A Planar Conformation and the Hydroxyl Groups in the B and C Rings Play a Pivotal Role in the Antioxidant Capacity of Quercetin and Quercetin Derivatives

Abstract
:1. Introduction
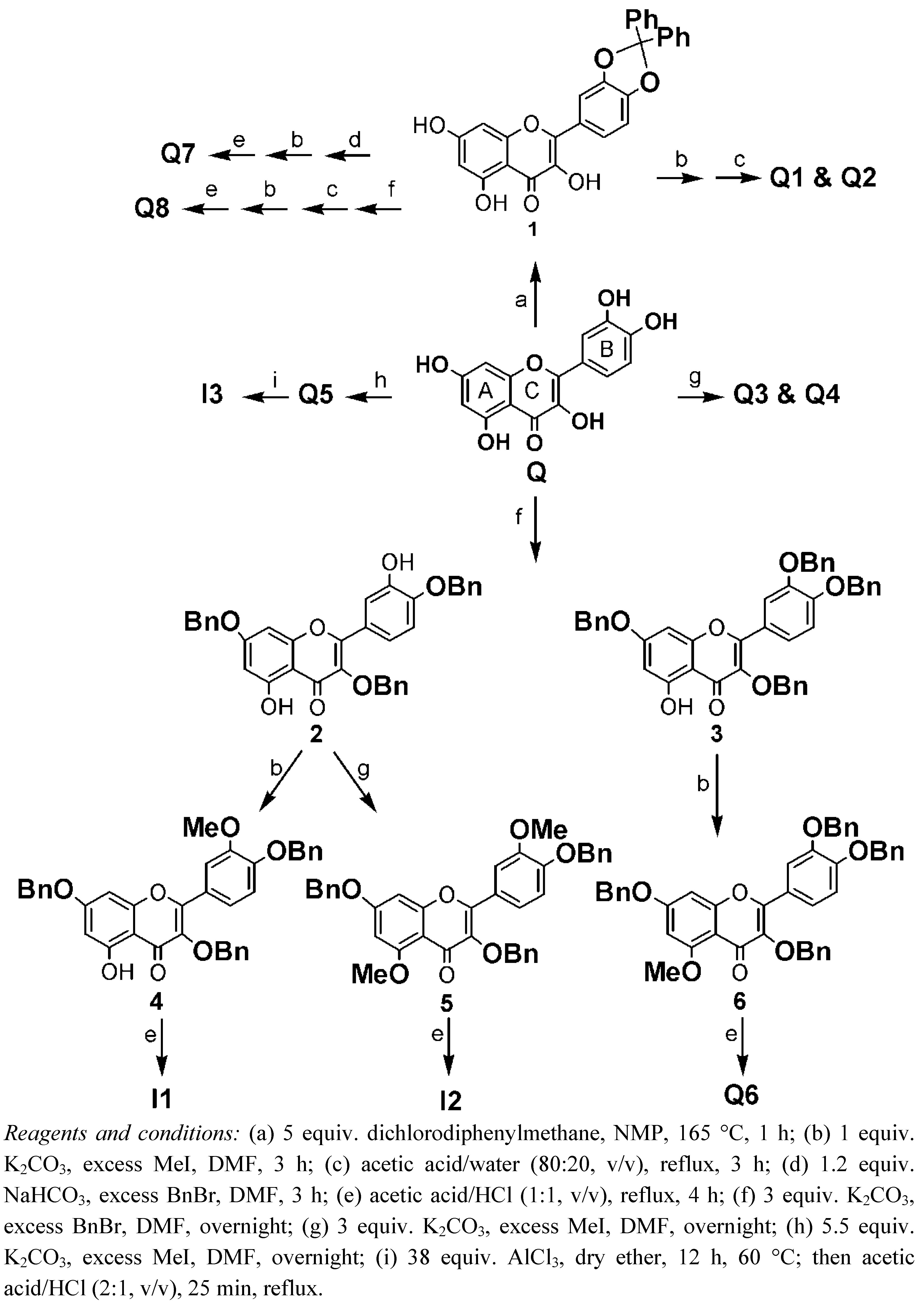
2. Synthesis


{kind=link}
{kind=link}

 |
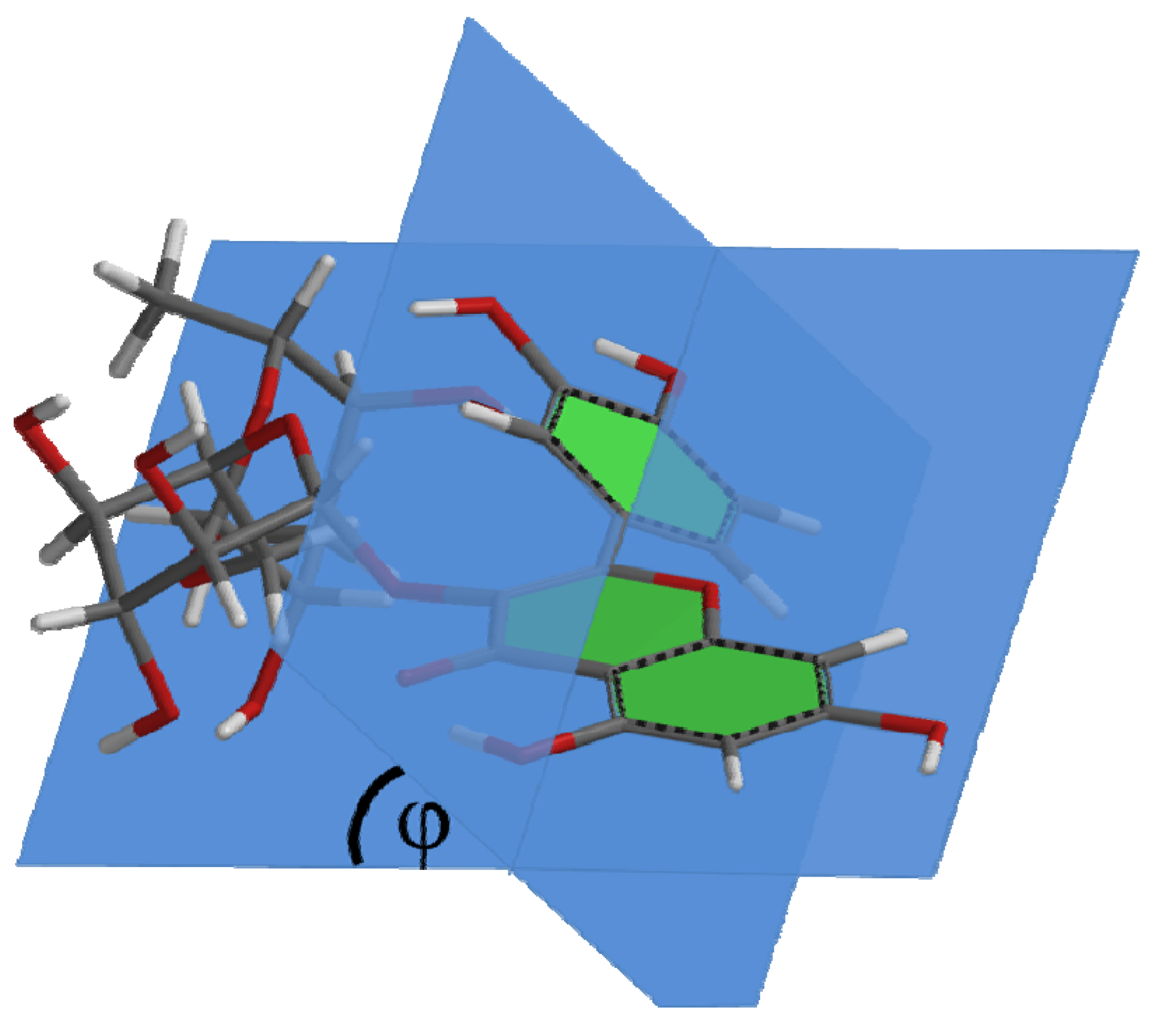
3. Results and Discussion
| Compound | Capacity | φ |
|---|---|---|
| Q | 8.6 | 0.29° |
| Q1 | 5.8 | 20° |
| R1 | 5.1 | 38° |

4. Experimental
4.1. Antioxidant Capacity
4.1.1. Chemicals
4.1.2. Scavenging of ABTS Radicals
4.2. Molecular Quantum Calculations
4.3. Synthesis
4.3.1. General
4.3.2. Protection of the Catechol Group of Quercetin. Synthesis of 2-(2,2-Diphenylbenzo [1,3]dioxol-5-yl)-3,5,7-trihydroxychromen-4-one (1)
4.3.3. Synthesis of Quercetin-3-OMe (Q1) and Quercetin-3,7-OMe (Q2)
4.3.4. Synthesis of Quercetin-3,7,4′-OMe (Q3) and Quercetin-3,7,3′,4′-OMe (Q4)
4.3.5. Synthesis of Quercetin-penta-OMe (Q5)
4.3.6. Synthesis of Quercetin-3,7,4′-OBn (2) and Quercetin-3,7,3′,4′-OBn (3)
4.3.7. Synthesis of Quercetin-3,7,4′-OBn-3′-OMe (4)
4.3.8. Synthesis of Quercetin-3,7,4′-OBn-5,3′-OMe (5)
4.3.9. Synthesis of Quercetin-3,7,3′,4′-OBn-5-OMe (6)
4.3.10. Debenzylation of 4, 5 and 6: Synthesis of Quercetin-3′-OMe (I1), Quercetin-5,3′-OMe (I2) and Quercetin-5-OMe (Q6), respectively (see section Section 4.3.17 “General procedure for debenzylation”)
4.3.11. Synthesis of 2-(2,2-Diphenylbenzo [1,3]dioxol-5-yl)-3-benzyloxy-5,7-dihydroxychromen-4-one (2*) and 2-(2,2-Diphenylbenzo [1,3]dioxol-5-yl)-3,7-dibenzyloxy-5-hydroxychromen-4-one (3*)
4.3.12. Monomethylation of 2*: Synthesis of 2-(2,2-Diphenylbenzo [1,3]dioxol-5-yl)-3-benzyloxy-5-hydroxy-7-methoxychromen-4-one (4*)
4.3.13. Debenzylation of 4*: Synthesis of Quercetin-7-OMe (Q7) (see Section 4.3.17 “General procedure for debenzylation”)
