), 2.78 (m, 6-CH2), 2.84 (m, COCH2CH2), 3.25 (m, CH2NH), 3.75 (under THF peaks, 17α-CH), 3.83 (s, 2 × COCH2NH), 6.56 (m, 2-CH, 4-CH and 3 × CH of 3-aminophenyl), 7.06 (m, 1-CH and 1 × CH of 3-aminophenyl); LRMS calculated for C32H43N4O5 [M-H]- 589.3, found 589.5 m/z.
16β-(N-3-Aminophenylacetyl-L-phenylalanine-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (99). Yellowish solid (20.4 mg, 28% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 2.98 and 3.17 (2m, CHCH2Ph and CH2NH), 3.43 (s, COCH2PhNH2), 3.75 (under THF peaks, 17α-CH), 3.92 (m, COCH2NH), 4.46 (m, CHCH2Ph), 6.58 (m, 2-CH, 4-CH and 3 × CH of 3-aminophenyl), 7.16 (m, 1-CH, 1 × CH of 3-aminophenyl, 5 × CH of phenyl); LRMS calculated for C40H49N4O5 [M-H]- 665.4, found 665.5 m/z.
16β-(N-4-Aminobenzoyl-L-4-nitro-phenylalanine-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (103). Yellowish solid (13.9 mg, 18% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (m, 18-CH3), 0.80 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 3.24 (m, CH2NH and CHCH2PhNO2), 3.75 (under THF peaks, 17α-CH), 3.92 (m, COCH2NH), 4.68 (m, CHCH2PhNO2), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 0.8 Hz, 2-CH), 6.65 (dd, J1 = 8.8 Hz, J2 = 2.2 Hz, 3′-CH and 5′-CH of 4-aminobenzoyl), 7.09 (d, J = 8.4 Hz, 1-CH), 7.54 (d, J = 8.6 Hz, 2′-CH and 6′-CH of 4-nitro-phenyl), 7.58 (m, 2′-CH and 6′-CH of 4-aminobenzoyl), 8.18 (m, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C39H48N5O7 [M+H]+ 698.3, found 698.2 m/z.
16β-(N-4-aminophenyl-butanoyl-L-4-nitro-phenylalanine-glycine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (109). Yellowish solid (25.5 mg, 32% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.80 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.41 (m, COCH2CH2), 2.54 (t, J = 7.8 Hz, CH2PhNH2), 2.78 (m, 6-CH2), 3.21 (m, CH2NH and CHCH2PhNO2), 3.75 (under THF peaks, 17α-CH), 3.91 (m, COCH2NH), 4.59 (m, CHCH2PhNO2), 6.49 (s, 4-CH), 6.55 (d, J = 8.4 Hz, 2-CH), 6.67 (m, 3′-CH and 5′-CH of 4-aminophenyl), 6.92 (m, 2′-CH and 6′-CH of 4-aminophenyl), 7.09 (d, J = 8.3 Hz, 1-CH), 7.51 (d, J = 8.6 Hz, 2′-CH and 6′-CH of 4-nitro-phenyl), 8.17 (d, J = 8.6 Hz, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C42H54N5O7 [M+H]+ 740.4, found 740.5 m/z.
16β-(N-3-Aminobenzoyl-glycine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (111). Yellowish solid (28.5 mg, 40% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 2.95 and 3.16 (2m, CHCH2Ph and CH2NH), 3.75 (under THF peaks, 17α-CH), 3.95 (m, COCH2NH), 4.60 (m, CHCH2Ph), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.5 Hz, J2 = 2.4 Hz, 2-CH), 6.88 (d, J = 7.6 Hz, 4′-CH of 3-aminophenyl), 7.05 to 7.35 (m, 1-CH, 2′-CH, 5′-CH and 6′-CH of 3-aminophenyl and 5 × CH of phenyl); LRMS calculated for C39H49N4O5 [M+H]+ 653.4, found 653.2 m/z.
16β-(N-4-Aminophenyl-propanoyl-glycine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (114). Yellowish solid (25.6 mg, 35% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.47 (m, CH2CH2CO), 2.79 (m, 6-CH2 and CH2CH2CO), 2.92 and 3.15 (2m, CHCH2Ph and CH2NH), 3.75 (under THF peaks, 17α-CH), 3.93 (m, COCH2NH), 4.57 (m, CHCH2Ph), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 2-CH), 6.67 (d, J = 8.3 Hz, 3′-CH and 5′-CH of 4-aminophenyl), 6.97 (d, J = 8.3 Hz, 2′-CH and 6′-CH of 4-aminophenyl), 7.09 (d, J = 8.4 Hz, 1-CH), 7.25 (m, 5 × CH of phenyl); LRMS calculated for C41H53N4O5 [M+H]+ 681.4, found 681.5 m/z.
