Eco-Friendly Methodology to Prepare N-Heterocycles Related to Dihydropyridines: Microwave-Assisted Synthesis of Alkyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylate and 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-dione

Abstract
:
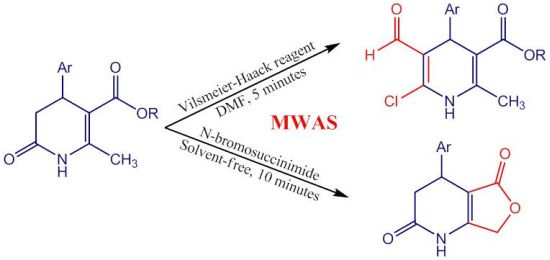
1. Introduction

2. Results and Discussion
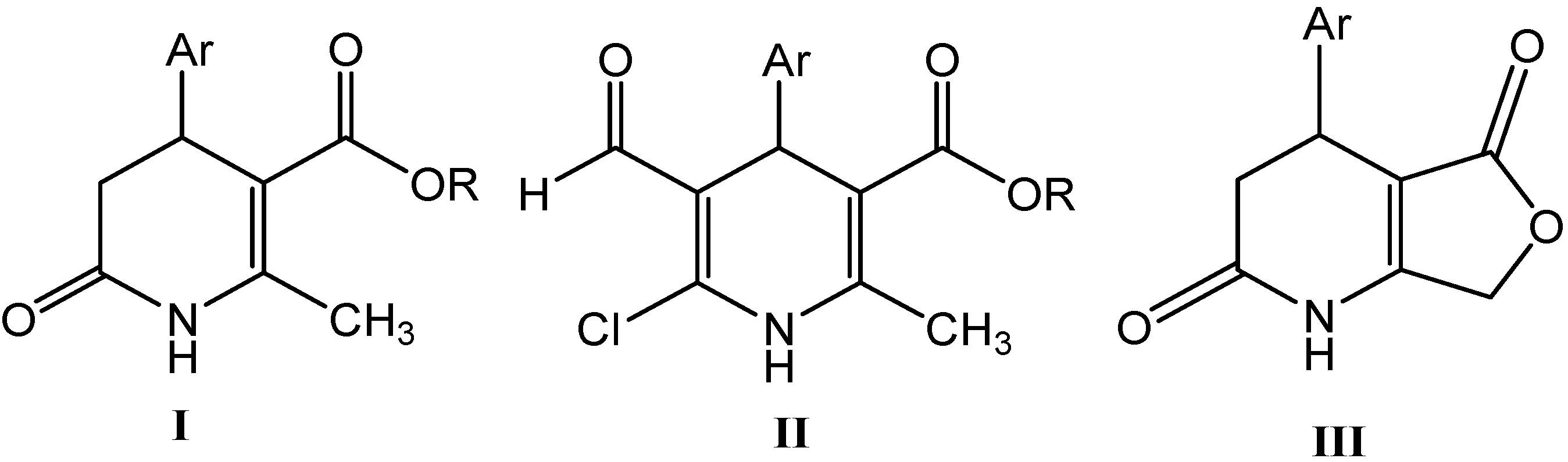
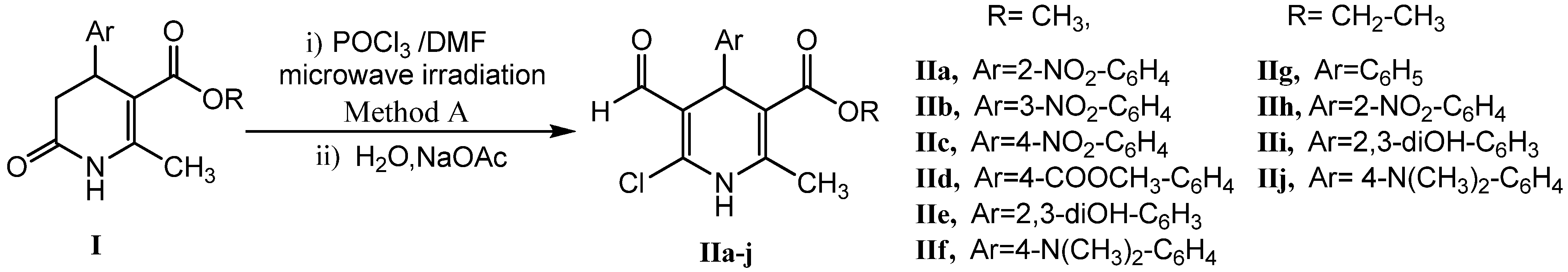
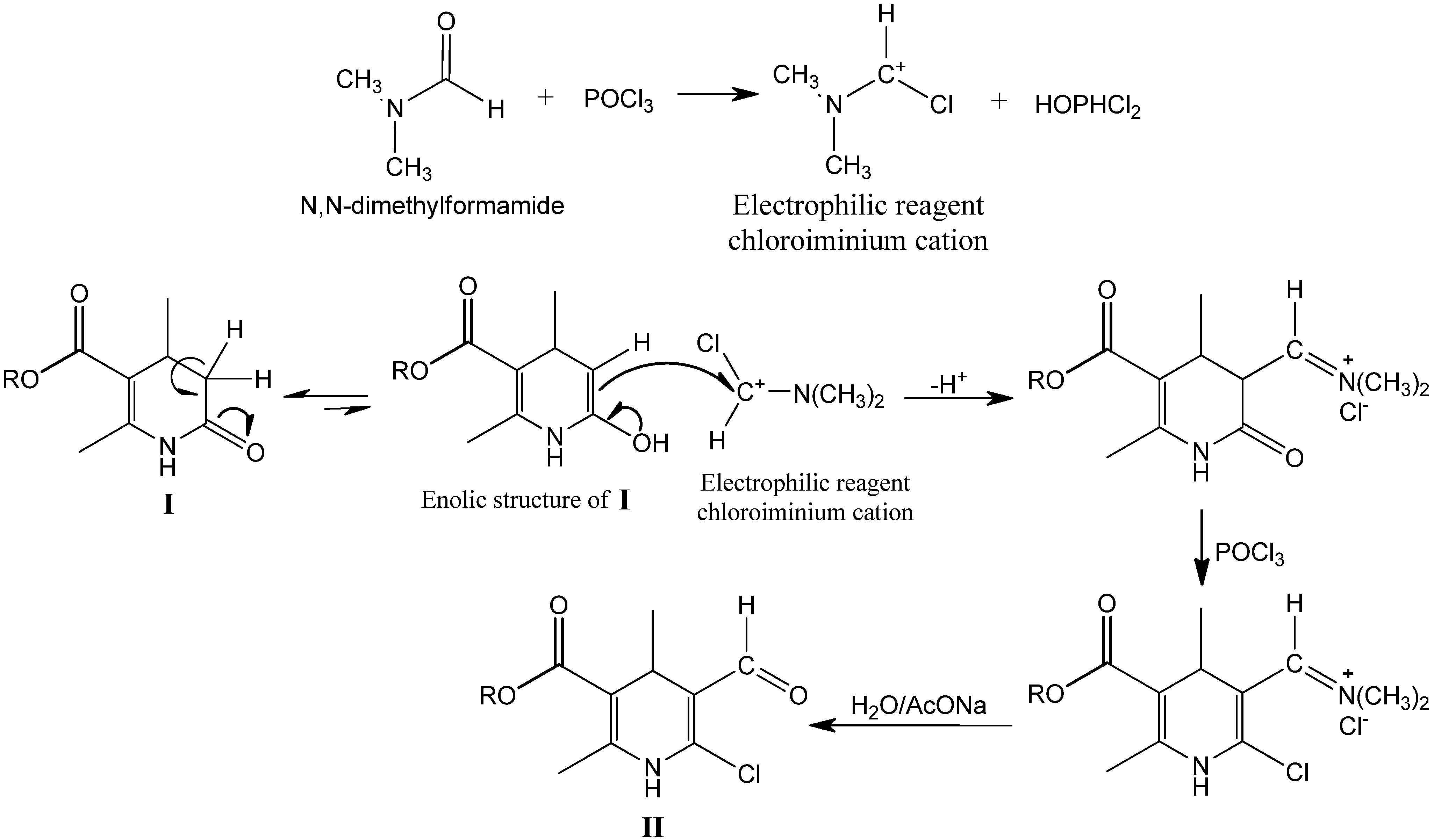
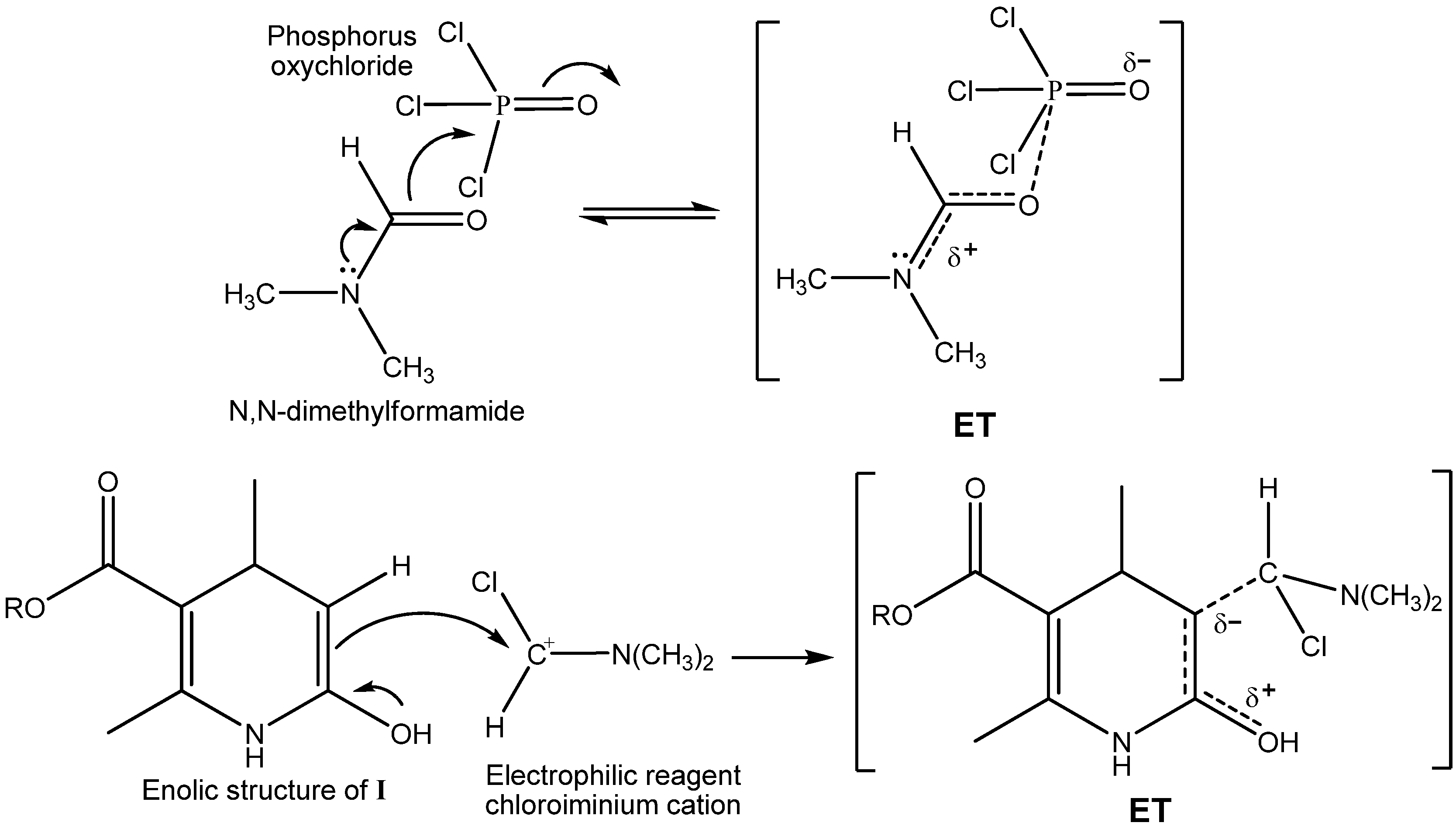
2.1. Microwave Assisted Synthesis (MWAS) of Methyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylates IIa–j


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | R | Ar | Method | T(°C) | t (min) | Yield (%) |
|---|---|---|---|---|---|---|
| IIa | CH3 | 2-NO2-C6H4 | A | 50 | 5 | 69 |
| B | RT | 1080 | 70 | |||
| IIb | CH3 | 3-NO2-C6H4 | A | 50 | 5 | 68 |
| B | RT | 1080 | 73 | |||
| IIc | CH3 | 4-NO2-C6H4 | A | 50 | 5 | 65 |
| B | RT | 1080 | 69 | |||
| IId | CH3 | 4-COOCH3-C6H4 | A | 50 | 5 | 63 |
| B | RT | 1080 | 73 | |||
| IIe | CH3 | 2,3-diOH-C6H4 | A | 50 | 5 | 70 |
| B | RT | 1080 | 63 | |||
| IIf | CH3 | 4- N(CH3)2-C6H4 | A | 50 | 5 | 60 |
| B | RT | 1080 | 68 | |||
| IIg | CH2CH3 | C6H5 | A | 50 | 5 | 62 |
| B | RT | 1080 | 75 | |||
| IIh | CH2CH3 | 2-NO2-C6H4 | A | 50 | 5 | 65 |
