Synthesis and Characterization of Polycarbonates by Melt Phase Interchange Reactions of Alkylene and Arylene Diacetates with Alkylene and Arylene Diphenyl Dicarbonates

Abstract
:1. Introduction
2. Results and Discussion
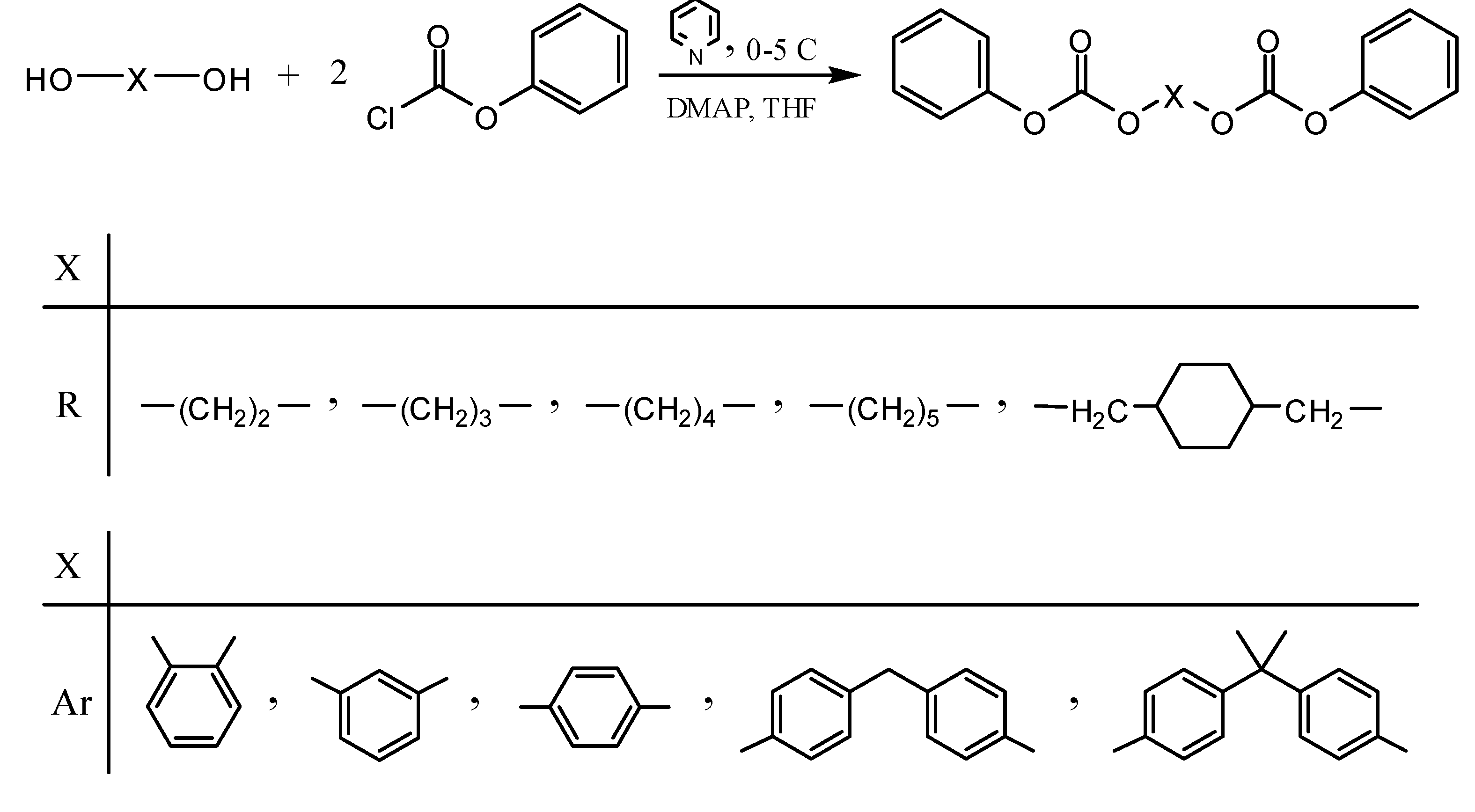
2.1. Synthesis and characterization of monomers


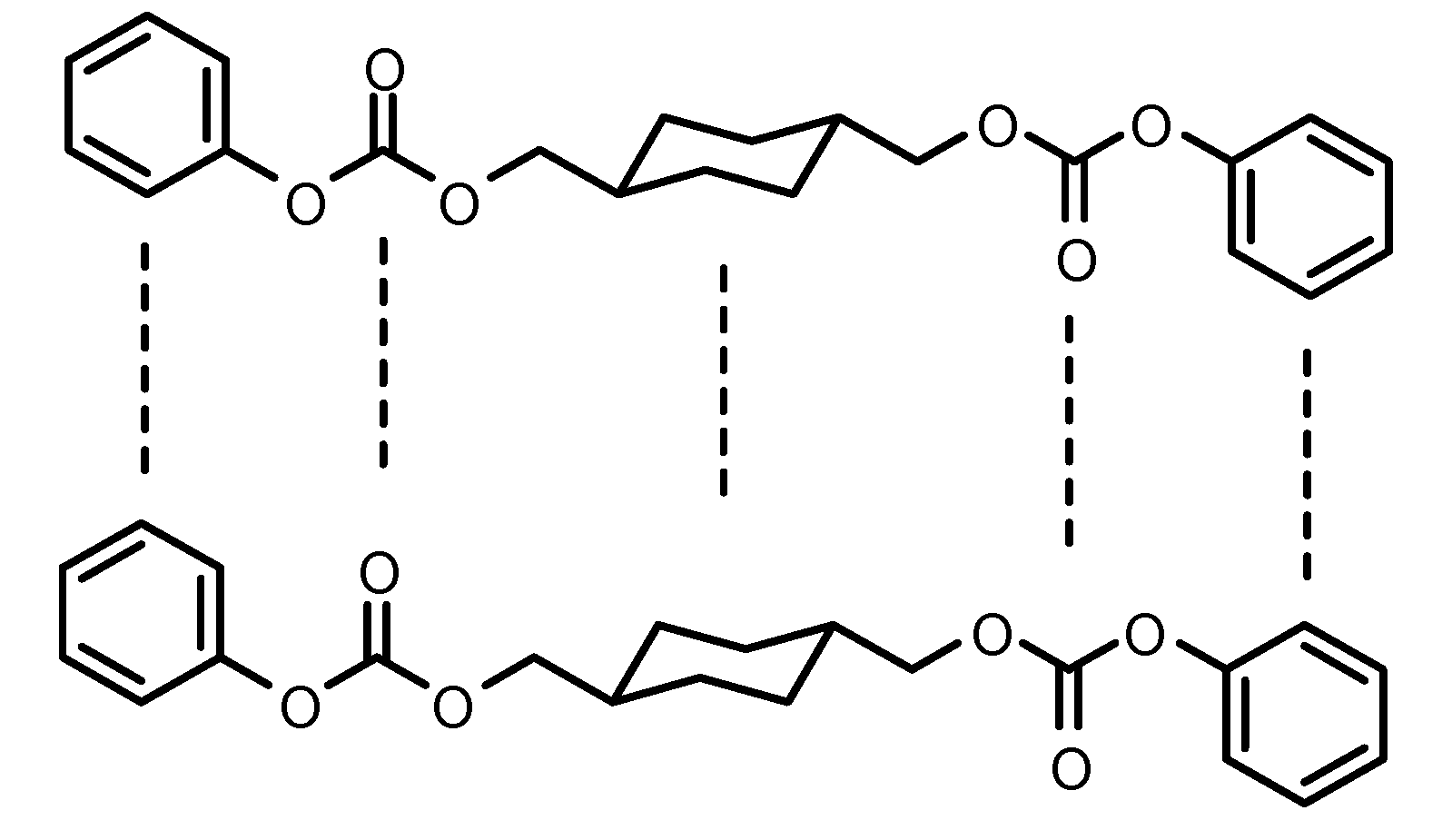{kind=link}
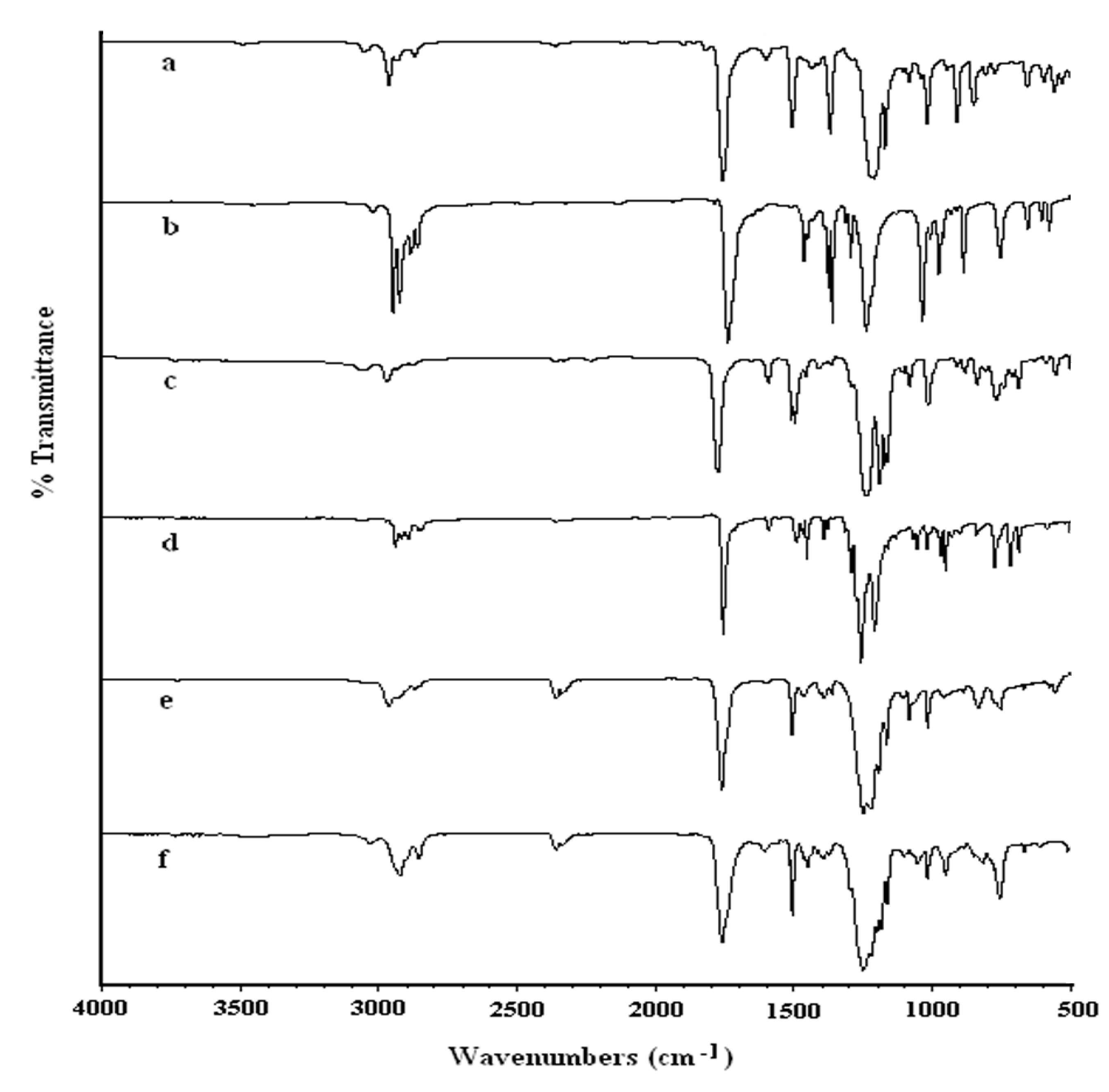{kind=link}
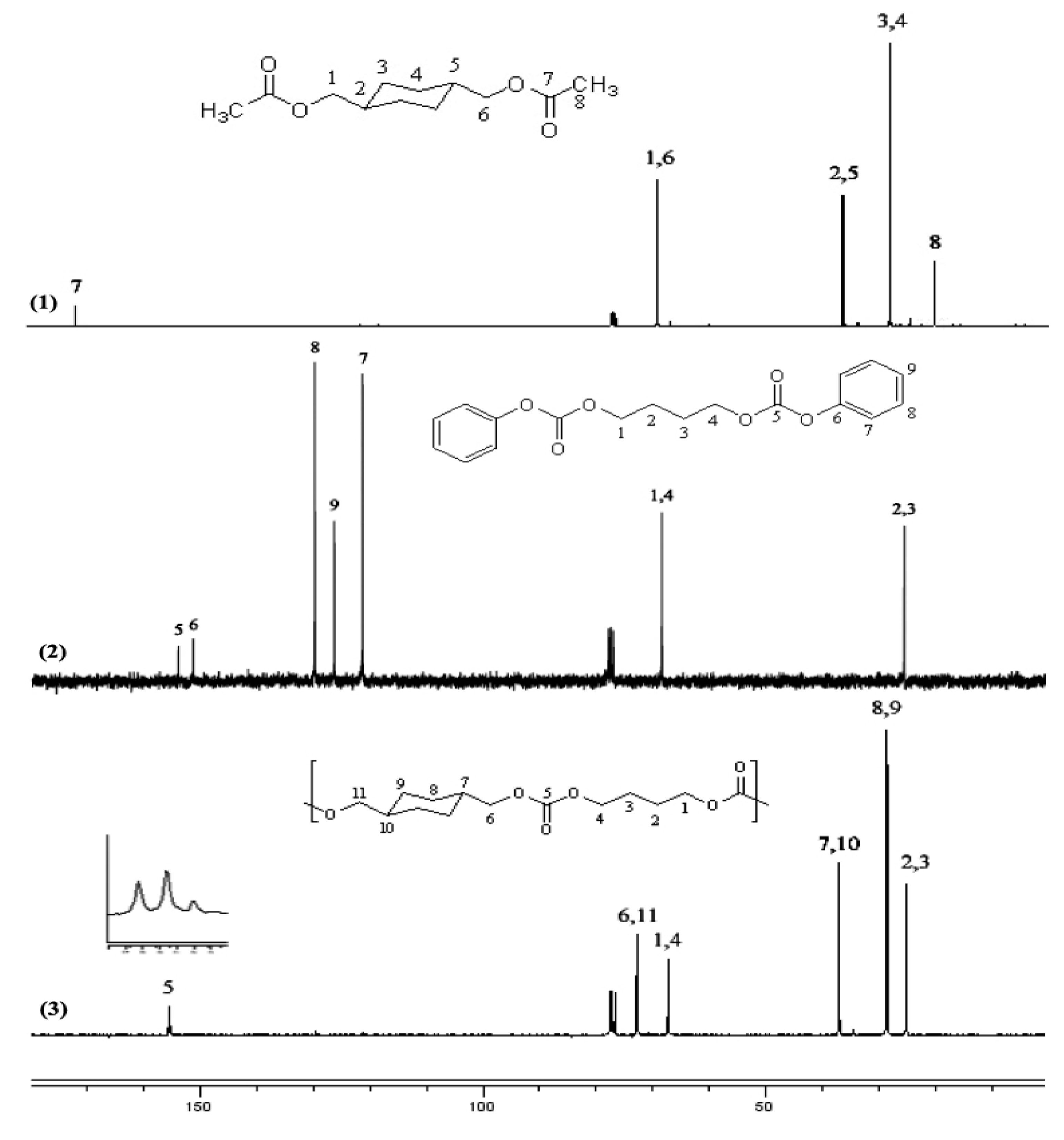{kind=link}
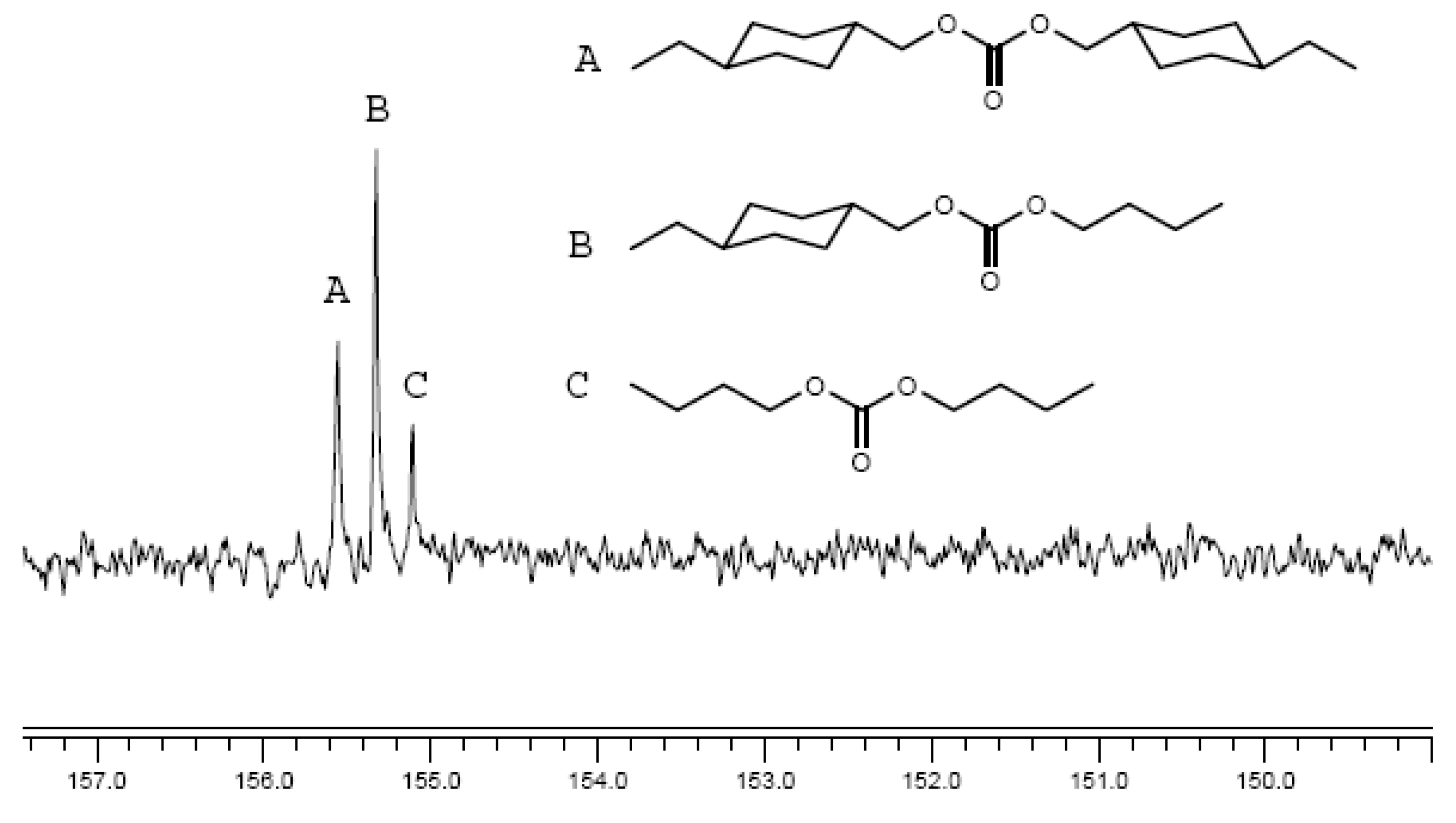{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield (%) | Tm (°C) | C-O-C (cm-1) | C=O (cm-1) | |
|---|---|---|---|---|