16β-(N-3-Aminobenzoyl-L-phenylalanine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (118). Yellowish solid (32.9 mg, 41% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 3.00 and 3.14 (2m, 2 × CHCH2Ph and CH2NH), 3.75 (under THF peaks, 17α-CH), 4.56 and 4.77 (2m, 2 × CHCH2Ph), 6.48 (d, J = 2.4 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 6.86 (d, J = 7.9 Hz, 4′-CH of 3-aminophenyl), 7.13 (m, 1-CH, 2′-CH, 5′-CH and 6′-CH of 3-aminophenyl and 10 × CH of phenyl); LRMS calculated for C46H55N4O5 [M+H]+ 743.4, found 743.2 m/z.
16β-(N-4-Aminophenyl-propanoyl-L-phenylalanine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (121). Yellowish solid (29.5 mg, 35% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.65 (t, J = 7.8 Hz, CH2CO), 2.78 (m, 6-CH2 and CH2CH2CO), 2.93 and 3.11 (2m, 2 × CHCH2Ph and CH2NH), 3.75 (under THF peaks, 17α-CH), 4.54 (m, 2 × CHCH2Ph), 6.48 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.5 Hz, 2-CH), 6.65 (d, J = 8.3 Hz, 3′-CH and 5′-CH of 4-aminophenyl), 6.90 (d, J = 8.2 Hz, 2′-CH and 6′-CH of 4-aminophenyl), 7.09 (d, J = 8.4 Hz, 1-CH), 7.22 (m, 10 × CH of phenyl); LRMS calculated for C48H59N4O5 [M+H]+ 771.4, found 771.6 m/z.
16β-(N-4-Aminophenyl-acetyl-L-4-nitro-phenylalanine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (126). Yellowish solid (27.6 mg, 32% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 2.94 and 3.14 (2m, CHCH2Ph, CHCH2PhNO2 and CH2NH), 3.33 (under MeOH peaks, COCH2PhNH2), 3.75 (under THF peaks, 17α-CH), 4.53 and 4.66 (2m, CHCH2Ph and CHCH2PhNO2), 6.48 (d, J = 2.4 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.2 Hz, 2-CH), 6.62 (d, J = 8.3 Hz, 3′-CH and 5′-CH of 4-aminophenyl), 6.88 (d, J = 8.4 Hz, 2′-CH and 6′-CH of 4-aminophenyl), 7.09 (d, J = 8.5 Hz, 1-CH), 7.23 (m, 2′-CH and 6′-CH of 4-nitro-phenyl and 5 × CH of phenyl), 8.03 (d, J = 8.7 Hz, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C47H56N5O7 [M+H]+ 802.4, found 802.4 m/z.
16β-(N-4-Aminophenyl-acetyl-glycine-L-4-nitro-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (133). Yellowish solid (20.1 mg, 26% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (m, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.79 (m, 6-CH2), 3.01 and 3.15 (2m, CHCH2PhNO2 and CH2NH), 3.45 (m, COCH2PhNH2), 3.75 (under THF peaks, 17α-CH and COCH2NH), 4.64 (m, CHCH2PhNO2), 6.50 (s, 4-CH), 6.55 (dd, J1 = 8.3 Hz, J2 = 2.5 Hz, 2-CH), 6.71 (m, 3′-CH and 5′-CH of 4-aminophenyl), 7.08 (m, 1-CH and 2′-CH and 6′-CH of 4-aminophenyl), 7.45 (d, J = 8.6 Hz, 2′-CH and 6′-CH of 4-nitro-phenyl), 8.17 (d, J = 8.7 Hz, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C40H50N5O7 [M+H]+ 712.4, found 712.5 m/z.
