Miniproteins as Phage Display-Scaffolds for Clinical Applications

Abstract
:1. Introduction

2. Properties of Miniprotein Scaffolds
- (i) The monomeric, small polypeptide chain has to be highly stable against enzymatic degradation, easy to engineer, and efficiently produced by recombinant expression or solid-phase peptide synthesis;
- (ii) The formation of a defined, rigid three-dimensional topology by secondary structural elements is mandatory;
- (iii) The tolerance to sequence variations or insertions within the recognition site has to conserve the protein folding or stability;
- (iv) The accessibility to a surface domain or binding pocket as a recognition site has to be ensured;
- (iv) A well-defined hydrophobic core that contributes to the high free energy of folding is advantageous.
2.1. Advantages of miniprotein scaffolds
2.2. Classification of miniprotein scaffolds

2.3. Scaffold library design for phage display screening
2.4. Miniprotein engineering
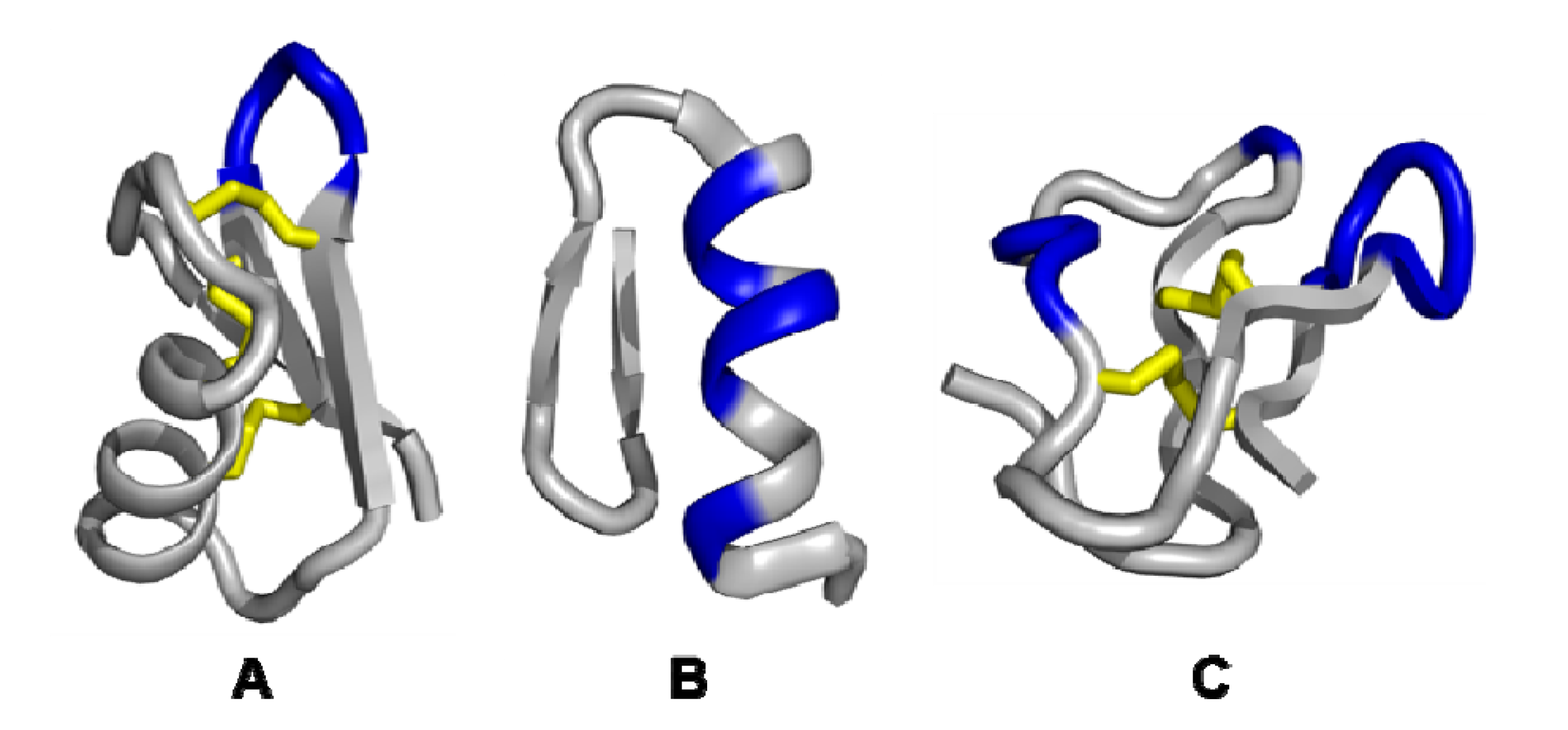
3. Examples of Miniprotein Display Scaffolds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold name | Acronym | Scaffold Category | Secondary structure motifs | Size (aa) | Random positions | Origin | Binding specificity (target) | References |
|---|---|---|---|---|---|---|---|---|
| EETI-II | Cyclotide | A | CSB / 3 SS | 28 | 6 aa / loop | Plant (Ecaballium elaterium) | Chymotrypsin, trypsin, integrins | [44,45] |
| Min-23 | Knottin | A | CSB / 2 SS | 23 | 8 -10 aa / β-turn | Rational design | MAbs, HIV-1 Nef, Tom70, AMA-1 | [40] |
| Scorpion toxin | Knottin | A | CSB / 3 SS | 37 | 4 aa / loop | Scorpion (Leiurus quinquestriatus hebraeus) | Acetylcholin receptor, MAbs | [46,47] |
| SFTI-I | Knottin | A | circular, 1 SS, 2 β-sheets | 14 | 6 -8 aa / loop | Plant (Helianthus annuus) | Trysin, chymotrypsin | [48,49] |
| Z domain | Affibody | B | 3 α-helical bundle | 58 | 13 aa / 2 α-helices | Bacteria (Staphylococcus aureus) | Taq polymerase, Her-2/neu, CD28 | [20,50,51] |