| B | RT | 1080 | 75 | |||
| IIi | CH2CH3 | 2,3-diOH-C6H4 | A | 50 | 5 | 63 |
| B | RT | 1080 | 71 | |||
| IIj | CH2CH3 | 4- N(CH3)2-C6H4 | A | 70 | 5 | 64 |
| B | RT | 1080 | 70 |


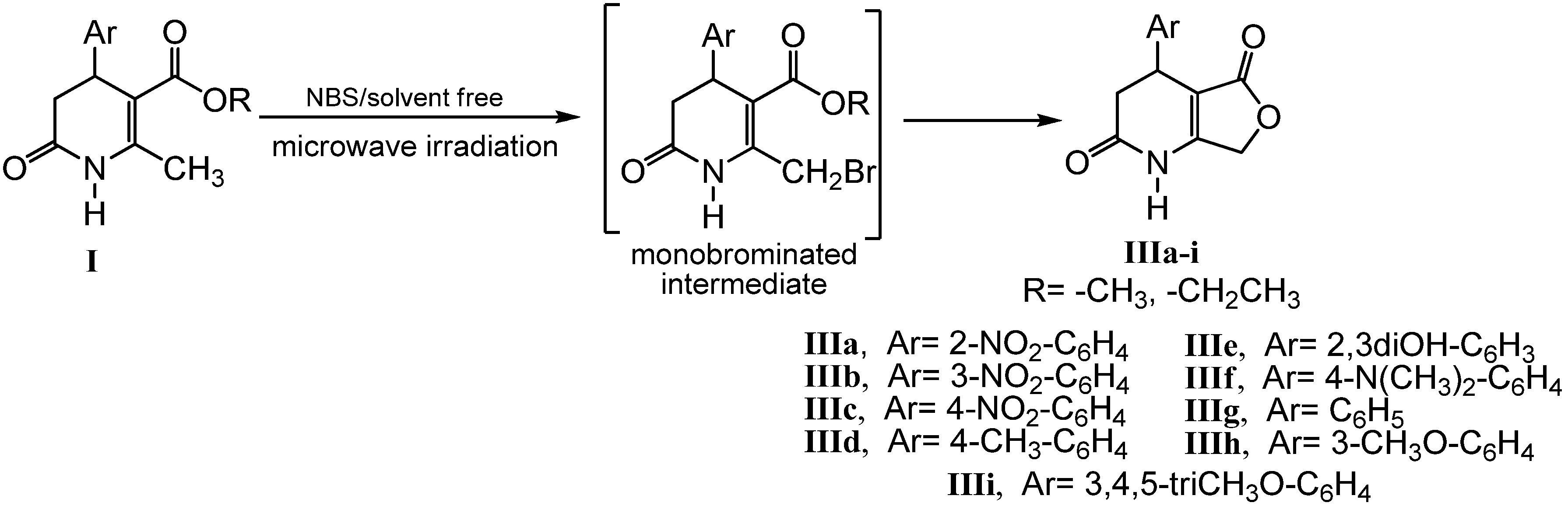
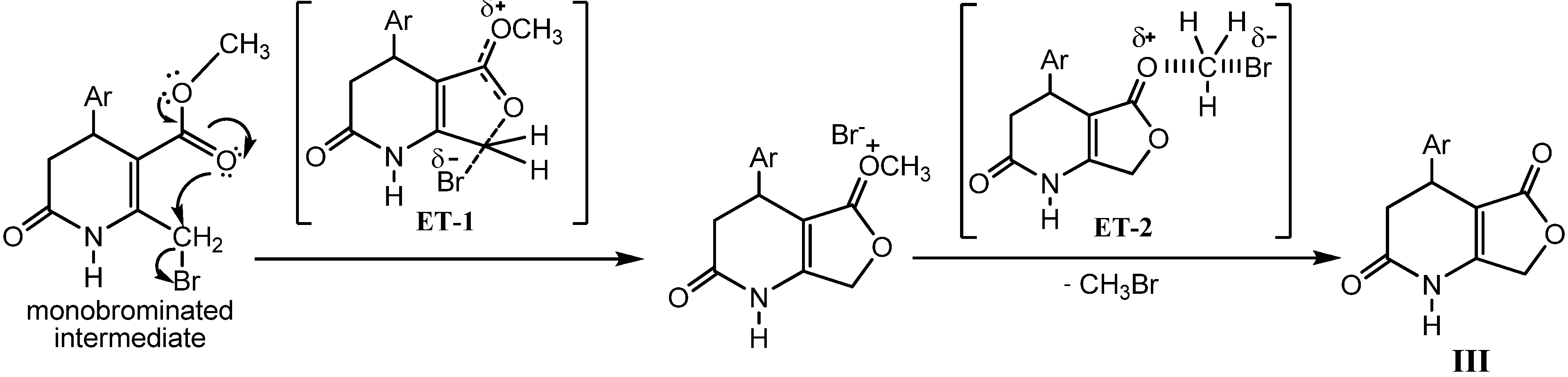
2.2. Microwave Assisted Synthesis (MWAS) of 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-diones IIIa–i

| Product | R | Ar | Method | T(°C) | t (min) | Yield (%) |
|---|---|---|---|---|---|---|
| IIIa | -CH3 | 2-NO2-C6H4 | A | 80 | 10 | 80 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 55 | ||
| -CH2CH3 | 62 | 720 | 60 | |||
| IIIb | -CH3 | 3-NO2-C6H4 | A | 80 | 10 | 79 |
| -CH2CH3 | 80 | 10 | 82 | |||
| -CH3 | B | 62 | 720 | 52 | ||
| -CH2CH3 | 62 | 720 | 58 | |||
| IIIc | -CH3 | 4-NO2-C6H4 | A | 80 | 10 | 82 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 55 | ||
| -CH2CH3 | 62 | 720 | 57 | |||
| IIId | -CH3 | 4-CH3-C6H4 | A | 80 | 10 | 89 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 61 | ||
| -CH2CH3 | 62 | 720 | 56 | |||
| IIIe | -CH3 | 2,3-diOH-C6H4 | A | 80 | 10 | 72 |
| -CH2CH3 | 80 | 10 | 79 | |||
| -CH3 | B | 62 | 720 | 52 | ||
| -CH2CH3 | 62 | 720 | 55 | |||
| IIIf | -CH3 | 4-N(CH3)2-C6H4 | A | 80 | 10 | 84 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 59 | ||
| -CH2CH3 | 62 | 720 | 53 | |||
