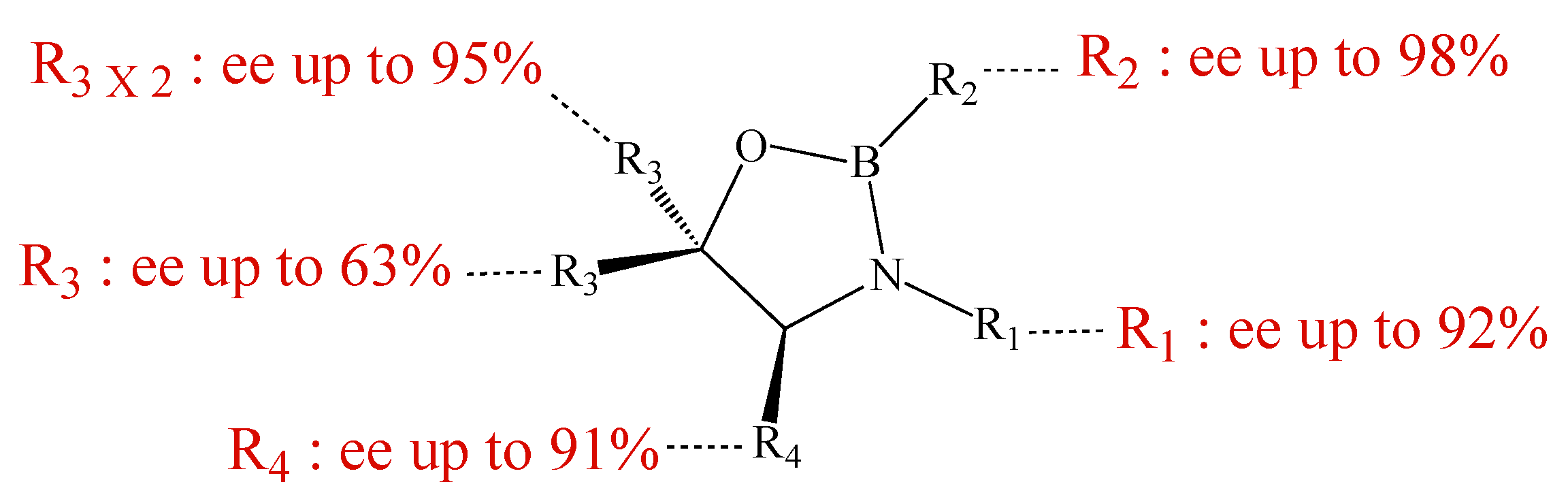
3.2. Preparation of the homogeneous catalysts
(4S,5R)-4,5-Diphenyl-1,3,2-oxazaborolidine (
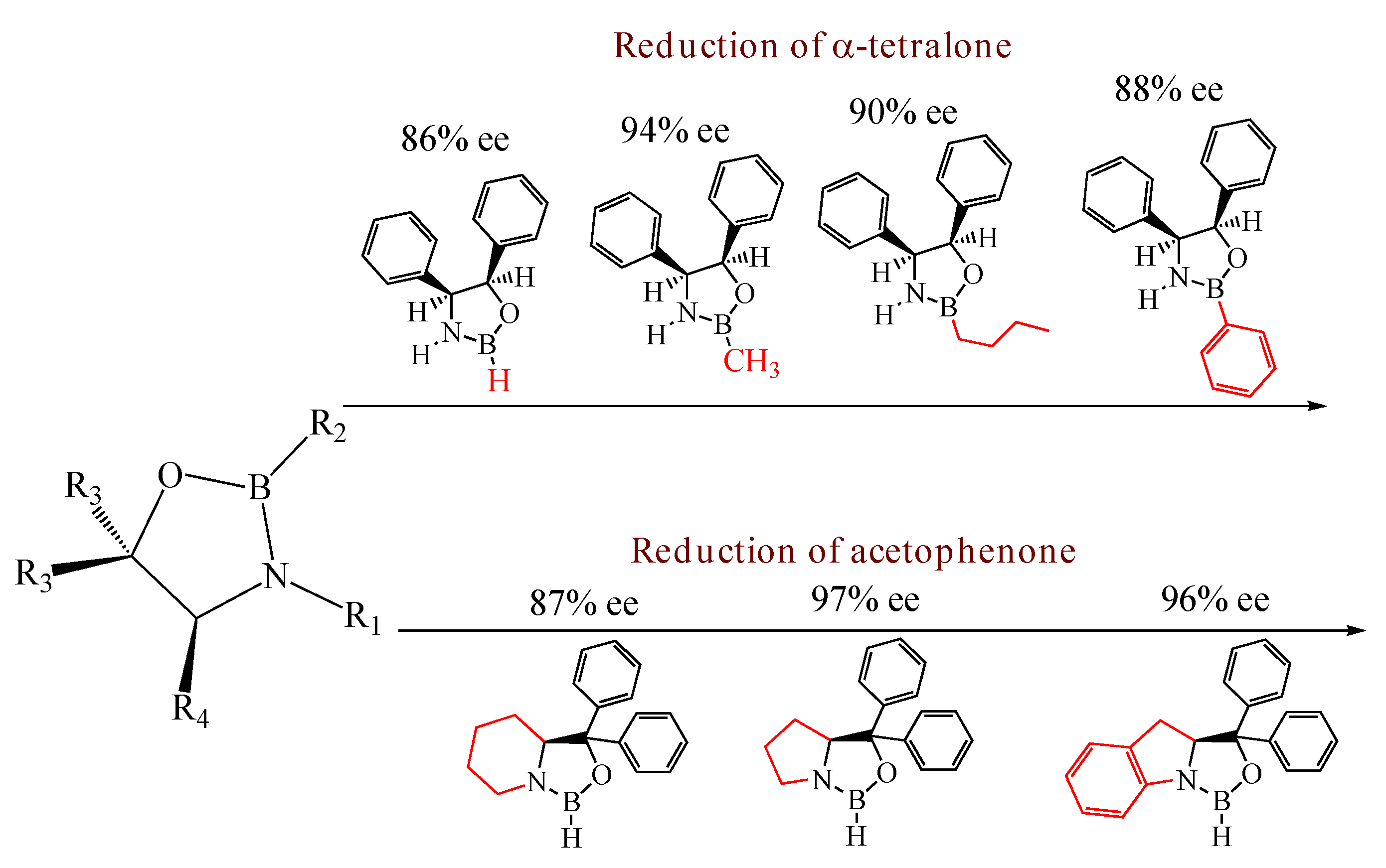
Table 3, entry 1). (1
S,2
R)-(+)-2-Amino-1,2-diphenyl-ethanol (1 g, 4.69 mmol) is dissolved in anhydrous THF (10 mL), then a solution of borane in THF (9.38 mL, 1 M, 9.38 mmol) is added at -78 ºC for 30 min. The resulting solution is gradually warmed to 30 ºC and stirred at 30 ºC during 10 h. The excess of borane is eliminated under vacuum. IR (KBr), 3,070, 3,028, 2,931, 2,867, 1,599, 1,452 cm
-1,
1H-NMR (250 MHz, CDCl
3) δ ppm, 7.40–7.29 (m, 10 H, Ar-H), 5.13–5.11 (d, 1 H, C-H), 4.39–4.37 (d, 1H, C-H), 2.18–2.12 (s, 1 H, N-H), yield 100%. See procedure described by Itsuno
et al. [
1,
8].
(4S,5R)-2,4,5-Triphenyl-1,3,2-oxazaborolidine (
Table 3, entry 2). (1
S,2
R)-(+)-2-Amino-1,2-diphenyl-ethanol (1 g, 4.69 mmol) and phenylboronic acid (4.69 mmol) are combined with anhydrous toluene (40 mL) and water is removed using a Dean-Stark apparatus for 24 h. The solvent is removed under vacuum to provide the oxazaborolidine as a colorless oil. IR (KBr), 3,070, 3,028, 2,931, 2,867, 1,599, 1,452 cm
-1,
1H-NMR(250 MHz, CDCl
3) δ ppm, 7.99–7.96 (d, 1 H, Ar-H), 7.40–7.30 (m, 3 H, Ar-H), 7.20–7.10 (m, 9 H, Ar-H), 4.79–4.76 (d, 7Hz, 1 H, C-H), 4.12–4.10 (d, 7Hz, 1 H, C-H), 2.18–2.12 (s, 1 H, N-H), yield 100%. See procedure described by Quallich
et al. [
3].