| Zinc finger | - | B | α-helix / β-sheet / Zn2+ | 26 | 5 aa / α-helix | Frog (Xenopus laevis) | MAbs | [52] |
| CBD | Knottin | C | CSB / 3 SS | 36 | 11 + 7 aa / 3 loops | Fungus (Trichoderma reesei) | Alkaline phospathase, α-amylase | [53,54] |
| DX-88 | Kunitz domain | C | α-helix / 2 β-sheets / 3 SS | 58 | 5 + 4 aa / 2 loops | Human (type VI collagen) | Plasma kalikrein | [32,55,56,57] |
| Tendamistat | Knottin | C | β-sandwich / 2 SS | 74 | 3 + 3 + 6 aa / 3loops | Bacteria (Streptomyces tendae) | α-Amylase, MAbs, integrins | [58,59] |
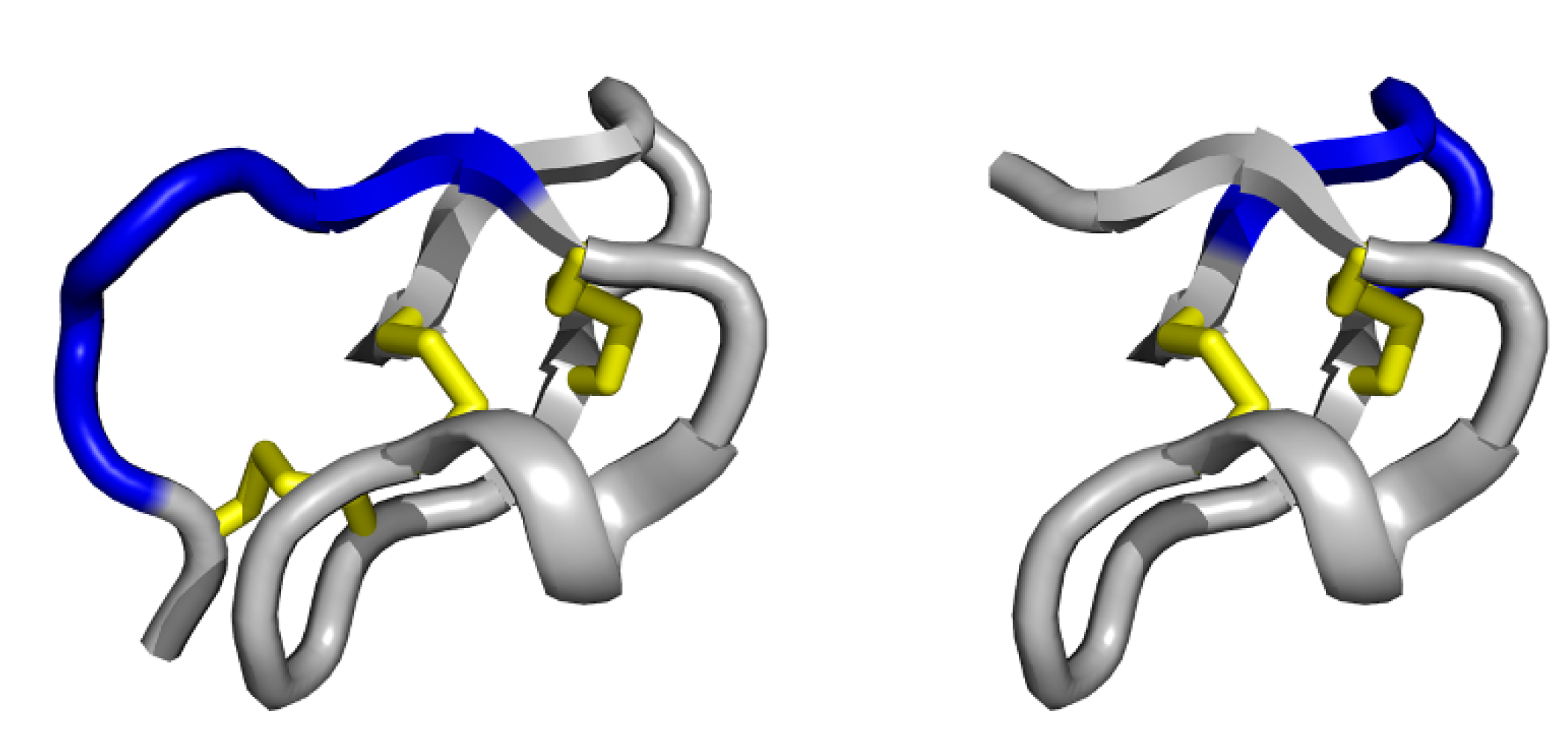
3.1. Single exposed loop scaffolds

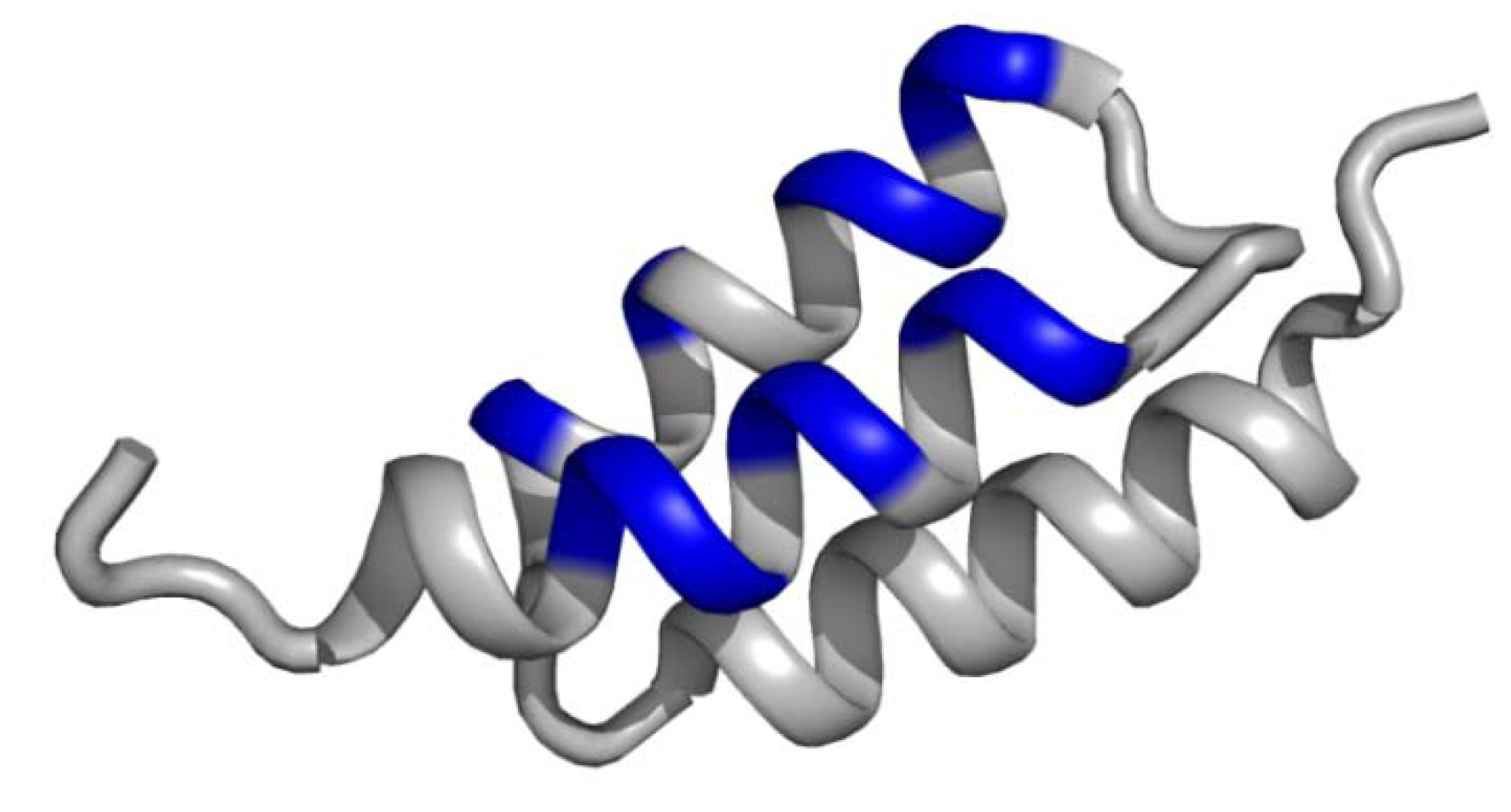
3.2. Non-contiguous surface domain scaffolds

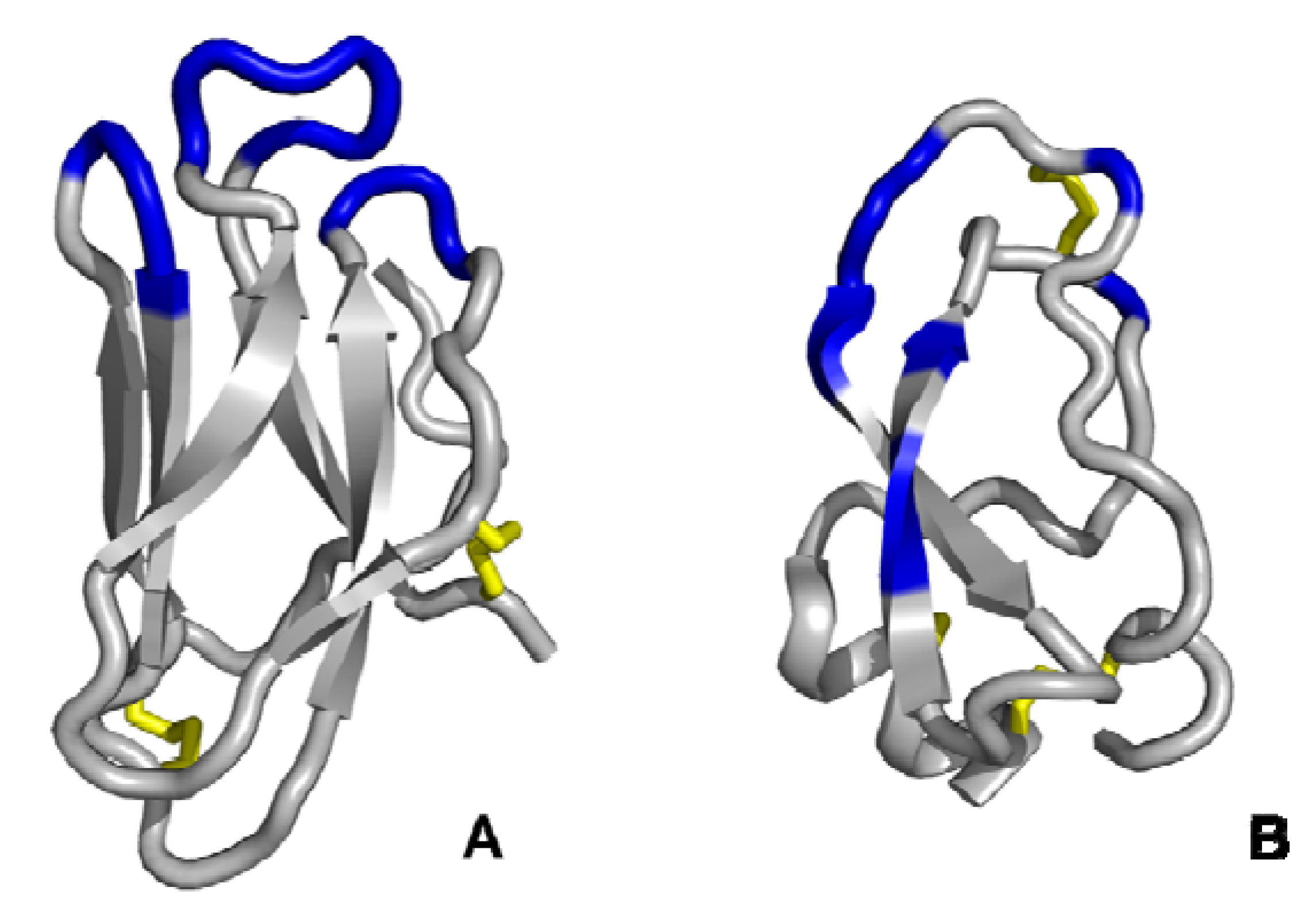
3.3. Multiple discontinuous domain scaffolds

4. Conclusions
Acknowledgements
References
- Skerra, A. Imitating the humoral immune response. Curr. Opin. Chem. Biol. 2003, 7, 683–693. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Pluckthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. [Google Scholar] [CrossRef]
- Descotes, J. Immunotoxicity of monoclonal antibodies. mAbs 2009, 1, 104–111. [Google Scholar] [CrossRef]
- Wu, A.M.; Senter, P.D. Arming antibodies: Prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar]
- Huang, L.; Muyldermans, S.; Saerens, D. Nanobodies: Proficient tools in diagnostics. Expert Rev Mol Diagn 2010, 10, 777–785. [Google Scholar] [CrossRef]
- Van Bockstaele, F.; Holz, J.B.; Revets, H. The development of nanobodies for therapeutic applications. Curr. Opin. Investig. Drugs 2009, 10, 1212–1224. [Google Scholar]
- Skerra, A. Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 2007, 18, 295–304. [Google Scholar]
- Skerra, A. Engineered protein scaffolds for molecular recognition. J. Mol. Recognit. 2000, 13, 167–187. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar]
- Young, K.H. Yeast two-hybrid: So many interactions, (in) so little time. Biol. Reprod. 1998, 58, 302–311. [Google Scholar] [CrossRef]