| IIIg | -CH3 | C6H5 | A | 80 | 10 | 84 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 59 | ||
| -CH2CH3 | 62 | 720 | 61 | |||
| IIIh | -CH3 | 3-CH3O-C6H4 | A | 80 | 10 | 88 |
| -CH2CH3 | 80 | 10 | 85 | |||
| -CH3 | B | 62 | 720 | 51 | ||
| -CH2CH3 | 62 | 720 | 58 | |||
| IIIi | -CH3 | 3,4,5-triCH3O-C6H4 | A | 80 | 10 | 83 |
| -CH2CH3 | 80 | 10 | 81 | |||
| -CH3 | B | 62 | 720 | 63 | ||
| -CH2CH3 | 62 | 720 | 59 |

3. Experimental
3.1. General
3.2. General Procedure for the MWAS of 4-Arylsubstituted alkyl 1,4,5,6-Tetrahydro-2-methyl-6-oxopyridine-3-carboxylates I
3.3. General Procedure for the MWAS of Methyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylates IIa–j
3.4. General Procedure for the MWAS of 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-diones IIIa–i
4. Conclusions
Acknowledgements
References and Notes
- Wess, G.; Urmann, M.; Sickenberger, B. Medicinal chemistry: Challenges and opportunities. Angew. Chem. Int. Ed. 2001, 40, 3341–3350. [Google Scholar] [CrossRef]
- Muegge, I. Pharmacophore features of potential drugs. Chem.-Eur. J. 2002, 8, 1976–1981. [Google Scholar] [CrossRef]
- Muñoz, B.; Chen, C.; McDonald, I.A. Resin activation capture technology: Libraries from stabilized acyl-pyridinium on solid support. Biotechnol. Bioeng. 2000, 71, 78–84. [Google Scholar] [CrossRef]