(4S,5R)-2-(4-Butylphenyl)-4,5-diphenyl-1,3,2-oxazaborolidine (
Table 3, entry 3). See procedure described for (4S,5R)-2,4,5-triphenyl-1,3,2-oxazaborolidine. IR (KBr), 3,070, 3,028, 2,931, 2,867, 1,599, 1,452 cm
-1,
1H-NMR (250 MHz, CDCl
3) δ ppm, 7.96–7.95 (d, 1 H, Ar-H), 7.60–7.50 (d, 1 H, Ar-H), 7.30–7.10 (m, 11 H, Ar-H), 7.10–6.90 (d, 1 H, Ar-H), 4.77–4.74 (d, 7Hz, 1 H, C-H), 4.13–4.11 (d, 7Hz, 1 H, C-H), 2.61–2.55 (t, 7Hz, 2 H, C-H), 2.10–2.08 (s, 1 H, N-H), 1.60–1.50 (m, 7Hz, 2 H, C-H), 1.35–1.25 (m, 7.5 Hz, 2 H, C-H), 0.89–0.84 (t, 8Hz, 3 H, C-H), 2.18–2.12 (s, 1 H, N-H).
(4S,5R)-4,5-Diphenyl-2-(4-(2-(triethoxysilyl)ethyl)phenyl)-1,3,2-oxazaborolidine (
Table 3, entry 4). 4-vinylphenylboronic acid (1 g, 6.75 mmol) is dissolved in anhydrous ethanol (20 mL) then triethoxysilane (1.66 g, 10.1 mmol) and Karsted’s catalyst (0.1 equiv. with respect to the complex) is added. The solution color changed to yellow and it was heated at 50 ºC for 12 h, at which point the solution becomes brown. The oxazaborolidine were prepared using the same procedure as described for (4
S,5
R)-2,4,5-triphenyl-1,3,2-oxazaborolidine. IR (KBr), 3,070, 3,028, 2,931, 2,867, 1,599, 1,452 cm
-1,
1H-NMR (250 MHz, CDCl
3) δ ppm, 7.99–7.96 (d, 2 H, Ar-H), 7.74-7.55 (m, 3 H, Ar-H), 7.20–7.10 (m, 9 H, Ar-H), 5.23–5.20 (d, 7 Hz, 1H, C-H), 4.79–4.70 (d, 7 Hz, 1 H, C-H), 3.85–3.70 (q, 8 Hz, 6 H, C-H), 2.63–2.57 (t, 7 Hz, 2 H, C-H), 1.99–1.98 (s, 1 H, N-H), 1.20–1.15 (t, 8 Hz, 9 H, C-H), 0.92–0.88 (t, 7 Hz, 2H, C-H).
(4S,5R)-4,5-Diphenyl-3-propyl-1,3,2-oxazaborolidine (
Table 4, entry 2). 1-bromopropane (0.123 mL, 1.35 mmol), (1
R,2
S)-2-amino-1,2-diphenylethanol (0.29 g, 1.35 mmol) and triethylamine (0.63 mL, 4.5 mmol) are mixed and heated at reflux during for 18 h. After cooling at 28 ºC, distilled water is added until neutral pH, then the organic phase is recovered with ethyl acetate without further purification. The product is then analyzed by GC/MS.
1H-NMR (250 MHz, CDCl
3) δ ppm, 7.40–7.29 (m, 10 H), 5.12 (d, 8 Hz, 1 H), 4.37 (d, 8 Hz, 1 H), 3.6 (s, 1 H), 2.55 (t, 7 Hz, 2 H), 1.45 (q, 7 Hz, 2 H), 0.9 (t, 7 Hz, 3 H).