- Roberts, R.W. Totally in vitro protein selection using mRNA-protein fusions and ribosome display. Curr. Opin. Chem. Biol. 1999, 3, 268–273. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMol Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Kolmar, H. Engineered cystine-knot miniproteins for diagnostic applications. Expert Rev. Mol. Diagn. 2010, 10, 361–368. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef]
- Batra, S.K.; Jain, M.; Wittel, U.A.; Chauhan, S.C.; Colcher, D. Pharmacokinetics and biodistribution of genetically engineered antibodies. Curr. Opin. Biotechnol. 2002, 13, 603–608. [Google Scholar] [CrossRef]
- Russeva, M.G.; Adams, G.P. Radioimmunotherapy with engineered antibodies. Expert. Opin. Biol. Ther. 2004, 4, 217–231. [Google Scholar] [CrossRef]
- Rudin, M.; Weissleder, R. Molecular imaging in drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 123–131. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A.; Nilsson, F.Y.; Feldwisch, J.; Wennborg, A.; Abrahmsen, L. Affibody molecules: Potential for in vivo imaging of molecular targets for cancer therapy. Expert Opin. Biol. Ther. 2007, 7, 555–568. [Google Scholar] [CrossRef]
- Haberkorn, U.; Eisenhut, M.; Altmann, A.; Mier, W. Endoradiotherapy with peptides - status and future development. Curr. Med. Chem. 2008, 15, 219–234. [Google Scholar]
- Zoller, F.; Eisenhut, M.; Haberkorn, U.; Mier, W. Endoradiotherapy in cancer treatment - basic concepts and future trends. Eur. J. Pharmacol. 2009, 625, 55–62. [Google Scholar] [CrossRef]
- Marshall, G.R. Three-dimensional structure of peptide--protein complexes: Implications for recognition. Curr. Opin. Structl. Biol. 1992, 2, 904–919. [Google Scholar] [CrossRef]
- Nygren, P.A.; Skerra, A. Binding proteins from alternative scaffolds. J. Immunol. Methods 2004, 290, 3–28. [Google Scholar] [CrossRef]
- Virnekas, B.; Ge, L.; Pluckthun, A.; Schneider, K.C.; Wellnhofer, G.; Moroney, S.E. Trinucleotide phosphoramidites: Ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucl. Acids Res. 1994, 22, 5600–5607. [Google Scholar] [CrossRef]