- Pelish, H.E.; Westwood, N.J.; Feng, Y.; Kirchhausen, T.; Shair, M.D. Use of biomimetic diversity-oriented synthesis to discover galanthamine-like molecules with biological properties beyond those of the natural product. J. Am. Chem. Soc. 2001, 123, 6740–6741. [Google Scholar] [CrossRef]
- Ding, S.; Gray, N.S; Wu, X.; Ding, Q.; Schultz, P.G. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J. Am. Chem. Soc. 2002, 124, 1594–1596. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Simon, C.; Constantieux, T.; Rodríguez, J. Utilisation of 1,3-dicarbonyl derivatives in multicomponent reactions. Eur. J. Org. Chem. 2004, 24, 4957–4980. [Google Scholar]
- Hatamjafari, F. New protocol to synthesize spiro-1,4-dihydropyridines by using a multicomponent reaction of cyclohexanone, ethyl cyanoacetate, isatin, and primary amines under microwave irradiation. Syn. Commun. 2006, 36, 3563–3570. [Google Scholar] [CrossRef]
- Orru, R.V.A.; de Greef, M. Recent advances in solution-phase multicomponent methodology for the synthesis of heterocyclic compounds. Synthesis 2003, 1471–1499. [Google Scholar]
- Yamamoto, T.; Niwa, S.; Ohno, S.; Onishi, T.; Matsueda, H.; Koganei, H.; Uneyama, H.; Fujita, S.; Takeda, T.; Kito, M.; et al. Structure-activity relationship study of 1,4-dihydropyridine derivatives blocking N-type calcium channels. Bioorg. Med. Chem. Lett. 2006, 16, 798–805. [Google Scholar] [CrossRef]