16β-(N-3-Aminophenyl-propanoyl-L-phenylalanine-L-4-nitro-phenylalanine-aminopropyl)-3,17β-di-hydroxy-estra-1,3,5(10)-triene (143). Yellowish solid (28.8 mg, 33% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.46 (m, CH2CO), 2.76 (m, 6-CH2 and CH2CH2CO), 3.10 (m, CHCH2Ph, CHCH2PhNO2 and CH2NH), 3.75 (under THF peaks, 17α-CH), 4.60 (m, CHCH2Ph and CHCH2PhNO2), 6.55 (m, 2-CH, 4-CH, 2′-CH, 4′-CH and 6′-CH of 3-aminophenyl), 7.14 (m, 1-CH, 5′-CH of 3-aminophenyl and 5 × CH of phenyl), 7.46 (d, J = 8.7 Hz, 2′-CH and 6′-CH of 4-nitro-phenyl), 8.16 (d, J = 8.6 Hz, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C48H58N5O7 [M+H]+ 816.4, found 816.4 m/z.
16β-(N-4-Aminophenyl-butanoyl-L-phenylalanine-L-4-nitro-phenylalanine-aminopropyl)-3,17β-di-hydroxy-estra-1,3,5(10)-triene (144). Yellowish solid (32.8 mg, 36% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.79 (m, 6-CH2, CH2PhNH2), 3.12 (m, CHCH2Ph, CHCH2PhNO2 and CH2NH), 3.75 (under THP peaks, 17α-CH), 4.60 (m, CHCH2Ph and CHCH2PhNO2), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 2-CH), 6.66 (m, 3′-CH and 5′-CH of 4-aminophenyl), 6.87 (d, J = 8.2 Hz, 2′-CH and 6′-CH of 4-aminophenyl), 7.09 (d, J = 8.5 Hz, 1-CH), 7.23 (m, 5 × CH of phenyl), 7.46 (d, J = 8.6 Hz, 2′-CH and 6′-CH of 4-nitro-phenyl), 8.15 (d, J = 8.7 Hz, 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C49H58N5O7 [M-H]- 828.4, found 828.3 m/z.
16β-(N-3-aminophenyl-acetyl-L-4-nitro-phenylalanine-L-4-nitro-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (148). Yellowish solid (27.4 mg, 30% yield); 1H-NMR (400 MHz, methanol-d4) δ 0.78 (s, 18-CH3), 0.85 to 2.40 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 3.01 and 3.16 (2m, 2 × CHCH2PhNO2, and CH2NH), 3.38 (m, CH2PhNH2), 3.75 (under THF peaks, 17α-CH), 4.65 (m, 2 × CHCH2PhNO2), 6.50 (m, 2-CH, 4-CH and 2′-CH, 4′-CH and 6′-CH of 3-aminophenyl), 6.96 (m, 5′-CH of 3-aminophenyl), 7.09 (d, J = 8.6 Hz, 1-CH), 7.39 (m, 2 × 2′-CH and 6′-CH of 4-nitro-phenyl), 8.10 (m, 2 × 3′-CH and 5′-CH of 4-nitro-phenyl); LRMS calculated for C48H55N6O9 [M+H]+ 847.4, found 847.3 m/z.
4.2.13. Synthesis of 2-(3′-bromo-phenyl)-acetic acid tert-butyl ester (153)
3-Bromophenylacetic acid (152) (2.5 g, 11.6 mmol) was dissolved in tert-butanol (120 mL) under an argon atmosphere at room temperature. Then, di-tert-butyl dicarbonate (5.33 mL, 23.2 mmol) and DMAP (426 mg, 3.48 mmol) were added. After 24 h, the reaction mixture was evaporated under reduced pressure and the crude product was purified by flash chromatography (hexanes/EtOAc, 97:3 to 95:5) to provide ester 153 (2.95 g, 94% yield) in the form of a colourless oil. IR (film) 1734 (C=O, ester); 1H-NMR (400 MHz, acetone-d6) δ 1.44 (s, (CH3)3CO), 3.58 (s, CH2COO), 7.30 (sbr, 5′-CH and 6′-CH), 7.45 and 7.51 (2sbr, 2′-CH and 4′-CH); 13C-NMR (75 MHz, acetone-d6) δ 28.1 (3×), 42.1, 81.1, 122.5, 129.1, 130.5, 131.0, 133.1, 138.6, 170.6; LRMS calculated for C12H14BrO2 [M-H]- 269.0, found 269.0 and 271.1 m/z.