3.3. Preparation of the heterogeneous catalyst
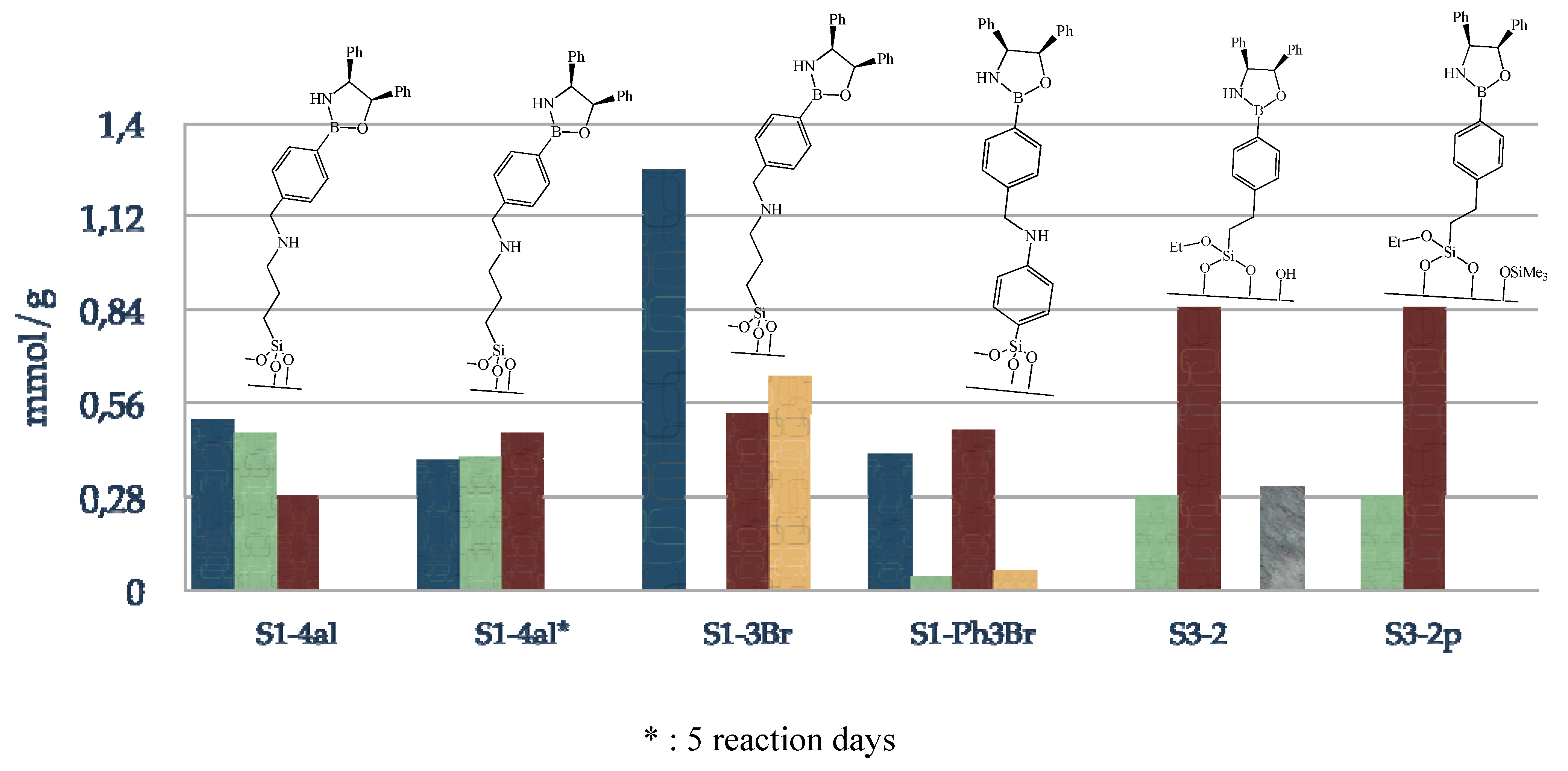
3.3.1. Functionalization of the Grace silica (S1-1)
Grace Davison silica (1 g) was activated at 150 ºC under vacuum for 3 h. After cooling the silica under argon, anhydrous toluene (40 mL) and 3-aminopropyltriethoxysilane (0.89 g, 4 mmol) is added and the mixture heated at 130 ºC for 12 h; the ethanol formed during the reaction is removed by the Dean-Stark method. The solid is filtered with toluene (3 × 20 mL), methanol (3 × 20 mL), dichloromethane (3 × 20 mL), diethyl ether (3 × 20 mL), then washed in a Soxhlet apparatus for 24 h (dichloromethane/diethylether = 1/1). Finally the solid is dried at 70 ºC for 3 h.