| Diphenyl dicarbonates | ||||
| 1,2- Ethanediol | 92.7 | 94 | 1209, 1257 | 1757 |
| 1,3-Propanediol | 87.5 | 52 | 1210, 1242 | 1761 |
| 1,4-Butanediol | 89.8 | 82 | 1205, 1260 | 1754 |
| 1,5-Pentanediol | 94.4 | 52 | 1210, 1252 | 1760 |
| 1,4-Cyclohexanedimethanol | 90.2 | 145–150 | 1208, 1260 | 1755 |
| Catechol | 83.3 | 100–103 | 1197, 1245 | 1787 |
| Resorcinol | 85.1 | 126 | 1128, 1163, 1234 | 1775 |
| Hydroquinone | 87.1 | 124 | 1271 | 1769 |
| 4,4'-Bis(hydroxyphenyl)methane | 91.6 | 97–100 | 1158, 1184, 1253 | 1768 |
| Bisphenol A | 84.4 | 90 | 1170, 1189, 1235 | 1775 |
| Diacetates | ||||
| Bisphenol A diacetate | — | 89–91 | 1170, 1210 | 1755 |
| 1,4-Cyclohexanedimethanol diacetate | 87 | 70 | 1239 | 1741 |

2.1.1. Infrared spectroscopy

2.1.2. NMR spectroscopy of monomers

| Chemical shift δ (ppm) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||||||||||||
| Dicarbonate | ||||||||||||||||||||||
 | 1H | 4.08 | 1.91 | 1.0- 17. | 1.0- 17. | 1.91 | 4.08 | — | — | a | a | a | ||||||||||
| 13C | 73.5 | 37.0 | 28.6 | 28.6 | 37.0 | 73.5 | 153.9 | 151.2 | 121.1 | 129.6 | 126 | |||||||||||
 | 1H | - | a | a | a | a | — | — | — | a | a | a | ||||||||||
| 13C | 142.2 | 123 | 126.6 | 126.6 | 123 | 142.2 | 151.3 | 151 | 121 | 129.7 | 127.3 | |||||||||||
 | 1H | 4.0 | — | a | a | — | — | — | a | a | a | |||||||||||