4.3.14. Tribenzylation of Compound 1 Followed by Selective Deprotection of the Catechol Moiety: Synthesis of Quercetin-3,7-OBn (5*)
4.3.15. Monomethylation of 5*: Synthesis of Compound Quercetin-3,7-OBn-4′-OMe (6*)
4.3.16. Debenzylation of 6*: Synthesis of Quercetin-4′-OMe (Q8) (see section Section 4.3.17 “General procedure for debenzylation”)
4.3.17. General Procedure for Debenzylation: Synthesis of Quercetin-3′-OMe (I1), Quercetin-5,3′-OMe (I2), Quercetin-5-OMe (Q6), Quercetin-7-OMe (Q7) and Quercetin-4′-OMe (Q8)
4.3.18. Quercetin-5,7,3′,4′-OMe (I3)
5. Conclusions
References
- Hertog, M.G.; Hollman, P.C.; Katan, M.B. Content of potentially anticarcinogenic flavonoids of 28 vegetables and 9 fruits commonly consumed in The Netherlands. J. Agric. Food Chem. 1992, 40, 2379–2383. [Google Scholar] [CrossRef]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Crozier, A.; Lean, M.E.; McDonald, M.S.; Black, C. Quantitative analysis of the flavonoid content of commercial tomatoes, onions, lettuce, and celery. J. Agric. Food Chem. 1997, 45, 590–595. [Google Scholar] [CrossRef]
- Justesen, U.; Knuthsen, P.; Leth, T. Quantitative analysis of flavonols, flavones, and flavanones in fruits, vegetables and beverages by high-performance liquid chromatography with photo-diode array and mass spectrometric detection. J. Chromatogr. A 1998, 799, 101–110. [Google Scholar] [CrossRef]
- Willems, A.M.; Bruynzeel, A.M.; Kedde, M.A.; van Groeningen, C.J.; Bast, A.; van der Vijgh, W.J. A phase I study of monohydroxyethylrutoside in healthy volunteers. Cancer Chemother. Pharmacol. 2006, 57, 678–684. [Google Scholar] [CrossRef]
- Bruynzeel, A.M.; Niessen, H.W.; Bronzwaer, J.G.; van der Hoeven, J.J.; Berkhof, J.; Bast, A.; van der Vijgh, W.J.; van Groeningen, C.J. The effect of monohydroxyethylrutoside on doxorubicin-induced cardiotoxicity in patients treated for metastatic cancer in a phase II study. Br. J. Cancer 2007, 97, 1084–1089. [Google Scholar] [CrossRef]