- Krumpe, L.R.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. Trinucleotide cassettes increase diversity of T7 phage-displayed peptide library. BMC Biotechnol. 2007, 7, 65. [Google Scholar] [CrossRef]
- Rodi, D.J.; Soares, A.S.; Makowski, L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J. Mol. Biol. 2002, 322, 1039–1052. [Google Scholar] [CrossRef]
- Pannekoek, H.; Meijer, M.; Gaardsvoll, H.; Zonneveld, A.J. Functional display of proteins, mutant proteins, fragments of proteins and peptides on the surface of filamentous (bacterio) phages: A review. Cytotechnology 1995, 18, 107–112. [Google Scholar]
- Hanes, J.; Pluckthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- McLafferty, M.A.; Kent, R.B.; Ladner, R.C.; Markland, W. M13 bacteriophage displaying disulfide-constrained microproteins. Gene 1993, 128, 29–36. [Google Scholar] [CrossRef]
- Sidhu, S.S. Engineering M13 for phage display. Biomol. Eng. 2001, 18, 57–63. [Google Scholar]
- Roberts, B.L. Directed evolution of a protein: Selection of potent neutrophil elastase inhibitors displayed on M13 fusion phage. Proc. Natl. Acad. Sci. USA 1992, 89, 2429–2433. [Google Scholar] [CrossRef]
- Sabatino, G.; Papini, A.M. Advances in automatic, manual and microwave-assisted solid-phase peptide synthesis. Curr. Opin. Drug Discov. Devel. 2008, 11, 762–770. [Google Scholar]
- Hackenberger, C.P.; Schwarzer, D. Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem. Int. Ed. Eng. 2008, 47, 10030–10074. [Google Scholar] [CrossRef]
- Baslé, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar]
- Brechbiel, M.W. Bifunctional chelates for metal nuclides. Q J. Nucl. Med. Mol. Imaging 2008, 52, 166–173. [Google Scholar]
- Walker, J.M. Basic Protein and Peptide Protocols; Springer: Berlin, Germany, 1994; Volume 32, pp. 441–448. [Google Scholar]
- Baltzer, L.; Nilsson, H.; Nilsson, J. De novo design of proteins - what are the rules? Chem. Rev. 2001, 101, 3153–3163. [Google Scholar] [CrossRef]
- Buchner, J.; Moroder, L. Oxidative Folding of Peptides and Proteins; RSC Publishing: Cambridge, UK, 2009. [Google Scholar]