| 13C | 40.6 | 138.8 | 129.6 | 121 | 149.5 | 152.2 | 151 | 121 | 130 | 126.4 | ||||||||||||
| Diacetate | ||||||||||||||||||||||
 | 1H | 1.65 | — | — | 7.20 | 6.97 | — | — | 2.27 | |||||||||||||
| 13C | 31.0 | 42.5 | 147.9 | 127.9 | 121 | 148.6 | 169.6 | 21.2 | ||||||||||||||
 | 1H | 3.81 | 1.74 | 1.0-1.7 | 1.0-1.7 | 1.74 | 3.81 | - | 1.98 | |||||||||||||
| 13C | 69.4 | 37 | 23.76 | 23.76 | 37 | 69.4 | 171.2 | 21 | ||||||||||||||
2.2. Synthesis and characterization of polycarbonates

2.2.1. Solution viscosity measurements
| Sample | Polycarbonate | Polym code | IR [v (cm-1)] C=O C-O | Inherent viscosity (ηinh) (dL/g) | Tg (°C) | |
|---|---|---|---|---|---|---|
| Series A | ||||||
| 1 | BPA–EGa) | 1A | — | — | — | — |
| 2 | BPA–PrD | 2A | 1,764 | 1,164, 1,194, 1,231 | 0.19 | 68.1 |
| 3 | BPA–BuD | 3A | 1,761 | 1,164, 1,195, 1,245 | 0.23 | 56.5 |
| 4 | BPA–PeD | 4A | 1,761 | 1,218, 1,250 | 0.24 | 49.5 |
| 5 | BPA–CAT a) | 5A | — | — | — | — |
| 6 | BPA–RESOL | 6A | 1,777 | 1,164, 1,203 | 0.24 | 83.1 |
| 7 | BPA–HQ | 7A | 1,772 | 1,163, 1,193, 1,228 | 0.26 | 97.0 |
| 8 | BPA–BHPM | 8A | 1,772 | 1,161, 1,188, 1,231 | 0.27 | 107.8 |
| Series B | ||||||
| 9 | CHDM-BuD | 1B | 1,743 | 1,254 | 0.43 | 13.0 |
| 10 | CHDM-CHDM | 2B | 1,743 | 1,252 | 0.36 | 38.7 |
