MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation

Abstract
:
1. Introduction
2. Computational Methods
2.1. Molecular dynamics simulation on a single monomer
2.2. Replica exchange molecular dynamics (REMD) simulations of the dimer at 280–405 K
2.3. Characterization of the dimer conformations and selection of the representative MD simulation
2.4. MMPBSA decomposition of the binding free energy, ΔGbind, and binding energy, ΔEbind
2.5. Software orchestration: Kepler scientific workflow
3. Results and Discussion
3.1. Decomposition of the binding free energy, ΔGbind

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation (1) | ΔGbind | <GC> | <GB> | <GA> |
|---|---|---|---|---|
| -15.9 (10.9) | -904.7 (36.2) | -443.0 (22.2) | -445.8 (19.5) | |
| Equation (4) | ΔGbind | <ΔEMM> | <ΔGsol> | |
| -15.9 (10.9) | -277.8 (55.7) | 261.9 (51.5) |
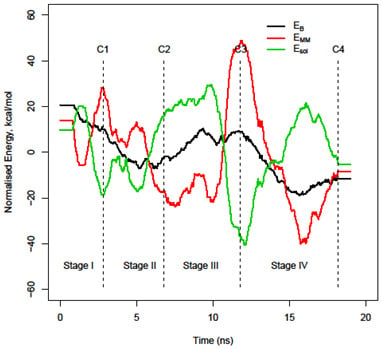
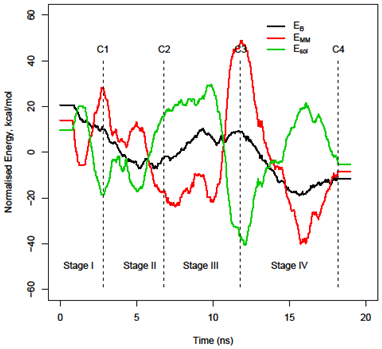
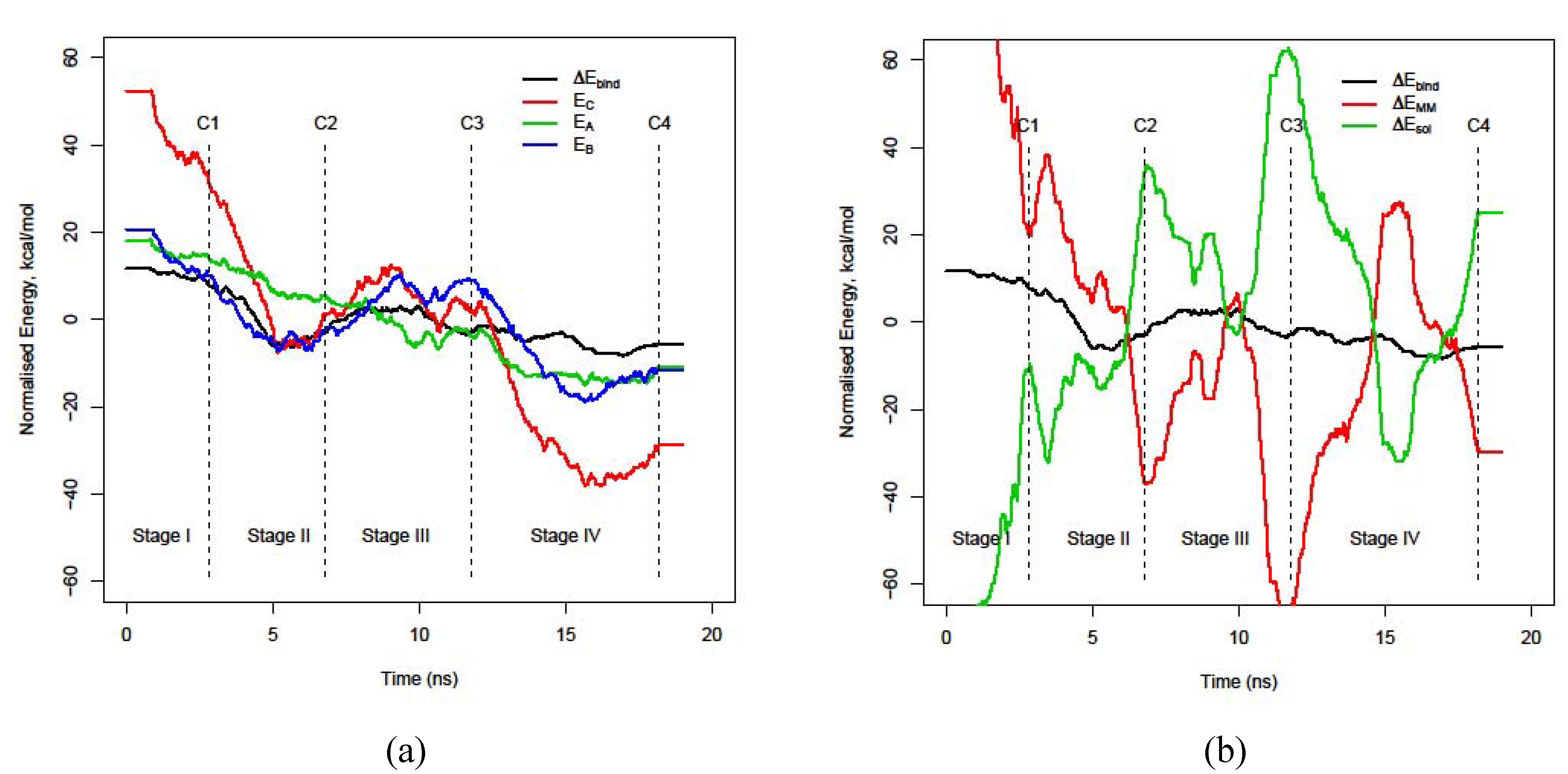
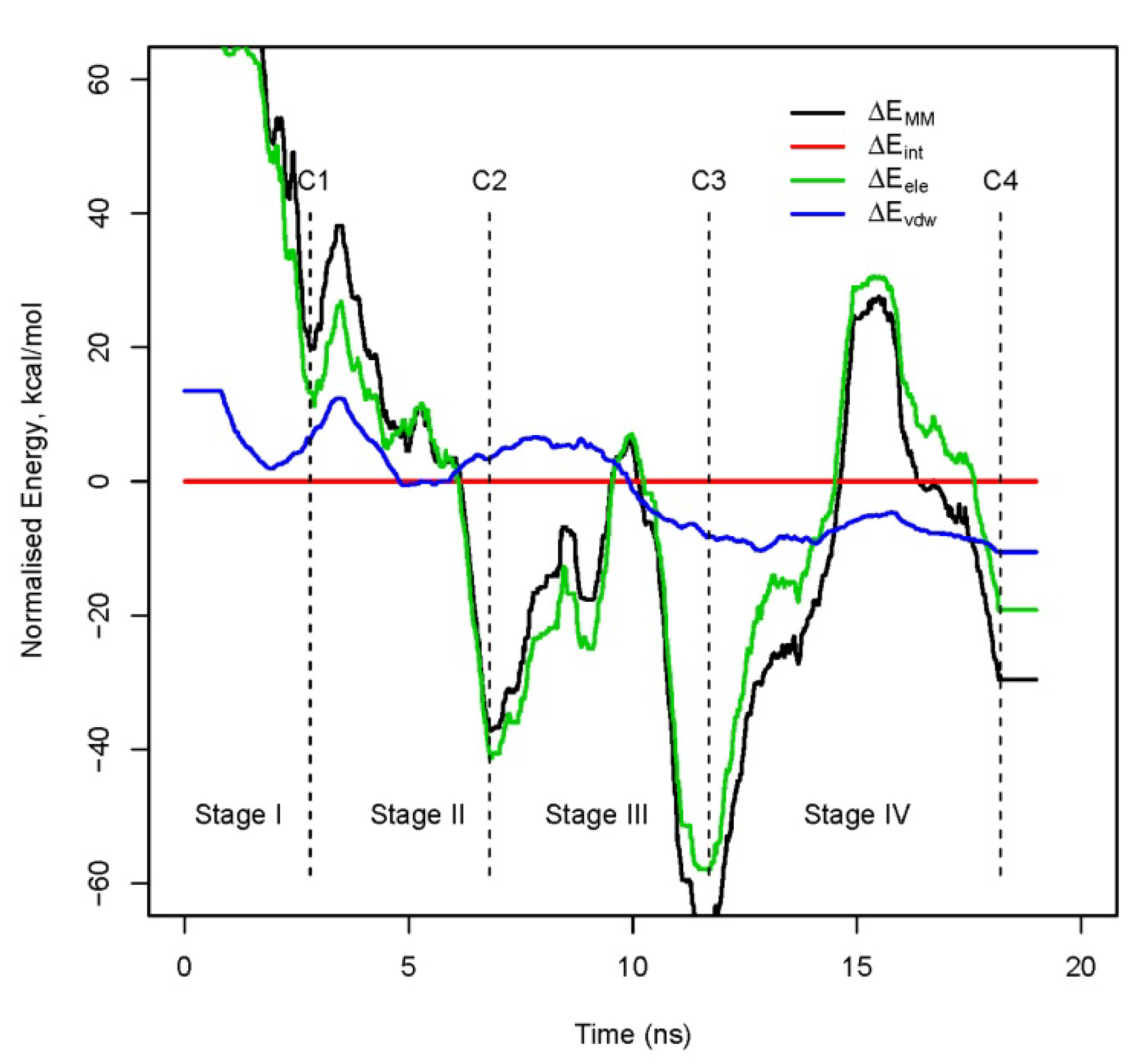
3.2. Decomposition of the binding energy, ΔEbind, over the course of the trajectory



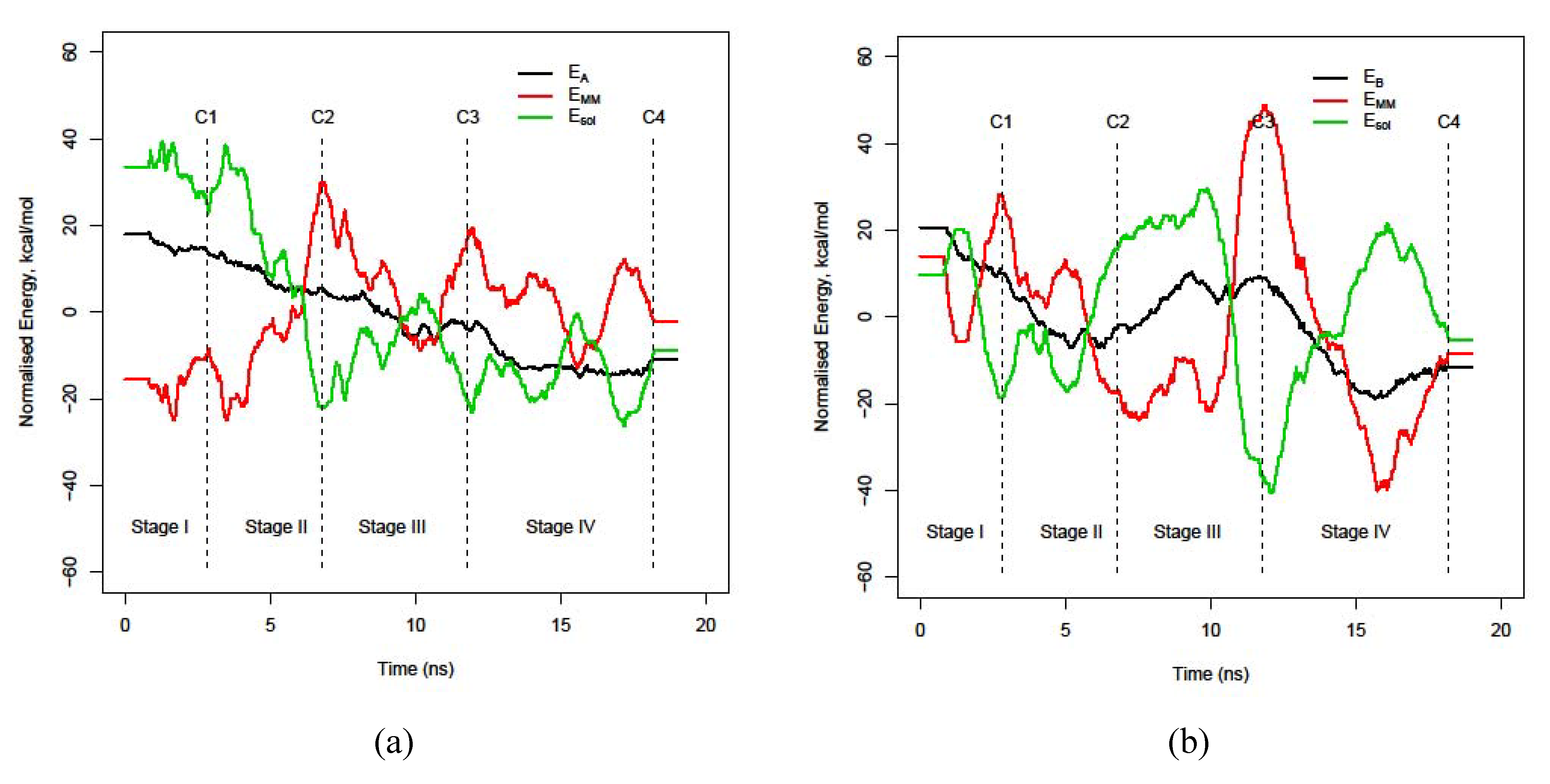
3.3. Energy stability of each monomer, EA and EB

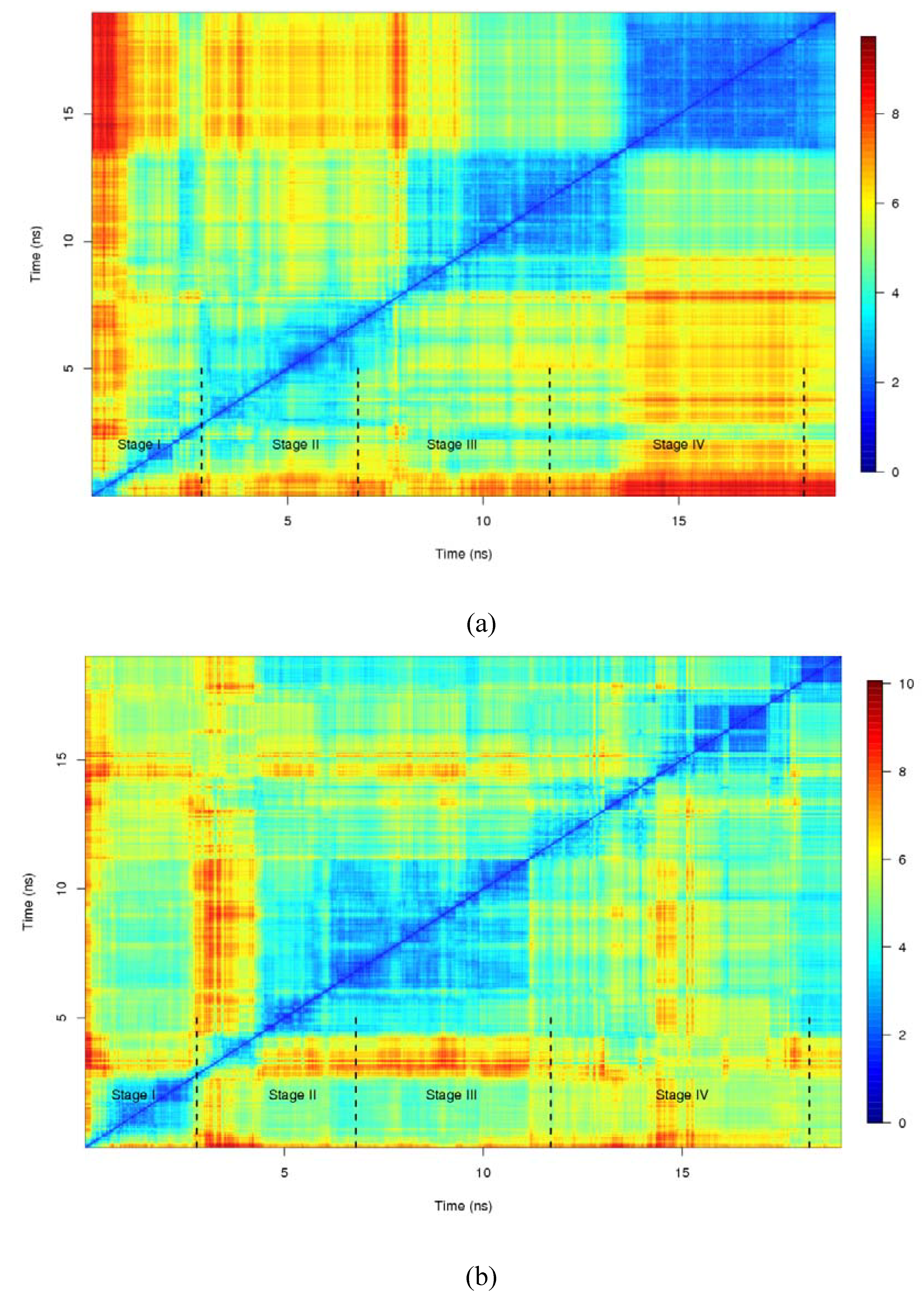
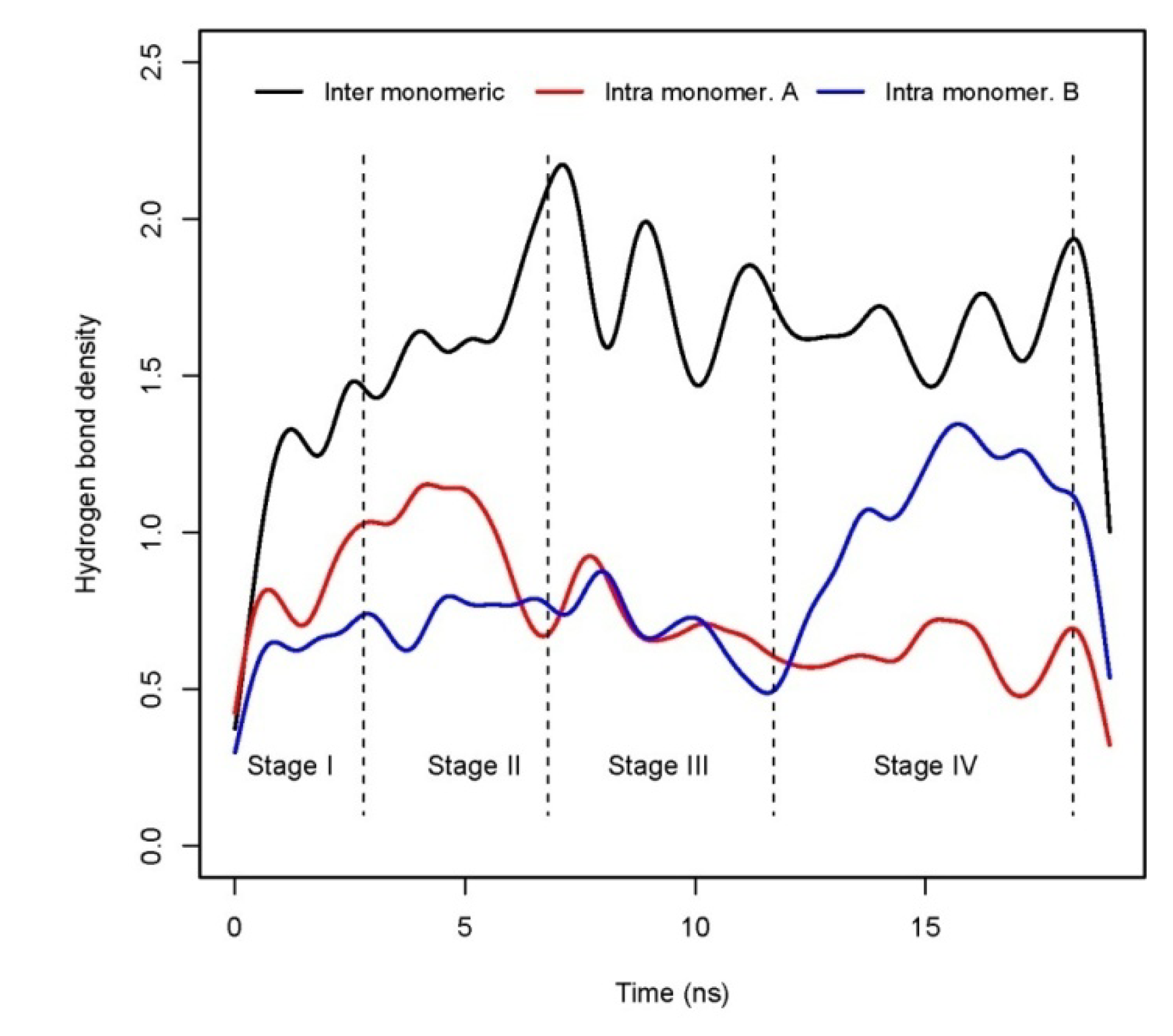
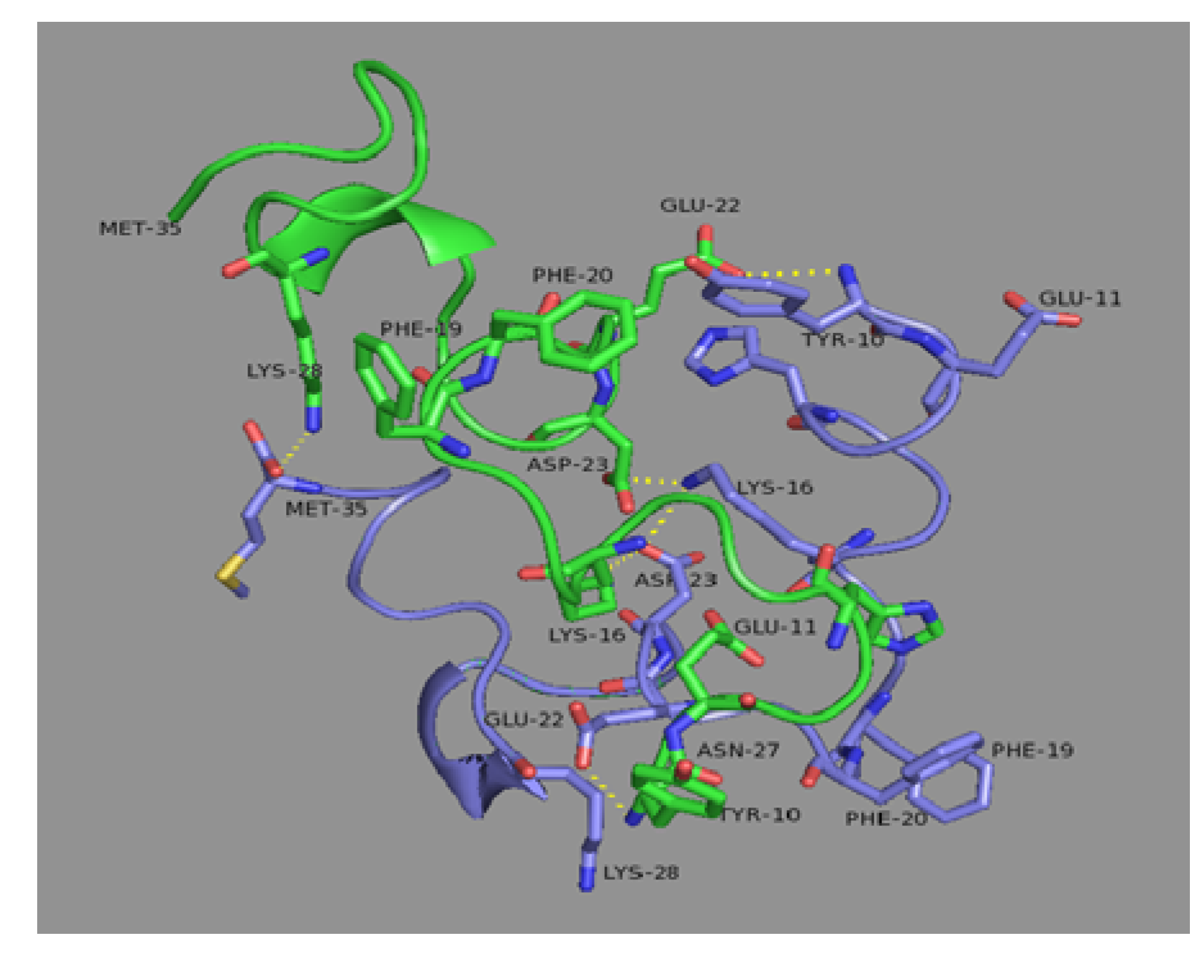