- Souriau, C.; Chiche, L.; Irving, R.; Hudson, P. New binding specificities derived from Min-23, a small cystine-stabilized peptidic scaffold. Biochemistry 2005, 44, 7143–7155. [Google Scholar] [CrossRef]
- Legendre, D. TEM-1 [beta]-lactamase as a scaffold for protein recognition and assay. Protein Sci. 2002, 11, 1506–1518. [Google Scholar]
- Schlehuber, S.; Skerra, A. Lipocalins in drug discovery: From natural ligand-binding proteins to [ldquo]anticalins[rdquo]. Drug Discov. Today 2005, 10, 23–33. [Google Scholar] [CrossRef]
- Nixon, A.E.; Wood, C.R. Engineered protein inhibitors of proteases. Curr. Opin. Drug Discov. Devel. 2006, 9, 261–268. [Google Scholar]
- Christmann, A.; Walter, K.; Wentzel, A.; Kratzner, R.; Kolmar, H. The cystine knot of a squash-type protease inhibitor as a structural scaffold for Escherichia coli cell surface display of conformationally constrained peptides. Protein Eng. 1999, 12, 797–806. [Google Scholar] [CrossRef]
- Silverman, A.P.; Levin, A.M.; Lahti, J.L.; Cochran, J.R. Engineered cystine-knot peptides that bind alpha(v)beta(3) integrin with antibody-like affinities. J. Mol. Biol. 2009, 385, 1064–1075. [Google Scholar] [CrossRef]
- Vita, C.; Roumestand, C.; Toma, F.; Menez, A. Scorpion toxins as natural scaffolds for protein engineering. Proc. Natl. Acad. Sci. USA 1995, 92, 6404–6408. [Google Scholar]
- Vita, C.; Vizzavona, J.; Drakopoulou, E.; Zinn-Justin, S.; Gilquin, B.; Menez, A. Novel miniproteins engineered by the transfer of active sites to small natural scaffolds. Biopolymers 1998, 47, 93–100. [Google Scholar] [CrossRef]
- Boy, R.G.; Mier, W.; Nothelfer, E.M.; Altmann, A.; Eisenhut, M.; Kolmar, H.; Tomaszowski, M.; Kramer, S.; Haberkorn, U. Sunflower trypsin inhibitor 1 derivatives as molecular scaffolds for the development of novel peptidic radiopharmaceuticals. Mol. Imaging Biol. 2010, 12, 377–385. [Google Scholar] [CrossRef]