- Rodríguez, H.; Martin, O.; Ochoa, E.; Suárez, M.; Reyes, O.; Garay, H.; Albericio, F.; Martin, N. Solid-phase synthesis and structural study of substituted 1,4,5,6-tetrahydro-6-oxopyridine-3-carboxylic acids. QSAR Comb. Sci. 2006, 25, 921–927. [Google Scholar] [CrossRef]
- lvarez, A.; Suárez, M.; Verdecia, Y.; Ochoa, E.; Barrie, B.; Pérez, R.; Díaz, M.; Martínez-Álvarez, R.; Molero, D.; Seoane, C.; et al. Synthesis and structural study of semicarbazone-containing 1,4-dihydropyridine. Heterocycles 2006, 68, 1631–1649. [Google Scholar] [CrossRef]
- Nekooeian, A.A.; Khalili, A.; Javidnia, K.; Mehdipour, A.R.; Miri, R. Antihypertensive effects of some new nitroxyalkyl 1,4-dihydropyridine derivatives in rat model of two-kidney, one-clip hypertension. Iran. J. Pharmaceut. Res. 2009, 8, 193–199. [Google Scholar]
- Polshettiwar, V.; Varma, R.S. Microwave-assisted synthesis of bio-active heterocycles in aqueous media. RSC Green Chem. Ser. 2010, 7, 91–122. [Google Scholar]
- Sivamurugan, V.; Vinu, A.; Palanichamy, M.; Murugesan, V. Rapid and cleaner synthesis of 1,4-dihydropyridines in aqueous medium. Heteroatom Chem. 2006, 17, 267–271. [Google Scholar] [CrossRef]