- Kay, C.D. Aspects of anthocyanin absorption, metabolism and pharmacokinetics in humans. Nutr. Res. Rev. 2006, 19, 137–146. [Google Scholar] [CrossRef]
- Spencer, J.P.; Kuhnle, G.G.; Williams, R.J.; Rice-Evans, C. Intracellular metabolism and bioactivity of quercetin and its in vivo metabolites. Biochem. J. 2003, 372, 173–181. [Google Scholar] [CrossRef]
- Spencer, J.P.; Abd-el-Mohsen, M.M.; Rice-Evans, C. Cellular uptake and metabolism of flavonoids and their metabolites: Implications for their bioactivity. Arch. Biochem. Biophys. 2004, 423, 148–161. [Google Scholar] [CrossRef]
- Mladenka, P.; Macakova, K.; Filipsky, T.; Zatloukalova, L.; Jahodar, L.; Bovicelli, P.; Silvestri, I.P.; Hrdina, R.; Saso, L. In vitro analysis of iron chelating activity of flavonoids. J. Inorg. Biochem. 2011, 105, 693–701. [Google Scholar] [CrossRef]
- Hermans, N.; Cos, P.; Maes, L.; De Bruyne, T.; Vanden Berghe, D.; Vlietinck, A.J.; Pieters, L. Challenges and pitfalls in antioxidant research. Curr. Med. Chem. 2007, 14, 417–430. [Google Scholar] [CrossRef]
- Bouktaib, M.; Lebrun, S.; Atmani, A.; Ronaldo, C. Hemisynthesis of all the O-monomethylated analogues of quercetin including the major metabolites, through selective protection of phenolic functions. Tetrahedron 2002, 58, 10001–10009. [Google Scholar]
- van Acker, F.A.; Hageman, J.A.; Haenen, G.R.; van Der Vijgh, W.J.; Bast, A.; Menge, W.M. Synthesis of novel 3,7-substituted-2-(3',4'-dihydroxyphenyl)flavones with improved antioxidant activity. J. Med. Chem. 2000, 43, 3752–3760. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- van den Berg, R.; Haenen, G.R.; van den Berg, H.; Bast, A. Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures. Food Chem. 1999, 66, 511–517. [Google Scholar] [CrossRef]