3.4. Root mean square deviation (RMSD) and hydrogen bond analysis


| No. | residue-residue | Type of hydrogen bond | Occupation, % | Comments |
|---|---|---|---|---|
| 1 | Glu22(A)-N/terminus(B) | Inter monomeric | 34.4 | |
| 2 | Lys16(B)-Asp23(B) | Intra monomer B | 23.5 | Equivalent to Lys16(A)-Asp23(A) |
| 3 | Asp23(A)-Lys16(B) | Inter monomeric | 23.4 | |
| 4 | Lys16(A)-Asp23(A) | Intra monomer A | 22.1 | Equivalent to Lys16(B)-Asp23(B) |
| 5 | Lys16(A)-Asp23(B) | Inter monomeric | 17.0 |
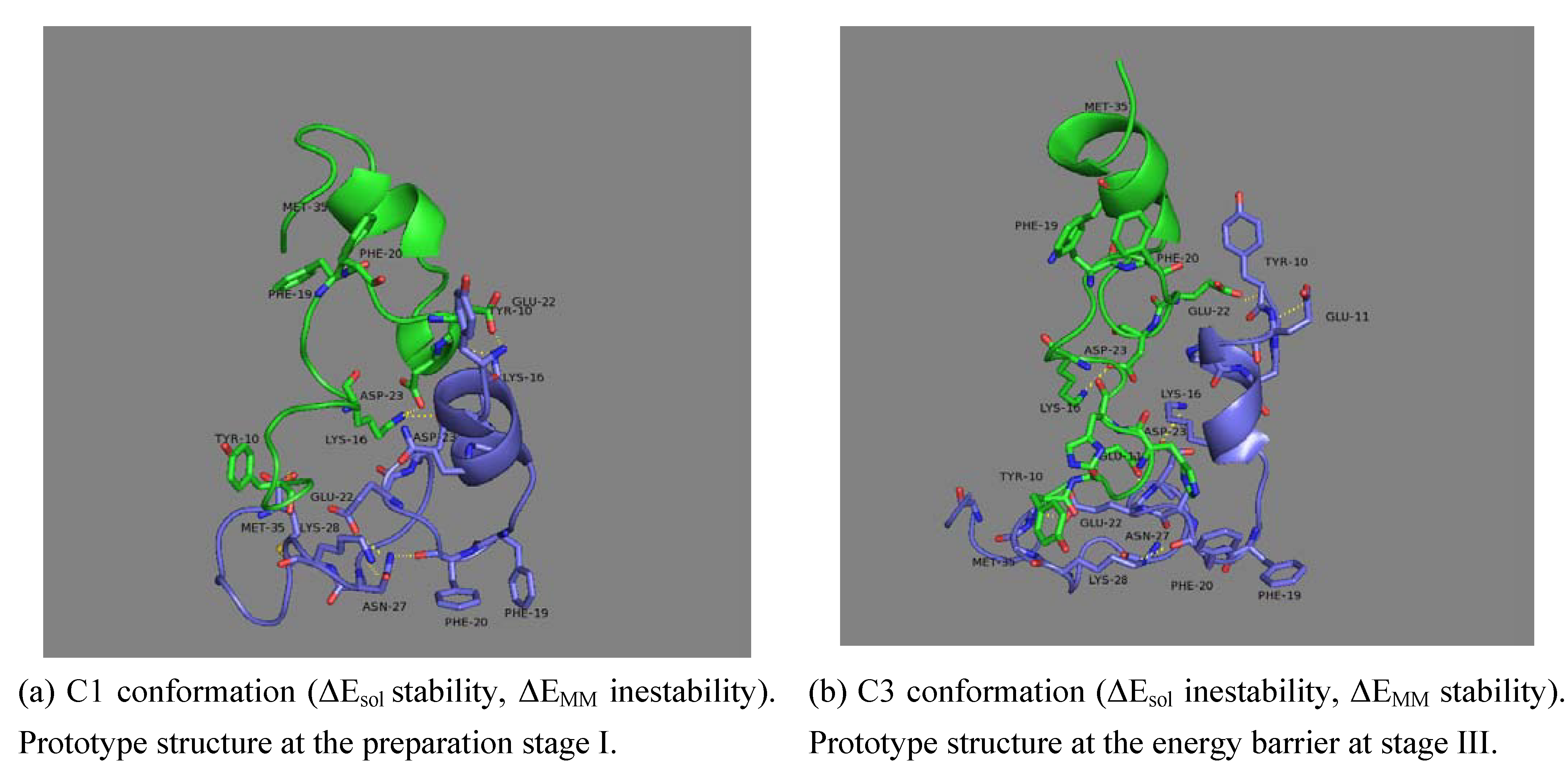
3.5. Geometrical description of the final dimer conformation

4. Conclusions
References
- Gasparini, L.; Rusconi, L.; Huaxi, X.; Del Soldato, P.; Ongini, E. Modulation β-Amyloid metabolism by non-steroidal antiinflammatory drugs in neuronal cell cultures. J. Neurochem. 2004, 88, 337–348. [Google Scholar]
- Ho, L.; Luterman, J.D.; Haroutuman, V.; Purohit, D.; Aisen, P.S.; Willis, F.; Naslund, J.; Buxbaum, J.D.; R., Pasineti, G.M. Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progresion of Alzheimer´s disease dementia. Arch. Neurol. 2001, 58, 487–492. [Google Scholar]
- McGeer, P.L. Cyclooxygenase-2 inhibitors rationales and therapeutic potential for Alzheimer’s disease. Drugs Aging 2000, 17, 1–11. [Google Scholar]