- Korsinczky, M.L.J.; Schirra, H.J.; Rosengren, K.J.; West, J.; Condie, B.A.; Otvos, L.; Anderson, M.A.; Craik, D.J. Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J. Mol. Biol. 2001, 311, 579–591. [Google Scholar] [CrossRef]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Stahl, S.; Uhlen, M.; Nygren, P.A. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar]
- Bianchi, E.A. Conformationally homogeneous combinatorial peptide library. J. Mol. Biol. 1995, 247, 154–160. [Google Scholar] [CrossRef]
- Lehtio, J.; Teeri, T.T.; Nygren, P.A. Alpha-amylase inhibitors selected from a combinatorial library of a cellulose binding domain scaffold. Proteins 2000, 41, 316–322. [Google Scholar]
- Smith, G.P. Small binding proteins selected from a combinatorial repertoire of knottins displayed on phage. J. Mol. Biol. 1998, 277, 317–332. [Google Scholar] [CrossRef]
- Dennis, M.S.; Lazarus, R.A. Kunitz domain inhibitors of tissue factor-factor VIIa. II. Potent and specific inhibitors by competitive phage selection. J. Biol. Chem. 1994, 269, 22137–22144. [Google Scholar]
- Ley, A.C.; Markland, W.; Ladner, R.C. Obtaining a family of high-affinity, high-specificity protein inhibitors of plasmin and plasma kallikrein. Mol. Divers. 1996, 2, 119–124. [Google Scholar]
- Markland, W.; Ley, A.C.; Ladner, R.C. Iterative optimization of high-affinity protease inhibitors using phage display. 2. Plasma kallikrein and thrombin. Biochemistry 1996, 35, 8058–8067. [Google Scholar] [CrossRef]
- Li, R.; Hoess, R.H.; Bennett, J.S.; DeGrado, W.F. Use of phage display to probe the evolution of binding specificity and affinity in integrins. Protein Eng. 2003, 16, 65–72. [Google Scholar] [CrossRef]
- McConnell, S.J.; Hoess, R.H. Tendamistat as a scaffold for conformationally constrained phage peptide libraries. J. Mol. Biol. 1995, 250, 460–470. [Google Scholar]
- Craik, D.J.; Clark, R.J.; Daly, N.L. Potential therapeutic applications of the cyclotides and related cystine knot mini-proteins. Expert Opin. Investig. Drugs 2007, 16, 595–604. [Google Scholar] [CrossRef]