- Narsaiah, A.; Venkat, N.B. Glycerine-CeCl37H2O: An efficient recyclable reaction medium for the synthesis of Hantzsch Pyridines. Asian J. Chem. 2010, 22, 8099–8106. [Google Scholar]
- Kranjc, K.; Kocevar, M. Microwave-assisted organic synthesis. General considerations and transformations of heterocyclic compounds. Curr. Org. Chem. 2010, 14, 1050–1074. [Google Scholar] [CrossRef]
- Sekhon, B.S. Microwave-assisted pharmaceutical synthesis. An overview. Int. J. Pharm. Tech. Res. 2010, 2, 827–833. [Google Scholar]
- Santagada, V.; Frecentese, F.; Perissutti, E.; Fiorino, F.; Severino, B.; Caliendo, G. Microwave assisted synthesis: A new technology in drug discovery. Mini-Rev. Med. Chem. 2009, 9, 340–358. [Google Scholar]
- Rodríguez, H.; Suárez, M.; Pérez, R.; Petit, A.; Loupy, A. Solvent-free synthesis of 4-aryl substituted 5-alkoxycarbonyl-6-methyl-3,4-dihydropyridones under microwave irradiation. Tetrahedron Lett. 2003, 44, 3709. [Google Scholar] [CrossRef]
- Rodríguez, H.; Coro, J.; Lam, A.; Salfrán, E.; Rodríguez-Salarrichs, J.; Suárez, M.; Albericio, F.; Martin, N. High-throughput preparation of alkyl 4-aryl substituted-2-methyl-6-thioxo-1,4,5,6-tetrahydropyridine-3-carboxylates under microwave irradiation. ARKIVOC 2011, ix, 125–141. [Google Scholar]
- CEM Tomorrow’s Science Today. Available online: http://www.cem.com/content656.html (accessed on 16 November 2011).