- Austen, B.M.; Paleologou, K.E.; Ali, S.A.E.A.; Qureshi, M.M.; Allsop, D.; El-Agnaf, O.M.A. Designing Peptide Inhibitors for Oligomerization and Toxicity of Alzheimer’s β-Amyloid Peptide. Biochem. 2008, 47, 1984–1992. [Google Scholar]
- Pouplana, R.; Lozano, J.J.; Pérez, C.; Ruiz, J. Structure-based QSAR study on differential inhibition of human Prostaglandin Endoperoxide H Synthase-2 (COX-2) by nonsteroidal antiinflammatory drugs. J. Comput.-Aided Mol. Des. 2002, 16, 683–710. [Google Scholar]
- Ruiz, J.; Pérez, C.; Pouplana, R. QSAR Study of dual Cyclooxygenase and 5-Lipoxygenase inhibitors 2,6 diterbutylphenol derivatives. Bioorg. Med. Chem. 2003, 11, 4207–4216. [Google Scholar]
- Pasinetti, G.M. Cyclooxygenase and Alzheimer´s disease: implications for preventive initiatives to slow the progression of clinical dementia. Arch. Geront. Geriat. 2001, 33, 13–28. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M; Milton, S.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Yong, W.; Lomakin, A.; Kirkitadze, M.D.; Teplow, D.B.; Chen, S.H.; Benedek, G.B. Structure determination of micelle-like intermedaites in amyloid beta-protein fibril assembly by using small angle neutron scattering. Proc. Natl. Acad. Sci. USA 2002, 99, 150–154. [Google Scholar]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid fibril formation by A-β(16−22), a seven-residue fragment of the Alzheimer's beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar]