- Castillo, J.; Benavente-Garcia, O.; Lorente, J.; Alcaraz, M.; Redondo, A.; Ortuno, A.; Del Rio, J.A. Antioxidant activity and radioprotective effects against chromosomal damage induced in vivo by X-rays of flavan-3-ols (Procyanidins) from grape seeds (Vitis vinifera): Comparative study versus other phenolic and organic compounds. J. Agric. Food Chem. 2000, 48, 1738–1745. [Google Scholar]
- Miller, N.J.; Rice-Evans, C.A. Factors influencing the antioxidant activity determined by the ABTS.+ radical cation assay. Free Radic. Res. 1997, 26, 195–199. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar]
- Williamson, G.; Plumb, G.W.; Garcia-Conesa, M.T. Glycosylation, esterification and polymerization of flavonoids and hydroxycinnamates: Effects on antioxidant properties. Basic Life Sci. 1999, 66, 483–494. [Google Scholar]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and prooxidant behavior of flavonoids: structure-activity relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Heijnen, C.G.; Haenen, G.R.; Vekemans, J.A.; Bast, A. Peroxynitrite scavenging of flavonoids: structure activity relationship. Environ. Toxicol. Pharmacol. 2001, 10, 199–206. [Google Scholar] [CrossRef]
- Haenen, G.R.; Paquay, J.B.; Korthouwer, R.E.; Bast, A. Peroxynitrite scavenging by flavonoids. Biochem. Biophys. Res. Commun. 1997, 236, 591–593. [Google Scholar] [CrossRef]
- Heijnen, C.G.; Haenen, G.R.; van Acker, F.A.; van der Vijgh, W.J.; Bast, A. Flavonoids as peroxynitrite scavengers: the role of the hydroxyl groups. Toxicol. In Vitro 2001, 15, 3–6. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. A comparative study of the antioxidant power of flavonoid catechin and its planar analogue. J. Agric. Food Chem. 2007, 55, 7944–7949. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Walle, T. Methylation of dietary flavones increases their metabolic stability and chemopreventive effects. Int. J. Mol. Sci. 2009, 10, 5002–5019. [Google Scholar] [CrossRef]