4.2.14. Synthesis of 2-(3′-vinyl-phenyl)-acetic acid tert-butyl ester (154)
A suspension of ester 153 (7.00 g, 25.8 mmol), tributyl(vinyl)tin (18.9 mL, 64.5 mmol), Pd2(dba)3 (2.36 g, 2.58 mmol) and P(t-Bu)3 (10% w/v in hexanes) (11.5 mL, 5.67 mmol) in anhydrous toluene (26 mL) was stirred for 16 h under argon atmosphere at room temperature. Then, diethyl ether (130 mL) and KF.2H2O (12.9 g) was added and the mixture was stirred for 30 min in order to quench the reaction. The suspension was filtered through a pad of celite, washed with diethyl ether, and the filtrate was evaporated to dryness. The crude product was purified by flash chromatography (hexanes/EtOAc, 97:3) to provide 154 (4.97 g, 88% yield) in the form of a colourless oil. IR (film) 1732 (C=O, ester); 1H-NMR (400 MHz, acetone-d6) δ 1.44 (s, (CH3)3CO), 3.56 (s, CH2COO), 5.25 (dd, J1 = 10.9 Hz, J2 = 0.8 Hz, CH of CH2=), 5.82 (dd, J1 = 17.6 Hz, J2 = 0.9 Hz, CH of CH2=), 6.76 (dd, J1 = 17.6 Hz, J2 = 10.9 Hz, CH=), 7.30 (m, 4 × CH of phenyl); 13C-NMR (100 MHz, acetone-d6) δ 28.1 (3×), 42.7, 80.7, 114.1, 125.3, 127.9, 129.3, 129.5, 136.1, 136.3, 137.7, 170.7.
4.2.15. Synthesis of 2-[3′-(2″-hydroxy-ethyl)-phenyl]-acetic acid tert-butyl ester (155)
To a solution of vinyl 154 (4.89 g, 22.4 mmol) dissolved in dry THF (450 mL) at 0 °C was added 1.0 M borane-THF complex (100 mL, 100 mmol) under an argon atmosphere. The mixture was stirred at 0 °C for 3 h. Then, 4 M aqueous NaOAc solutions (28.0 mL, 112 mmol) and 30% (w/v) H2O2 (12.7 mL, 112 mmol) were added. After 2 h at 0 °C, the reaction was quenched by water and the extraction was performed with EtOAc. The organic phase was washed with brine, dried over MgSO4, and evaporated to dryness. The crude compound was purified by flash chromatography (hexanes/EtOAc, 7:3 to 6:4) to give alcohol 155 (2.36 g, 45% yield) in the form of a colourless oil. IR (film) 3435 (OH), 1732 (C=O, ester); 1H-NMR (400 MHz, acetone-d6) δ 1.43 (s, (CH3)3CO), 2.81 (t, J = 7.0 Hz, CH2CH2OH), 3.51 (s, CH2COO), 3.68 (m, OH), 3.75 (t, J = 6.8 Hz, CH2OH), 7.18 (m, 4 × CH of phenyl); 13C-NMR (75 MHz, acetone-d6) δ 28.0 (3×), 40.0, 42.7, 63.7, 80.5, 127.4, 128.0, 128.8, 130.5, 135.7, 140.4, 171.0; LRMS calculated for C14H19O3 [M-H]- 235.1, found 235.1 m/z.
4.2.16. Synthesis of 2-[3′-(tert-butoxycarbonylmethyl)-phenyl]-acetic acid (156)
Alcohol 155 (500 mg, 2.11 mmol) was dissolved in dry DCM (42 mL) under an argon atmosphere. Dess-Martin periodinane (988 mg, 2.33 mmol) was then added and the reaction was stirred at room temperature. After 1 h, the mixture was evaporated to dryness. The crude product was filtered through a pad of silica gel using hexanes/EtOAc, 9:1 as eluent to afford the aldehyde (385 mg, 78% yield) in the form of a colourless oil. 1H-NMR (400 MHz, acetone-d6) δ 1.43 (s, (CH3)3CO), 3.55 (s, CH2COO), 3.75 (d, J = 2.1 Hz, CH2CHO), 7.21 (m, 2′-CH, 4′-CH, 6′-CH), 7.33 (t, J = 7.3 Hz, 5′-CH), 9.74 (t, J = 2.2 Hz, CHO). The aldehyde (372 mg, 1.59 mmol) was dissolved in t-BuOH (22 mL) and 2-methyl-2-butene (9 mL). An oxidative solution freshly prepared by dissolving NaClO2 (1.59 g) and NaH2PO4 (1.59 g) in H2O (15.9 mL) was added and the reaction mixture was stirred for 15 min at room temperature. The reaction was quenched by addition of water and the extraction was performed with EtOAc. The organic phase was washed with brine, dried over MgSO4 and evaporated to dryness under reduced pressure to afford carboxylic acid 156 (350 mg, 88% yield) in the form of a colourless oil. IR (film) 3700–2300 (OH, COOH), 1731 (C=O, ester), 1713 (C=O, acide); 1H-NMR (400 MHz, acetone-d6) δ 1.44 (s, (CH3)3CO), 3.54 (s, CH2COO), 3.63 (s, CH2COOH), 7.23 (m, 4 × CH of phenyl); 13C-NMR (100 MHz, acetone-d6) δ 27.8 (3×), 40.8, 42.5, 80.4, 128.1, 128.2, 128.8, 130.7, 135.5, 135.7, 170.7, 172.4; LRMS calculated for C14H17O4 [M-H]- 249.1, found 249.1 m/z.