- Zhang, S.; Iwata, K.; Lachenmann, M.J.; Peng, J.W.; Li, S.; Simson, E.R.; Lu, Y.; Felix, A.M.; Maggio, J.E.; Lee, J.P. The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 2000, 130, 130–141. [Google Scholar]
- Ma, B.Y.; Nussinov, R. Stabilities and conformations of Alzheimer's β-amyloid peptide oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): Sequence effects. Proc. Natl. Acad. Sci. USA 2002, 99, 14126–14131. [Google Scholar]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Terenius, L.; and Nordstedt, C. Arrest of β-Amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar]
- Fraser, P.E.; Nguyen, J.T.; Surewiez, W.K.; Kirschner, D.A. Ph dependent structural transitions of Alzheimer's amyloid peptides. Biophys. J. 1991, 60, 1190–1201. [Google Scholar]
- Kim, W.; Hecht, M.H. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer’s Ab42 peptide. Proc Natl. Acad. Sci. USA 2006, 103, 15824–15829. [Google Scholar]
- Hwang, W.; Zhang, S.; Kamm, R.D.; Karplus, M. Kinetic control of dimer structure formation in amyloid fibrillogenesis. Proc Natl. Acad. Sci. USA 2004, 101, 12916–12921. [Google Scholar]
- Reddy, G.; Straub, J.E.; Thirumalai, D. Influence of Preformed Asp23-Lys28 salt bridge on the conformational fluctuations of Monomers and Dimers of Aβ peptides with implications for Rates of Fibril Formation. J. Phys. Chem. 2009, 113, 1162–1172. [Google Scholar]
- Habicht, G.; Haupt, C.; Friedrich, R.P.; Hortschansky, P.; Sachse, C.; Meinhardt, J.; Wieligmann, K.; Gellermann, G.P.; Brodhun, M.; Gotz, J.; Halbhuber, K.J.; Röcken, C.; Horn, U.; Fändrich, M. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation y stabillizing Aβ protofibrills. Proc. Nat. Acad. Sci. USA 2007, 104, 19232–19237. [Google Scholar]
- Sato, T.; Kienlen-Campard, P.; Ahmed, M.; Liu, W.; Li, H.; Elliott, J.I.; Aimoto, S.; Constantinescu, S.N.; Octave, J.N.; Smith, S.O. Inhibitors of amyloid toxicity based on beta-sheet packing of Aβ40 and Aβ42. Biochemistry 2006, 45, 5503–5516. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W.; Merz, K.M.; Wang, B.; Hayik, S.; Roitberg, A.; Seabra, G.; Kolossváry, I.; Wong, K.F.; Paesani, F.; Vanicek, J.; Wu, X.; Brozell, S.R.; Steinbrecher, T.; Gohlke, H.; Yang, L.; Tan, C.; Mongan, J.; Hornak, V.; Cui, G.; Mathews, D.H.; Seetin, M.G.; Sagui, C.; Babin, V. and Kollman P.A. AMBER 10; University of California at San Francisco: San Francisco, CA, USA, 2008. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: an N logN method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Jang, S.; Shin, S. Amyloid β-peptide oligomerizationin silico: dimer and trimer. J. Phys. Chem. B 2006, 110, 1955–1958. [Google Scholar]