- Craik, D.J.; Simonsen, S.; Daly, N.L. The cyclotides: Novel macrocyclic peptides as scaffolds in drug design. Curr. Opin. Drug Discov. Develop. 2002, 5, 251–260. [Google Scholar]
- Wentzel, A.; Christmann, A.; Adams, T.; Kolmar, H. Display of passenger proteins on the surface of Escherichia coli K-12 by the enterohemorrhagic E. coli intimin EaeA. J. Bacteriol. 2001, 183, 7273–7284. [Google Scholar] [CrossRef]
- Le Nguyen, D.; Heitz, A.; Chiche, L.; Castro, B.; Boigegrain, R.A.; Favel, A.; Coletti-Previero, M.A. Molecular recognition between serine proteases and new bioactive microproteins with a knotted structure. Biochimie 1990, 72, 431–435. [Google Scholar] [CrossRef]
- Craik, D.J.; Cemazar, M.; Daly, N.L. The chemistry and biology of cyclotides. Curr. Opin. Drug Discov. Develop. 2007, 10, 176–184. [Google Scholar]
- Craik, D.J.; Cemazar, M.; Wang, C.K.; Daly, N.L. The cyclotide family of circular miniproteins: Nature's combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar]
- Heitz, A.; Le-Nguyen, D.; Chiche, L. Min-21 and min-23, the smallest peptides that fold like a cystine-stabilized beta-sheet motif: Design, solution structure, and thermal stability. Biochemistry 1999, 38, 10615–10625. [Google Scholar] [CrossRef]
- Heitz, A.; Chiche, L.; Le-Nguyen, D.; Castro, B. Folding of the Squash Trypsin Inhibitor EETI II. Eur. J. Biochem. 1995, 233, 837–846. [Google Scholar]
- Reiss, S.; Sieber, M.; Oberle, V.; Wentzel, A.; Spangenberg, P.; Claus, R.; Kolmar, H.; Losche, W. Inhibition of platelet aggregation by grafting RGD and KGD sequences on the structural scaffold of small disulfide-rich proteins. Platelets 2006, 17, 153–157. [Google Scholar] [CrossRef]
- Kimura, R.H.; Levin, A.M.; Cochran, F.V.; Cochran, J.R. Engineered cystine knot peptides that bind alphavbeta3, alphavbeta5, and alpha5beta1 integrins with low-nanomolar affinity. Proteins 2009, 77, 359–369. [Google Scholar] [CrossRef]
- Kimura, R.H.; Cheng, Z.; Gambhir, S.S.; Cochran, J.R. Engineered knottin peptides: A new class of agents for imaging integrin expression in living subjects. Cancer Res. 2009, 69, 2435–2442. [Google Scholar] [CrossRef]