- Suárez, M.; Verdecia, Y.; Illescas, B.; Martínez-Alvarez, R.; Álvarez, A.; Ochoa, E.; Seoane, C.; Kayali, N; Martin, N. Synthesis and study of novel fulleropyrrolidines bearing biologically active 1,4-dihydropyridines. Tetrahedron 2003, 59, 9179–9186. [Google Scholar] [CrossRef]
- Suárez, M.; Martin, N.; Martínez, R.; Verdecia, Y.; Molero, D.; Alba, L.; Seoane, C.; Ochoa, E. 1H and 13C spectral assignment of o-chloroformyl substituted 1,4-dihydropyridine derivatives. Magn. Reson. Chem. 2002, 40, 303–306. [Google Scholar] [CrossRef]
- Verdecia, Y.; Suárez, M.; Morales, A.; Rodríguez, E.; Ochoa, E.; González, L.; Martín, N.; Quinteiro, M.; Seoane, C.; Soto, J.L. Synthesis of methyl 4-aryl-6-methyl-4,7-dihydro-1H-pyrazolo[3,4-b]pyridine-5-carboxylates from methyl 4-aryl-6-methyl-2-oxo-1,2,3,4-tetrahydropyridine-5-carboxylates. J. Chem. Soc. Perkin Trans. 1 1996, 947–951. [Google Scholar]
- Ruiz, E.; Rodríguez, H.; Coro, J.; Niebla, V.; Rodríguez, A.; Martínez, R.; Novoa, H.; Suárez, M.; Martín, N. Efficient sonochemical synthesis of alkyl 4-aryl-6-chloro-5-formyl-2-methyl-1,4-dihydropyridines-3-carboxylate derivatives. Ultrason. Sonochem. 2011, 8, 32–36. [Google Scholar]
- Gupton, J.T.; Banner, E.J.; Sartin, M.D.; Coppock, M.B.; Hempel, J.E.; Kharlamova, A.; Fisher, D.C.; Giglio, B.C.; Smith, K.L.; Keough, M.J.; et al. The application of vinylogous iminium salt derivatives and microwave accelerated Vilsmeier-Haack reactions to efficient relay syntheses of the polycitone and storniamide natural products. Tetrahedron 2008, 64, 5246–5253. [Google Scholar]
- Raghavendra, M.; Bhojya Naik, H.S.; Sherigara, B.S. One pot synthesis of some new 2-hydrazino-[1,3,4]thiadiazepino[7,6-b]quinolines under microwave irradiation conditions. ARKIVOK 2006, 15, 153–159. [Google Scholar]
- Al-Saleh, B.; Hilmy, N.M.; El-Apasery, M.A.; Elnagdi, M.H. Microwaves in organic synthesis: synthesis of pyridazinones, phthalazinones and pyridopyridazinones from 2-oxo-arylhydrazones under microwave irradiation. J. Heterocycl. Chem. 2006, 43, 1575–1581. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. Nonthermal effects of microwaves in organic synthesis. In Microwave in Organic Synthesis, 1st; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2002; pp. 61–110. [Google Scholar]
- Eissa, A.A.M.; Farag, N.A.H.; Soliman, G.A.H. Synthesis, biological evaluation and docking studies of novel benzopyranone congeners for their expected activity as anti-inflammatory, analgesic and antipyretic agents. Bioorg. Med. Chem. 2009, 17, 5059–5070. [Google Scholar] [CrossRef]
- 32 Chang, C.C.; Cao, S.; Kang, S.; Kai, L.; Tian, X.; Pandey, P.; Dunne, S.F.; Luan, C.-H.; Surmeier, D.J.; Silverman, R.B. Antagonism of 4-substituted 1,4-dihydropyridine-3,5-dicarboxylates toward voltage-dependent L-type Ca2+ channels CaV1.3 and CaV1.2. Bioorg. Med. Chem. 2010, 18, 3147–3158. [Google Scholar]