- Urbanc, B.; Cruz, L.; Ding, F.; Sammond, D.; Khare, S.; Buldyrev, S.V.; Stanley, H.E.; Dokholyan, N.V. Molecular Dynamics Simulation of β-Amyloid Dimer Formation. Biophys. J. 2004, 87, 2310–2321. [Google Scholar]
- Tarus, B.; Straub, J.; Thirumalai, D. Probing the initial stage of aggregation of the Aβ10−35 protein: Assesssing the propensity for peptide dimerization. J. Mol. Biol. 2005, 345, 1141–1158. [Google Scholar]
- Jang, S.; Shin, S.J. Computational Study on the Structural Diversity of Amyloid Beta Peptide (Aβ10−35) Oligomers. Phys. Chem. B 2008, 112, 3479–3483. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; Donini, O.; Cieplak, P.; Srinivasan, J.; Case, D.A.; Cheatham III, T.E. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E., III; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar]
- Gohlke, H.; Case, D.A. Converging Free Energy Estimates: MM-PB(GB)SA Studies on the Protein-Protein Complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar]
- Kuhn, B.; Kollman, P.A. Binding of a Diverse Set of Ligands to Avidin and Streptavidin: An Accurate Quantitative Prediction of Their Relative Affinities by a Combination of Molecular Mechanics and Continuum Solvent Models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M.; Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA, Find Similar. J. Comput. Chem. 2001, 31, 797–810. [Google Scholar]