- Duenas, M.; Gonzalez-Manzano, S.; Gonzalez-Paramas, A.; Santos-Buelga, C. Antioxidant evaluation of O-methylated metabolites of catechin, epicatechin and quercetin. J. Pharm. Biomed. Anal. 2010, 51, 443–449. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moalin, M.; Strijdonck, G.P.F.v.; Beckers, M.; Hagemen, G.J.; Borm, P.J.; Bast, A.; Haenen, G.R.M.M. A Planar Conformation and the Hydroxyl Groups in the B and C Rings Play a Pivotal Role in the Antioxidant Capacity of Quercetin and Quercetin Derivatives. Molecules 2011, 16, 9636-9650. https://doi.org/10.3390/molecules16119636
Moalin M, Strijdonck GPFv, Beckers M, Hagemen GJ, Borm PJ, Bast A, Haenen GRMM. A Planar Conformation and the Hydroxyl Groups in the B and C Rings Play a Pivotal Role in the Antioxidant Capacity of Quercetin and Quercetin Derivatives. Molecules. 2011; 16(11):9636-9650. https://doi.org/10.3390/molecules16119636
Chicago/Turabian StyleMoalin, Mohamed, Gino P. F. van Strijdonck, Maud Beckers, Geja J. Hagemen, Paul J. Borm, Aalt Bast, and Guido R. M. M. Haenen. 2011. "A Planar Conformation and the Hydroxyl Groups in the B and C Rings Play a Pivotal Role in the Antioxidant Capacity of Quercetin and Quercetin Derivatives" Molecules 16, no. 11: 9636-9650. https://doi.org/10.3390/molecules16119636
APA StyleMoalin, M., Strijdonck, G. P. F. v., Beckers, M., Hagemen, G. J., Borm, P. J., Bast, A., & Haenen, G. R. M. M. (2011). A Planar Conformation and the Hydroxyl Groups in the B and C Rings Play a Pivotal Role in the Antioxidant Capacity of Quercetin and Quercetin Derivatives. Molecules, 16(11), 9636-9650. https://doi.org/10.3390/molecules16119636