4.2.17. Synthesis of resin 158 for preparation of library D
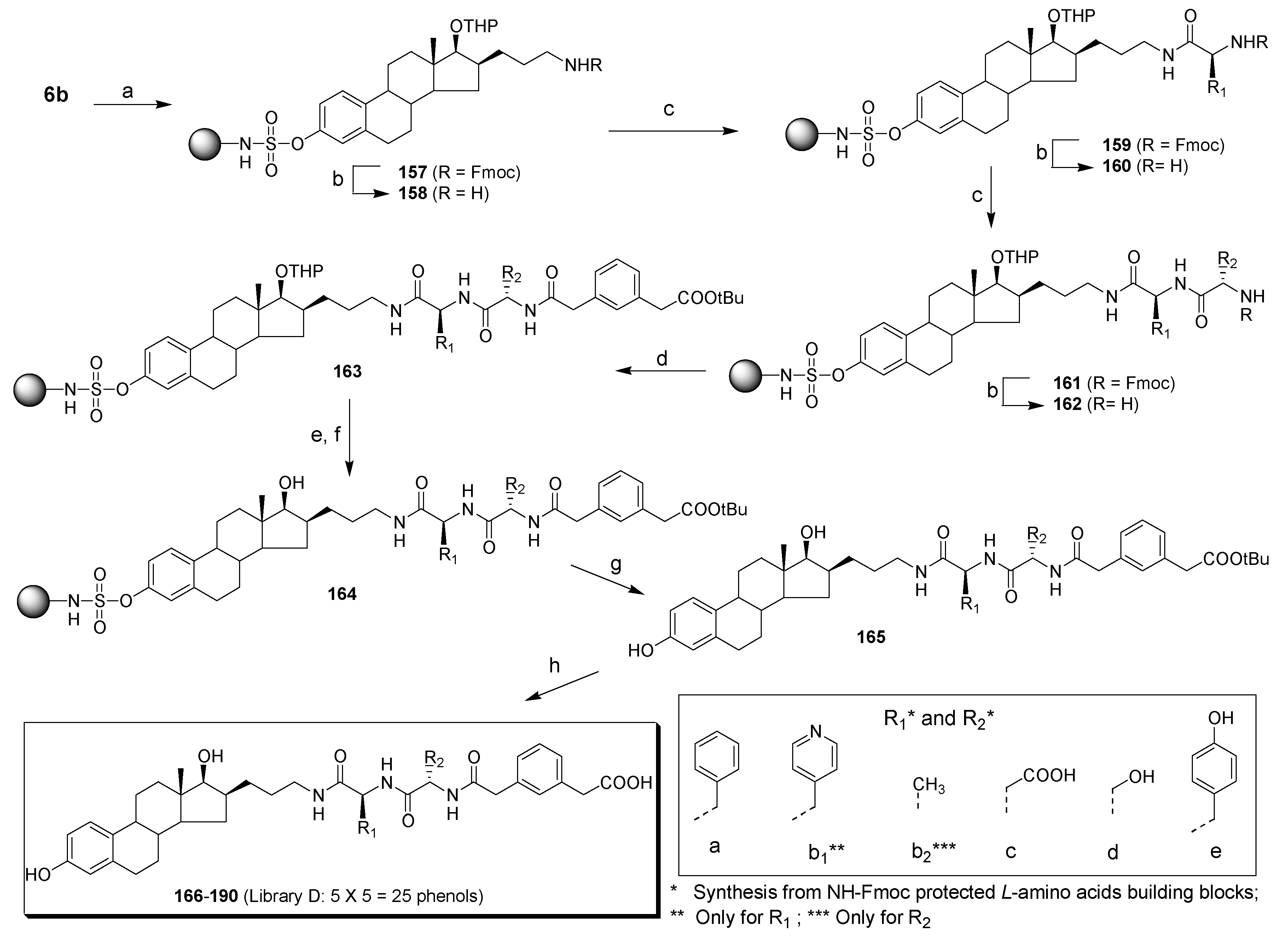
Two coupling reactions were run at the same time using a 50 mL (flask A) and a 25 mL (flask B) peptide flask. A solution of precursor 6b (2.50 g, 3.50 mmol) in dry DCM (30 mL) was prepared and 20.7 mL were added in flask A containing trityl chloride resin (3.0 g, 1.6 mmol/g theoretical loading) under an argon atmosphere. 9.3 mL of the solution of precursor 6b in DCM was added, under argon atmosphere to flask B containing trityl chloride resin (1.36 g, 1.6 mmol/g theoretical loading). After 5 min, DIPEA (7.6 mL in flask A and 3.4 mL in flask B) was added and the mixtures were shaken for 30 min at room temperature. Then, the argon inlets were removed and the mixtures were shaken for an additional 16 h. The resin was filtered and washed with DCM (3×) and MeOH (4x), and dried overnight under a vacuum to afford resin 157. The deprotection step was also run in 2 flasks, a 50 mL (flask A) and a 25 mL (flask B) peptide flask. Resin 157 was swollen in a solution of piperidine (20%) in DCM (25 mL in flask A and 10 mL in flask B) and shaken for 1 h at room temperature. The resin was then filtered and washed with DCM (3×) and MeOH (3×), and dried under a vacuum for 16 h to afford globally (flask A + flask B) 5.47 g or resin 158. Acidic mini cleavage (5% TFA/DCM, 10 min) of a sample of resin 158 and TLC analysis confirmed the complete deprotection of the amine. The loading yield, calculated by the mass increase, was 40%. It is noteworthy to mention that the loading yield could not be higher than 50% since 2 equivalents of trityl chloride resin were used for 1 equivalent of precursor 6b. Then 25 bottom fritted reaction vessels of a 40 solid-phase reaction block of ACT LabTech manual synthesizer were loaded with 200 mg of resin 158, which correspond to 0.102 mmol of compound 6b (without the Fmoc protective group).
4.2.18. Introduction of three levels of molecular diversity (library D)
The levels of molecular diversity were introduced by a similar procedure as described above in the preparation of libraries A, B and C. For the first level of molecular diversity, five stock solutions, each containing 2.5 equivalents (5x) of Fmoc-protected amino acid [Fmoc-L-Phe-OH (496 mg, 1.28 mmol), Fmoc-L-4-Pal-OH (Fmoc-L-4-pyridinealanine-OH) (497 mg, 1.28 mmol), Fmoc-L-Asp(OtBu)-OH (527 mg, 1.28 