| 11 | CHDM-BPA | 3B | 1,761 | 1,216, 1,249 | 0.29 | 90.4 |
| 12 | CHDM-BHPM | 4B | 1,760 | 1,185, 1,220, 1,251 | 0.24 | 56.9 |
2.2.2. Infrared spectroscopy
2.2.3. NMR spectra of polycarbonates


2.2.4. 13C-NMR spectral evidence of unit sequence randomization



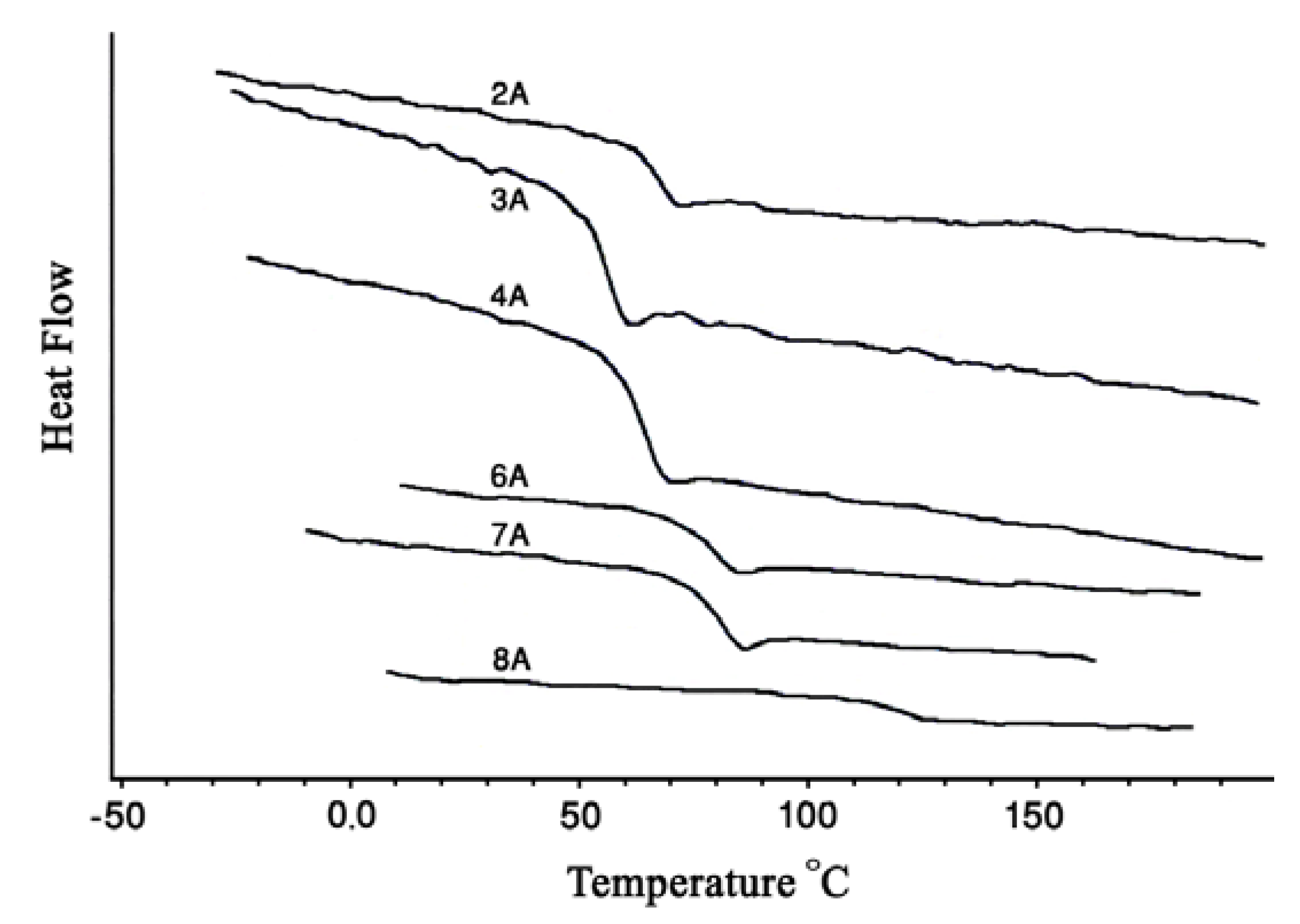
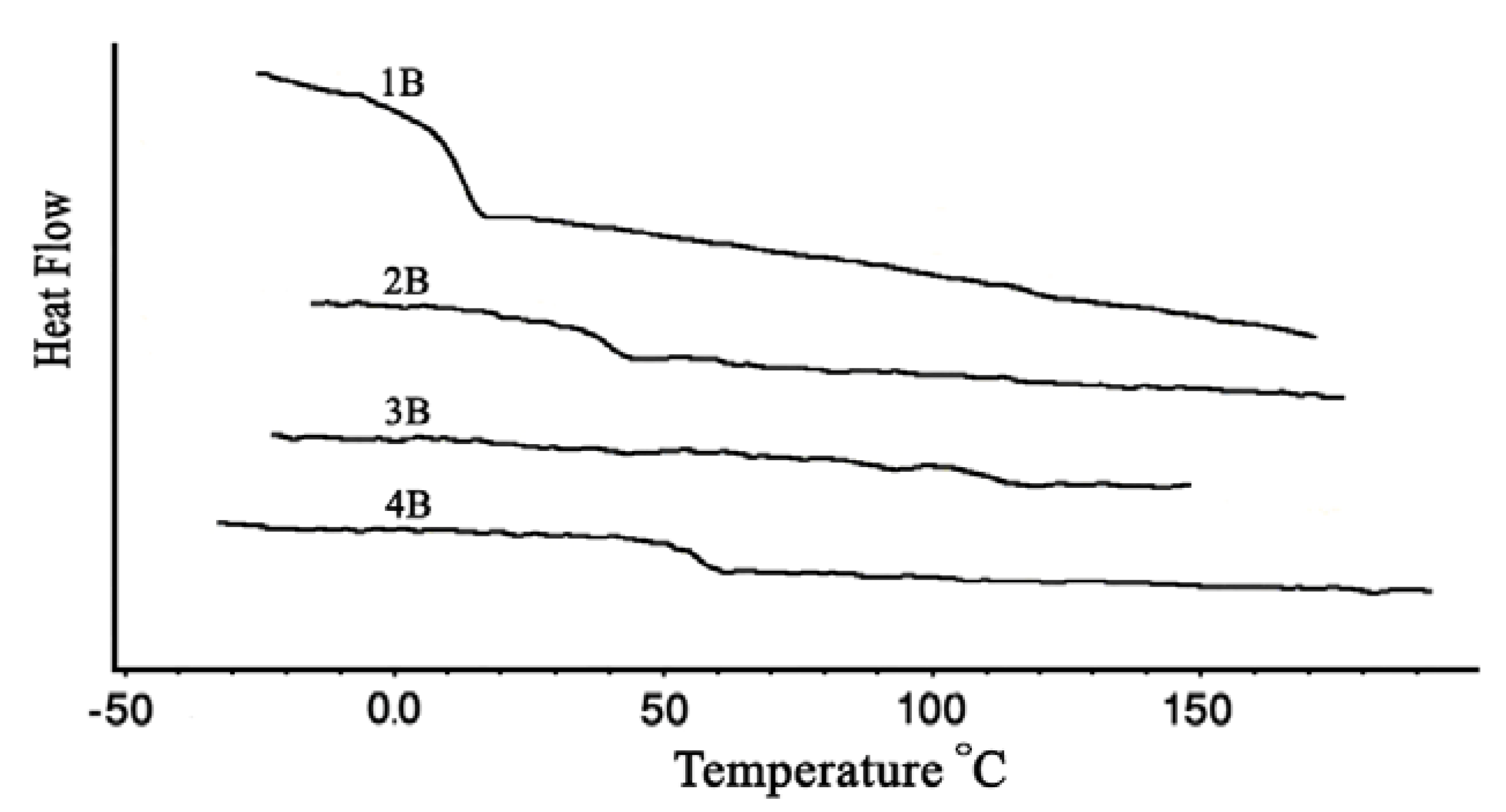
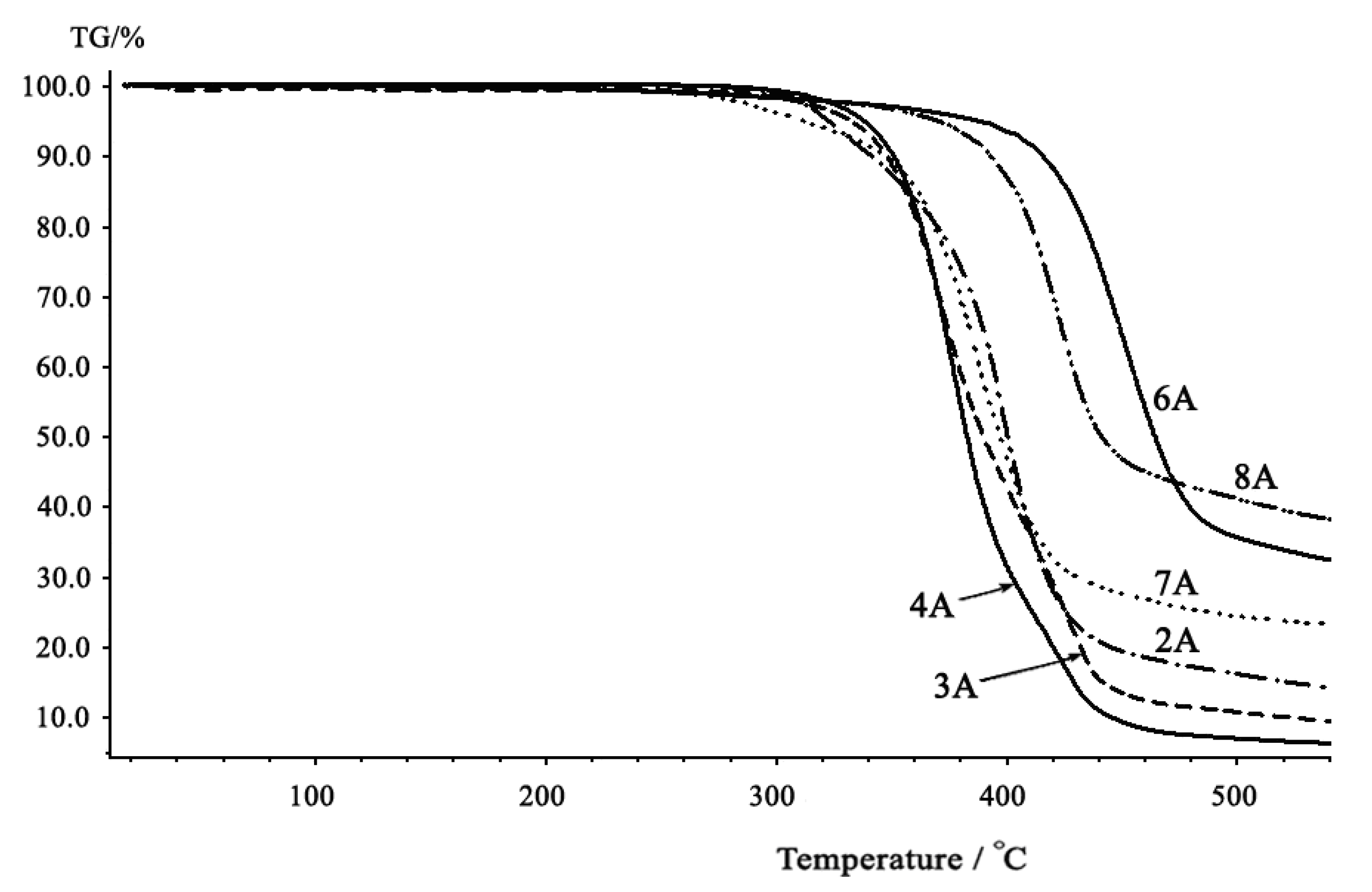
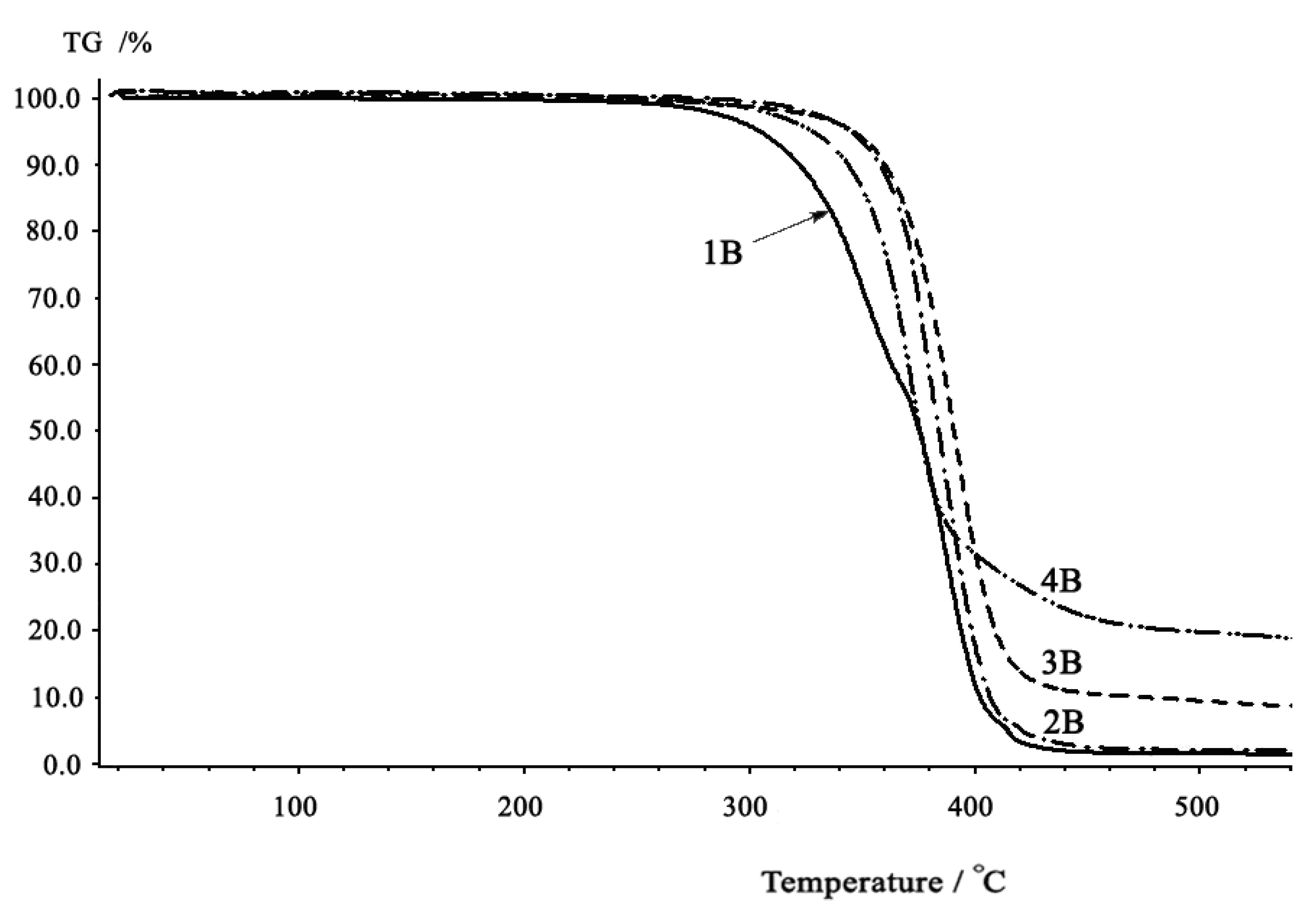
2.3. Thermal properties




3. Experimental
3.1. Materials
3.2. Monomer synthesis
3.3. Polycarbonate synthesis
- 1)
- Series A: was prepared from the reaction of BPA diacetate with alkylene and arylenediphenyldicarbonates.