- Sitkoff, D.; Sharp, K.; Honing, B. Accurate calculations of hydration free energies using macroscopic solvents. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar]
- Schlitter, J. Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem. Phys. Lett. 1993, 215, 617–621. [Google Scholar]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar]
- Liang, S.; Li, L.; Hsu, W.L.; Pilcher, M.N.; Uversky, V.; Zhou, Y.; Dunker, A.K.; Meroueh, S.O. Exploring the molecular design of protein interaction sites with molecular dynamics simulations and free energy calculations. Biochemistry 2009, 48, 399–414. [Google Scholar]
- R-Development-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2007. Available online: http://www.R-project.org, accessed on 13 April 2010.
- Deelman, E.; Gannon, D., Shields. Workflows and e-Science: An overview of workflow system features and capabilities. Future Gener. Comp. Sys. 2009, 25, 528–540. [Google Scholar]
- Altintas, I.; Berkley, C.; Jaeger, E.; Jones, M.; Ludäscher, B.; Mock, S. Kepler: an extensible system for design and execution of scientific workflows. In 16th International Conference on Scientific and Statistical Database Management, Santorini Island, Greece, 2004. IEEE publication number P2146.
- Borreguero, J.M.; Urbanc, B.; Lazo, N.D.; Bitan, G.; Buldyrev, S.V.; Teplow, D.B.; Stanley, H.E. Folding events in the 21−30 region of amyloid β-protein (Aβ): studied in silico. Proc. Natl. Acad. Sci. USA 2005, 102, 6015–6020. [Google Scholar]
- Massi, F.; Peng, J.; Lee, J.P.; Straub, J. Simulation study of the structure and dynamics of the Alzheimer’s amyloid peptide congener in solution. Biophys. J. 2001, 80, 31–44. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Campanera, J.M.; Pouplana, R. MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation. Molecules 2010, 15, 2730-2748. https://doi.org/10.3390/molecules15042730
Campanera JM, Pouplana R. MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation. Molecules. 2010; 15(4):2730-2748. https://doi.org/10.3390/molecules15042730
Chicago/Turabian StyleCampanera, Josep M., and Ramon Pouplana. 2010. "MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation" Molecules 15, no. 4: 2730-2748. https://doi.org/10.3390/molecules15042730
APA StyleCampanera, J. M., & Pouplana, R. (2010). MMPBSA Decomposition of the Binding Energy throughout a Molecular Dynamics Simulation of Amyloid-Beta (Aß10−35) Aggregation. Molecules, 15(4), 2730-2748. https://doi.org/10.3390/molecules15042730