mmol), Fmoc-L-Ser(Trt)-OH (729 mg, 1.28 mmol) or Fmoc-L-Tyr(2-Cl-Trt)-OH (870 mg, 1.28 mmol)], 2.5 equivalents (5x) of PyBrOP (595 mg, 1.28 mmol) and 2.5 equivalents (5x) of HOBt (175 mg, 1.28 mmol), were prepared in dry DMF (10 mL), and 5 equivalents (5x) of DIPEA (450 µL, 2.56 mmol) were added to each stock solution. To each of the 25 resins 158, was added 2.0 mL of the appropriate stock solution. The mixtures were shaken under an argon atmosphere for 4 h at room temperature, then filtered, washed with DMF (2×), DCM (2×) and MeOH (3×), and dried under a vacuum to afford 5 groups of resins of general structure 159. The completion of the coupling step was verified by acidic mini-cleavage and TLC analysis as described above. The cleavage of the Fmoc group of resins 159 was subsequently performed by a 1 h treatment with 2.0 mL of a solution of piperidine (20%) in DCM. The resins were then filtered, washed with DCM (3×), MeOH (3×) and DCM (1×), and dried under a vacuum to give 5 groups of resins of general structure 160. The second level of molecular diversity was introduced using the same building blocks as for the first level except that Fmoc-L-4-Pal-OH was replaced by Fmoc-L-Ala-OH (398 mg, 1.28 mmol). The procedure described above for the introduction of the first level of molecular diversity was applied without modification. A TLC analysis after acidic mini-cleavage of a sampling of resins 161 confirmed that the introduction of the second level of molecular diversity was completed. Resins of general structure 161 were then treated with 2.0 mL of a solution of piperidine (20%) in DCM for 1 h at room temperature. The resins were filtered, washed with DCM (3×), MeOH (3×) and DCM (1×), and dried to afford resins of general structure 162. The third level was introduced on resins 162 by coupling carboxylic acid building block 156. A stock solution of 156 (1.60 g, 6.40 mmol), PyBrOP (2.98 g, 6.40 mmol) and HOBt (865 mg, 6.40 mmol) was prepared in dry DMF (50 mL) and DIPEA (2.23 mL, 12.8 mmol) was added. To each of the 25 resins 162 was added 2.0 mL of the stock solution. The mixtures were shaken under argon for 4 h at room temperature, then filtered, washed with MeOH (1×), DMF (2×), DCM (3×), MeOH (3×) and DCM (1×), and dried under a vacuum to afford resins of general structure 163. The completion of the coupling reaction was verified by acidic mini-cleavage and TLC analysis.