- 2)
- Series B: was prepared from the reaction of 1,4-cyclohexanedimethylene diacetate with alkylene and arylenediphenyldicarbonates.
3.4. Monomer and polymer characterization
3.5. Thermal properties
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds are available from the authors.
References and Notes
- Brunelle, D.J. Polycarbonates. In Encyclopedia of Polymer Science and Technology, 3rd; Mark, H.F., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2004; Volume 7, pp. 397–426. [Google Scholar]
- Deshpande, M.M.; Jadhav, A.S.; Gunari, A.A.; Sehra, J.C.; Sivaram, S. Polycarbonate synthesis by melt phase carbonate-ester interchange reaction of bisphenol-A diacetate with Dimethyl carbonate. J. Polym. Sci. A Polym. Chem. 1995, 33, 701–705. [Google Scholar] [CrossRef]
- Clagett, D.; Shafer, S. Polycarbonates. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pregman Press: Oxford, UK, 1989; Volume 5, pp. 345–356. [Google Scholar]
- Freitag, D.; Grigo, U.; Mϋller, P.; Nouvertnẻ, W. Polycarbonates. In Encyclopedia of Polymer Science and Engineering; Mark, H.F., Overberger, C.G., Menges, G., Kroschwilz, J.I., Eds.; Wiley: New York, NY, USA, 1987; Volume 11, pp. 648–718. [Google Scholar]
- Pokharkar, V.; Sivaram, S. Poly(alkylene carbonate)s by the carbonate interchange reaction of aliphatic diols with dimethyl carbonate: Synthesis and characterization. Polymer 1995, 36, 4851–4854. [Google Scholar]
- Park, J.H.; Hyun, J.C.; Kim, W.N.; Kim, S.R.; Ryu, S.C. Extensional and complex viscosities of linear and branched polycarbonates. Macromol. Res. 2002, 10, 135–139. [Google Scholar] [CrossRef]
- Liu, Z.; Cunha, A.M.; Yi, X.-S.; Bernado, A.C. Key properties to understand the performance of polycarbonate reprocessed by injection molding. J. Appl. Polym. Sci. 2000, 77, 1393–1400. [Google Scholar]
- Jones, R.O.; Ballone, P. A combined density functional and Monte Carlo study of polycarbonate. Mat. Res. Soc. Symp. Proc. 2001, AA3.1/1-AA3.1/12, 677. [Google Scholar]
- Liaw, D.J.; Chang, P. Synthesis and characterization of aromatic and brominated aromatic polycarbonates by two-phase-transfer-catalyzed polycondensation of bisphenols with trichloromethylchloroformate. J. Appl. Polym. Sci. 1997, 63, 195–204. [Google Scholar] [CrossRef]
- Kricheldorf, H.; Luebbers, D. Polymers of carbonic acid. 3. Thermotropic polycarbonates derived from 4,4`-dihydroxybiphenyl and various diphenols. Macromolecules 1990, 23, 2656–2662. [Google Scholar] [CrossRef]
- Lee, J.; Song, C.; Kim, J.-I.; Kim, J.-H. Preparation of aromatic polycarbonates nanoparticles using supercritical carbon dioxide. J. Nanopart. Res. 2002, 4, 53–59. [Google Scholar] [CrossRef]
- Ballistreri, A.; Montaudo, G.; Scamporrino, E.; Puglisi, C.; Vitalini, D.; Ucinella, S. Intumescent flame retardants of polymers. IV. The polycarbonate- aromatic sulfonates system. J. Polym. Sci. A Polym. Chem. 1988, 26, 2113–2127. [Google Scholar] [CrossRef]
- Martin, E.H.; Brittain, J. A convenient laboratory preparation of aromatic polycarbonate. Polym. Bull. 2002, 47, 517–520. [Google Scholar] [CrossRef]
- Haba, O.; Itakura, I.; Ueda, M.; Kuze, S. Synthesis of polycarbonate from dimethyl carbonate and bisphenol-A through a nonphosgene process. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 2087–2093. [Google Scholar] [CrossRef]
- Skupinska, J.; Kuczynska, L.; Wielgosz, Z. Aromatic polycarbonates by direct synthesis from bisphenol A and carbon monoxide. React. Kinet. Catal. Lett. 2003, 80, 269–276. [Google Scholar] [CrossRef]
- Fraitag, D.; Fengler, G.; Morbitzer, L. Routes to new aromatic polycarbonates with special material properties. Angew. Chem. Int. Ed. Engl. 1991, 30, 1598–1610. [Google Scholar] [CrossRef]
- Kim, W.B.; Joshi, U.A.; Lee, J.S. Making polycarbonates without employing phosgene: An overview on catalytic chemistry of intermediate and precursor syntheses for polycarbonate. Ind. Eng. Chem. Res. 2004, 13, 1897–1914, and references therein. [Google Scholar]
- Ono, Y. Dimethyl carbonate for environmentally benign reactions. Catal.Today 1997, 35, 15–25. [Google Scholar] [CrossRef]
- Kricheldorf, H.; Lee, S.; Bettina, W. Polymers of carbonic acid, 12 spontaneous and hematin-initiated polymerizations of trimethylene carbonate and neopentylene carbonate. Macromol.Chem. Phys. 1996, 197, 1043–1054. [Google Scholar] [CrossRef]
- Thorat, S.D.; Philips, P.J.; Semenov, V.; Gakh, A. Physical properties of aliphatic polycarbonates made from CO2 and epoxides. J. Appl. Polym. Sci. 2003, 89, 1163–1176. [Google Scholar] [CrossRef]
- Vorgdanis, L.; Martens, B.; Uchtmann, H.; Hensel, F.; Heitz, W. Synthetic and thermodynamic investigations in the polymerization of ethylene carbonate. Makromol.Chem. 1990, 191, 465–472. [Google Scholar] [CrossRef]
- Sweileh, B.A.; Al-Hiari, Y.M.; Aiedeh, K.M. A new, nonphosgene route to poly(bisphenol A carbonate) by melt-phase interchange reactions of alkylenediphenyldicarbonates with bisphenol A. J. Appl. Polym. Sci. 2008, 110, 2278–2292. [Google Scholar] [CrossRef]
- Sun, S.; Chang, T. Studies on the thermotropic liquid crystalline polycarbonates. III. Synthesis and properties of fully aromatic liquid-crystalline polycarbonates. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2711–2719. [Google Scholar] [CrossRef]
- Sun, S.-J.; Chang, T.-C. Studies on the thermotropic crystalline polycarbonates. II. Synthesis and properties of fully aromatic liquid crystalline polycarbonates. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2237–2243. [Google Scholar]