- Morales, A.; Ochoa, E.; Suarez, M.; Verdecia, Y.; Gonzalez, L.; Martin, N.; Quinteiro, M.; Seoane, C.; Soto, J.L. Novel hexahydrofuro[3,4-b]-2(1H)-pyridones from 4-aryl substituted 5-alkoxycarbonyl-6-methyl-3,4-dihydropyridones. J. Heterocycl. Chem. 1996, 33, 103–107. [Google Scholar] [CrossRef]
- Ochoa, E.; Suárez, M.; Verdecia, Y.; Pita, B.; Martin, N.; Quinteiro, M.; Seoane, C.; Soto, J.L.; Duque, J.; Pomes, R. Structural study of 3,4-dihydropyridones and furo[3,4-b]-2(1H)-pyridones as potential calcium channel modulators. Tetrahedron 1998, 54, 12409–12420. [Google Scholar] [CrossRef]
- Suárez, M.; Martínez-Alvarez, R.; Martín, N.; Verdecia, Y.; Ochoa, E.; Alba, L.; Seoane, C.; Kayali, N. Electrospray ionisation and ion-trap fragmentation of substituted 3,4-dihydro-2(1H)-pyridin-2-ones. Rapid Commun. Mass Spectrom. 2002, 16, 749–754. [Google Scholar] [CrossRef]
- Curran, T. Wohl-Ziegler reaction. Name React. Homologations 2009, 1, 661–673. [Google Scholar]
- Young, S.D. Facile conversion of Hantzsch type 4-aryl-2,6-dimethyl-1,4-dihydropyridine-3,5-carboxylates into 4-aryl-2-methyl-5-oxo-1,4,5,7-tetrahydrofuro[3,4-b]pyridine-3-carboxylates. Synthesis 1984, 7, 617–618. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rodríguez, H.; Martin, O.; Suarez, M.; Martín, N.; Albericio, F. Eco-Friendly Methodology to Prepare N-Heterocycles Related to Dihydropyridines: Microwave-Assisted Synthesis of Alkyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylate and 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-dione. Molecules 2011, 16, 9620-9635. https://doi.org/10.3390/molecules16119620
Rodríguez H, Martin O, Suarez M, Martín N, Albericio F. Eco-Friendly Methodology to Prepare N-Heterocycles Related to Dihydropyridines: Microwave-Assisted Synthesis of Alkyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylate and 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-dione. Molecules. 2011; 16(11):9620-9635. https://doi.org/10.3390/molecules16119620
Chicago/Turabian StyleRodríguez, Hortensia, Osnieski Martin, Margarita Suarez, Nazario Martín, and Fernando Albericio. 2011. "Eco-Friendly Methodology to Prepare N-Heterocycles Related to Dihydropyridines: Microwave-Assisted Synthesis of Alkyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylate and 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-dione" Molecules 16, no. 11: 9620-9635. https://doi.org/10.3390/molecules16119620
APA StyleRodríguez, H., Martin, O., Suarez, M., Martín, N., & Albericio, F. (2011). Eco-Friendly Methodology to Prepare N-Heterocycles Related to Dihydropyridines: Microwave-Assisted Synthesis of Alkyl 4-Arylsubstituted-6-chloro-5-formyl-2-methyl-1,4-dihydropyridine-3-carboxylate and 4-Arylsubstituted-4,7-dihydrofuro[3,4-b]pyridine-2,5(1H,3H)-dione. Molecules, 16(11), 9620-9635. https://doi.org/10.3390/molecules16119620