4.2.19. Removal of protective groups (library D)
Resins 163 that were prepared with Fmoc-L-Ser(Trt)-OH or Fmoc-L-Tyr(2-Cl-Trt)-OH building blocks were treated with 2.0 mL of a solution of TFA/TIS (triisopropylsilane)/DCM, 2:1:97 for 10 min at room temperature. The resins were filtered and washed with DCM (3×), MeOH (3×) and DCM (3×). This procedure was performed twice in order to completely remove the trityl or the 2-Cl-trityl protective groups. The deprotection step was completed when the solution of acidic mini-cleavage of a sampling of resins 163 (after step e) did not become yellow anymore. The THP protective group was next removed in mild acid condition. A solution of p-TSA (666 mg, 3.5 mmol) dissolved in t-BuOH (25 mL) and DCM (25 mL) was prepared and 2.0 mL of this solution was added to each of the 25 resins 163 (after step e, if necessary). The mixtures were shaken for 24 h at room temperature. They were then filtered, washed with DCM (3×), MeOH (3×) and DCM (1×), and dried under a vacuum to afford resins 164.
4.2.20. Generation of E2 derivatives 165 by nucleophilic cleavage
To each of the 25 resins 164 was added 2.0 mL of a solution of DEA (30%) in THF. The mixtures were shaken for 45 h at room temperature. Then 1.5 mL of a solution of DEA (30%) in THF was added to each of the 25 resins and the mixtures were shaken for an additional 20 h. The mixtures were then filtered and washed with THF (3×). The filtrates were collected in preweighed tubes and evaporated in a Speedvac apparatus. Each compound was dissolved in THF, evaporated twice and dried under a vacuum pump in order to obtain the DEA-free product. The phenol derivatives 165 were obtained in the form of a yellowish viscous oil. A random sampling was done and 2 compounds were analyzed by 1H NMR to verify their structure.
16β-(N-[3′-(Acetylbutyl ester)]-phenylacetyl-L-tyrosine-L-phenylalanine-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (165: R1 = a, R2 = e). 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 1.44 (s, (CH3)3CO), 2.78 (m, 6-CH2), 2.93 and 3.12 (2m, CHCH2Ph, CHCH2PhOH and CH2NH), 3.46 and 3.48 (2s, COCH2Ph), 3.51 (s, CH2COOtBu), 3.75 (under THF peaks, 17α-CH), 4.52 (m, CHCH2Ph and CHCH2PhOH), 6.49 (d, J = 2.4 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 6.65 (d, J = 8.5 Hz, 3′-CH and 5′-CH of 4-hydroxyphenyl), 6.94 (d, J = 8.5 Hz, 2′-CH and 6′-CH of 4-hydroxyphenyl), 7.01 (d, J = 7.5 Hz, 1-CH), 7.14 (m, 9 × CH of phenyl).
16β-(N-[3′-(Acetylbutyl ester)]-phenylacetyl-L-serine-L-aspartic acid-aminopropyl)-3,17β-dihydroxy-estra-1,3,5(10)-triene (165: R1 = c, R2 = d). 1H-NMR (400 MHz, methanol-d4) δ 0.80 (s, 18-CH3), 0.85 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 1.45 (s, (CH3)3CO), 2.78 (m, 6-CH2 and CH2COOH), 3.15 (m, CH2NH), 3.54 (s, COCH2Ph), 3.62 (s, CH2COOtBu), 3.75 (under THF peaks, 17α-CH), 3.75 and 3.86 (under THF peaks and dd, J1 = 10.6 Hz, J2 = 5.5 Hz, CH2OH), 4.22 (t, J = 6.0 Hz, CHCH2OH), 4.75 (m, CHCH2COOH), 6.49 (d, J = 2.3 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 2-CH), 7.09 (d, J = 8.4 Hz, 1-CH), 7.24 (m, 4 × CH of phenyl).