- Sun, S.J.; Li, C.H.; Chang, T.C. Studies on the Thermotropic crystalline polycarbonates. I. synthesis and properties of thermotropic liquid crystalline poly(azomethine carbonates). Eur. Polym. J. 1993, 29, 951–955. [Google Scholar] [CrossRef]
- De Backer, C.; Ottenburgs, R.; Van Beylen, M.; Saymn, C. Synthesis and liquid- crystalline properties of thermotropichomopolycarbonates. Macromol. Chem. Phys. 1995, 196, 1495–1514. [Google Scholar] [CrossRef]
- De Backer, C.; Ottenburgs, R.; Van Beylen, M.; Saymn, C. Synthesis and liquid crystalline properties of thermotropiccopolycarbonates. Macromol. Chem. Phys. 1996, 197, 443–466. [Google Scholar] [CrossRef]
- Sato, M.; Kurosawa, K.; Nakatsuchi, K.; Okhatsu, Y. Synthesis and liquid-crystalline properties of thermotropic homo- and copolycarbonates. J. Polym. Sci. A Polym. Chem. 1988, 26, 3077–3088. [Google Scholar] [CrossRef]
- Sato, M.; Tomihisa, N.; Komatsu, F.; Yahata, T.; Shoji, Y. Synthesis and liquid-crystalline properties of Thermotropiccopolycarbonates composed of biphenyl and diphenylpropane moieties. Euro. Polym. J. 1990, 26, 467–470. [Google Scholar] [CrossRef]
- Sato, M.; Muraki, K.; Mukaida, K-I. Thermotropic liquid crystalline semirigid polycarbonates based on diphenyl ether and benzophenone units. Macromolecules 1994, 27, 4577–4582. [Google Scholar] [CrossRef]
- Sato, M.; Tada, Y.; Goto, Y.; Mukaida, K. Semi-rigid thermotropic liquid-crystalline copolycarbonates comprising main-chain hexafluoropentane segments. Macromol. Rap. Comm. 1995, 16, 139–146. [Google Scholar] [CrossRef]
- Sato, M.; Muraki, K.; Mukaida, K.-I. Synthesis and thermal properties of semi-rigid homo-, co- and terpolycarbonates containing naphthalene rings. Euro. Polym. J. 1995, 31, 867–873. [Google Scholar] [CrossRef]
- Liaw, D.-J.; Chang, P. Synthesis and characterization of copolycarbonates from melt transesterfication. Polymer 1996, 37, 2857–2863. [Google Scholar] [CrossRef]
- Liaw, D.-J.; Chen, P.-C. Preparation and polyesters derived from 4,4`-sulfonyl dibenzoyl chloride by solution polycondensation. J. Polym. Sci. Part. A Polym. Chem. 1996, 34, 885–891. [Google Scholar] [CrossRef]
- Tomoki, H.; Hirosuke, K. Halogen-free solventless manufacture of polyesters and polycarbonate-polyesters with excellent physical properties. Jpn. Kokai Tokkyo Koho JP 09235362, 1997. [Google Scholar]
- Kouichi, K.; Shigeki, K.; Noriyuki, K.; Masaya, O. Producing a transparent polycarbonate with heat and water resistance. Eur. Pat. Appl. EP 488182, 1992. [Google Scholar]
- Tatsuya, S.; Tsutomu, Y. Phosgene-free manufactre of high-molecular-weight polycarbonates. Jpn. Kokai Tokkyo Koho JP 02284917, 1990. [Google Scholar]
- Yoshitaka, N.; Yoshiyuki, Y. Preparation of aromatic poycarbonates without using phosgene. Jpn. Kokai Tokkyo Koho JP 02166119, 1990. [Google Scholar]
- Barry, H. Production of polycarbonates. Eur. Pat. Appl. EP 299773, 1989. [Google Scholar]
- Al-Hamouz, O.C.S.; Sweileh, B.A.; Al-Salah, H.A. Synthesis and characterization of polycarbonates by melt-phase interchange reactions with alkylene and arylenediphenyldicarbonates. J. Appl. Polym. Sci. 2006, 102, 3597–3609. [Google Scholar] [CrossRef]
- Li, X.Y.; Yee, A.F. Design of mechanically robust high-Tg polymers: Physical properties of glassy poly(ester carbonate)s with cyclohexylene rings in the backbone. Macromolecules 2003, 36, 9421–9429. [Google Scholar] [CrossRef]
- Sweileh, B.A.; Al-Hiari, Y.M. Synthesis and thermal properties of polycarbonates based on bisphenol A by single-phase organic solvent polymerization. J. Polym. Res. 2006, 13, 181–191. [Google Scholar] [CrossRef]
- Berti, C.; Bonora, V.; Pilati, F. Reactive blending of poly(ethylene terephthalate) and polycarbonate. 1. A reappraisal of the reactions occurring during melt-mixing. Makromol. Chem. 1992, 193, 1665–1677. [Google Scholar] [CrossRef]
- Liaw, D.-J.; Chang, P. Synthesis and characterization of polycarbonates based on bisphenol S by melt transesterification. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 2453–2460. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Sweileh, B.A.; Al-Hiari, Y.M.; Kailani, M.H.; Mohammad, H.A. Synthesis and Characterization of Polycarbonates by Melt Phase Interchange Reactions of Alkylene and Arylene Diacetates with Alkylene and Arylene Diphenyl Dicarbonates. Molecules 2010, 15, 3661-3682. https://doi.org/10.3390/molecules15053661
Sweileh BA, Al-Hiari YM, Kailani MH, Mohammad HA. Synthesis and Characterization of Polycarbonates by Melt Phase Interchange Reactions of Alkylene and Arylene Diacetates with Alkylene and Arylene Diphenyl Dicarbonates. Molecules. 2010; 15(5):3661-3682. https://doi.org/10.3390/molecules15053661
Chicago/Turabian StyleSweileh, Bassam A., Yusuf M. Al-Hiari, Mohammad H. Kailani, and Hani A. Mohammad. 2010. "Synthesis and Characterization of Polycarbonates by Melt Phase Interchange Reactions of Alkylene and Arylene Diacetates with Alkylene and Arylene Diphenyl Dicarbonates" Molecules 15, no. 5: 3661-3682. https://doi.org/10.3390/molecules15053661
APA StyleSweileh, B. A., Al-Hiari, Y. M., Kailani, M. H., & Mohammad, H. A. (2010). Synthesis and Characterization of Polycarbonates by Melt Phase Interchange Reactions of Alkylene and Arylene Diacetates with Alkylene and Arylene Diphenyl Dicarbonates. Molecules, 15(5), 3661-3682. https://doi.org/10.3390/molecules15053661