3.3.2. Immobilization of the boronic acid (S1-2Br)
S1-1 (1 g) was activated at 100 ºC under vacuum for 3 h. After cooling the silica under argon, boronic acid (3 mmol) dissolved in anhydrous toluene (40 mL) and triethylamine (1.01 g, 10 mmol) are added and the mixture then heated at 120 ºC for 36 h. The solid is filtered with toluene (3 × 20 mL), dichloromethane (3 × 20 mL), diethyl ether (3 × 20 mL), then washed with a Soxhlet apparatus for 24 h (dichloromethane/diethyl ether = 1/1). Finally the solid is dried at 70 ºC for 3 h (S1-2Cl or S1-2Br).
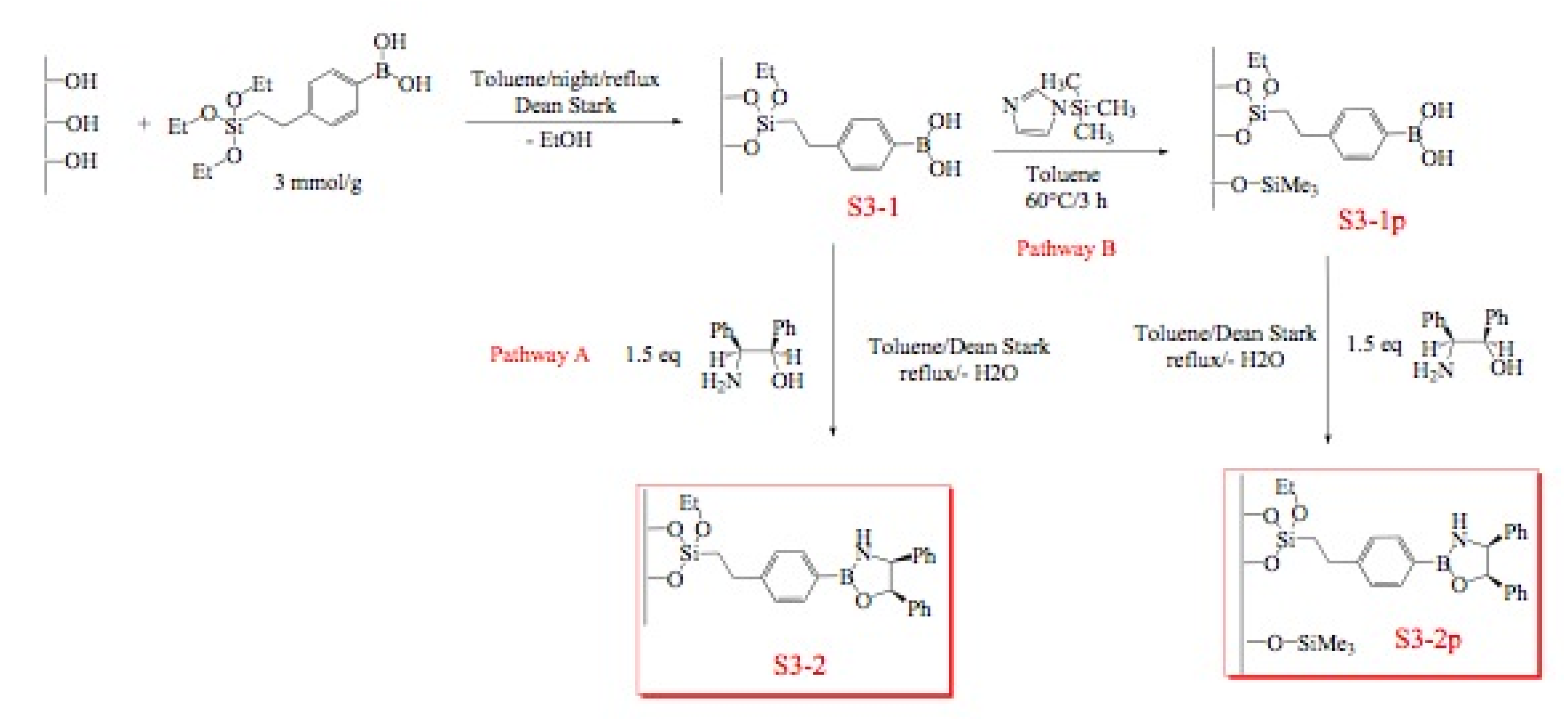
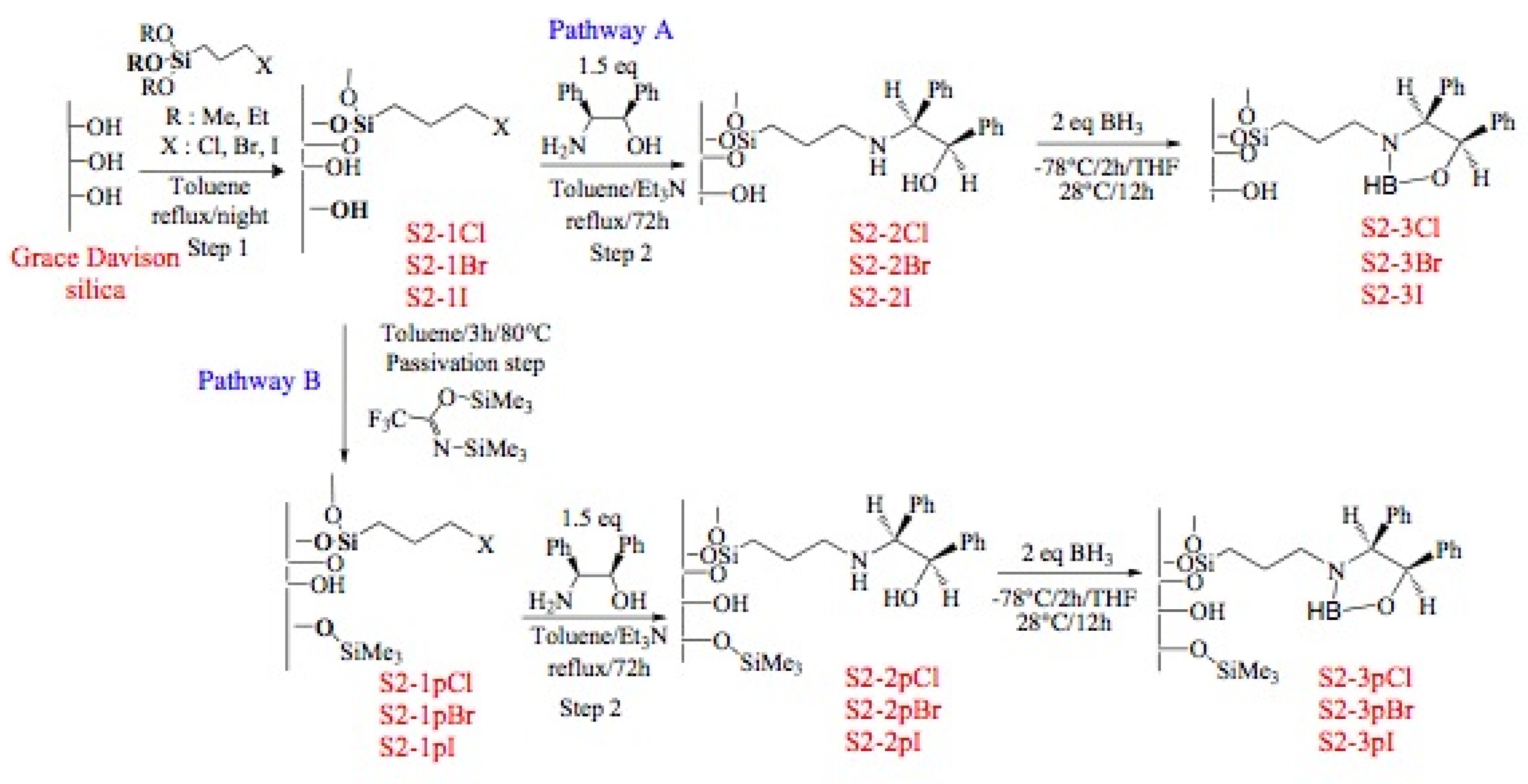
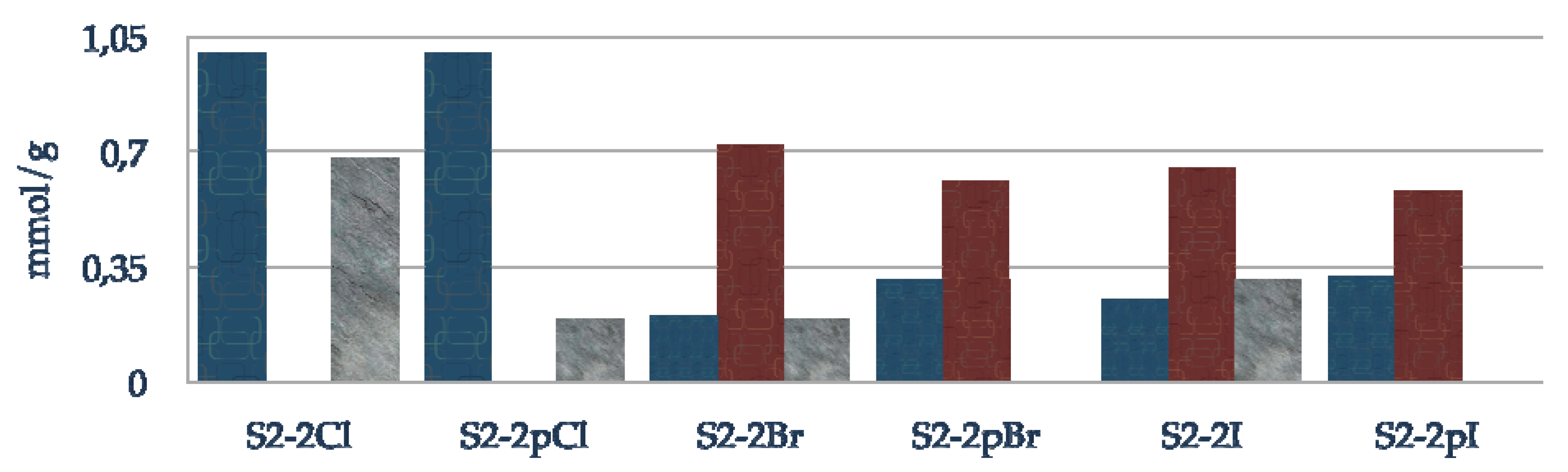
3.3.4. Functionalization of the Grace silica (S2-1X)