4.2.21. Generation of E2 derivatives (library D) by an acid treatment
In order to remove the
tert-butyl ester protective group, each compound
165 was put in a tube and treated for 3 h at room temperature with 2.0 mL of HCl (4M in dioxane). The reaction mixtures were then evaporated to dryness under reduced pressure and dried overnight under a vacuum pump. The crude products were dissolved in MeOH and preadsorbed on C-18 silica gel. Purification by a C-18 silica gel column (Honeywell Burdick & Jackson, solid phase systems C18 columns, size 2000 mg/8 mL (product # 9009) distributed by VWR) afforded carboxylic acid, methyl ester or a mixture of carboxylic acid and methyl ester derivatives
166–190 (2–31 mg, 2–33% overall yield from
158). LRMS and notes on
1H-NMR analysis of each library members are presented in
Table 4.
1H-NMR of a random sampling of four members is described as examples.
166 (methyl ester). 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.95 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.79 (m, 6-CH2), 2.90 and 3.10 (2m, 2 × CHCH2Ph and CH2NH), 3.45 and 3.46 (2s, COCH2Ph), 3.61 (s, CH2COOCH3), 3.67 (s, COOCH3), 3.71 (d, J = 9.8 Hz, 17α-CH), 4.53 (t, J = 7.3 Hz, COCHNH), 4.60 (m, COCHNH), 6.48 (d, J = 2.5 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.5 Hz, 2-CH), 7.01 (d, J = 7.5 Hz, 1-CH), 7.05 to 7.30 (m, 14 × CH of phenyl).
180 (methyl ester). 1H-NMR (400 MHz, methanol-d4) δ 0.80 (s, 18-CH3), 1.00 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.75 (m, 6-CH2), 2.80, 3.00 and 3.14 (3m, CHCH2COOH, CH2PhOH) and CH2NH), 3.54 (m, COCH2Ph), 3.63 and 3.66 (2s, CH2COOCH3), 3.68 (s, COOCH3), 3.71 (d, J = 9.7 Hz, 17α-CH), 4.48 (m, CHCH2PhOH), 4.64 (m, CHCH2COOH), 6.49 (d, J = 2.5 Hz, 4-CH), 6.55 (dd, J1 = 8.3 Hz, J2 = 2.6 Hz, 2-CH), 6.68 (m, 2 × CH of PhOH), 7.00 to 7.25 (m, 1-CH, 2 × CH of PhOH and 4 × CH of Ph).
184 (carboxylic acid). 1H-NMR (400 MHz, methanol-d4) δ 0.79 (s, 18-CH3), 0.95 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 3.20 (m, CH2NH), 3.60 and 3.63 (2s, COCH2Ph), 3.65 and 3.69 (2s, CH2COOH), 3.71 (d, J = 9.9 Hz, 17α-CH), 3.79 and 3.89 (2m, 2 × CH2OH), 4.39 and 4.45 (2m, 2 × CHCH2OH), 6.49 (d, J = 2.4 Hz, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.5 Hz, 2-CH), 7.09 (d, J = 8.4 Hz, 1-CH), 7.24 (m, 4 × CH of Ph).
185 (mix of methyl ester and carboxylic acid). 1H-NMR (400 MHz, methanol-d4) δ 0.80 (s, 18-CH3), 0.90 to 2.35 (m, CH and CH2 of steroid skeleton and alkyl chain), 2.78 (m, 6-CH2), 2.90 and 3.05 (2m, CH2PhOH), 3.17 (m, CH2NH), 3.52 and 3.53 (2s, COCH2Ph), 3.58, 3.63 and 3.65 (3s, CH2COOX, × = H or CH3), 3.68 (s, COOCH3), 3.72 and 3.79 (2m, 17α-CH and CH2OH), 4.33 (m, CHCH2PhOH), 4.60 (m, CHCH2OH), 6.49 (s, 4-CH), 6.55 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 2-CH), 6.68 (dd, J1 = 8.4 Hz, J2 = 1.8 Hz, 2 × CH of PhOH), 7.00 to 7.35 (m, 1-CH, 2 × CH of PhOH and 4 × CH of Ph).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