Grace silica (1 g) was activated at 150 ºC under vacuum for 3 h. After cooling the silica under argon, anhydrous toluene (40 mL) and 3-halogenopropyltriethoxysilane (0.89 g, 4 mmol) are added and the mixture heated at 130 ºC for 12 h, with the ethanol formed during the reaction being removed with a Dean-Stark apparatus. The solid is filtered with toluene (3 × 20 mL), methanol (3 × 20 mL), dichloromethane (3 × 20 mL), diethyl ether (3 × 20 mL), then washed with a Soxhlet apparatus for 24 h (dichloromethane/diethyl ether = 1/1). Finally the solid is dried at 70 ºC for 3 h (S2-1X).
3.3.5. Passivation step of residual silanols
S2-1X (1 g) was activated at 100 ºC under vacuum for 3 h. After cooling the silica under argon, anhydrous toluene (40 mL) and N,O-bis(trimethylsilyl)trifluoroacetamide (0.57 mL, 3 mmol) are added and the mixture heated at 80 ºC for 3 h. Then the solid is filtered with toluene (3 × 20 mL), methanol (3 × 20 mL), dichloromethane (3 × 20 mL), diethyl ether (3 × 20 mL), and washed with a Soxhlet apparatus for 24 h (dichloromethane/diethyl ether = 1/1). Finally the solid is dried at 70 ºC for 3 h (S2-1pX).
3.3.6. Immobilization of chiral amino alcohol
S2-1Br (1 g, 0.92 mmol) was activated at 100 ºC under vacuum for 3 h. After cooling the silica under argon, anhydrous toluene (30 mL), (1R,2S)-2-amino-1,2-diphenylethanol (0.29 g, 1.38 mmol) and triethylamine (0.64 mL, 4.6 mmol) are added and the mixture heated at reflux during 72 h. The solid is filtered with toluene (3 × 20 mL), methanol (3 × 20 mL), dichloromethane (3 × 20 mL), diethyl ether (3 × 20 mL), then washed with a Soxhlet apparatus for 24 h (dichloromethane/diethyl ether = 1/1). Finally the solid is dried at 70 ºC for 3 h (S2-1pX).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






