- Nielsen, C.H.; Kimura, R.H.; Withofs, N.; Tran, P.T.; Miao, Z.; Cochran, J.R.; Cheng, Z.; Felsher, D.; Kjaer, A.; Willmann, J.K.; Gambhir, S.S. PET imaging of tumor neovascularization in a transgenic mouse model with a novel 64Cu-DOTA-knottin peptide. Cancer Res. 2010, 70, 9022–9030. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Korsinczky, M.J.L.; Clark, R.J.; Foley, F.; Craik, D.J. Sunflower trypsin inhibitor-1, proteolytic studies on a trypsin inhibitor peptide and its analogs. J. Pept. Sci. 2010, 94, 665–672. [Google Scholar]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef]
- Nord, K.; Nilsson, J.; Nilsson, B.; Uhlen, M.; Nygren, P.A. A combinatorial library of an alpha-helical bacterial receptor domain. Protein Eng. 1995, 8, 601–608. [Google Scholar] [CrossRef]
- Nygren, P.A. Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008, 275, 2668–2676. [Google Scholar] [CrossRef]
- Ronnmark, J.; Hansson, M.; Nguyen, T.; Uhlen, M.; Robert, A.; Stahl, S.; Nygren, P.A. Construction and characterization of affibody-Fc chimeras produced in Escherichia coli. J. Immunol. Methods 2002, 261, 199–211. [Google Scholar] [CrossRef]
- Engfeldt, T.; Renberg, B.; Brumer, H.; Nygren, P.A.; Karlstrom, A.E. Chemical synthesis of triple-labelled three-helix bundle binding proteins for specific fluorescent detection of unlabelled protein. ChemBioChem 2005, 6, 1043–1050. [Google Scholar] [CrossRef]
- Nissen, F.; Kraft, T.E.; Ruppert, T.; Eisenhut, M.; Haberkorn, U.; Mier, W. Hot or not - the influence of elevated temperature and microwave irradiation on the solid phase synthesis of an affibody. Tetrahedron Lett. 2010, 51, 6216–6219. [Google Scholar]
- Tolmachev, V. Imaging of HER-2 overexpression in tumors for guiding therapy. Curr. Pharm. Des. 2008, 14, 2999–3019. [Google Scholar]
- Wikman, M.; Steffen, A.C.; Gunneriusson, E.; Tolmachev, V.; Adams, G.P.; Carlsson, J.; Stahl, S. Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng. Des. Sel. 2004, 17, 455–462. [Google Scholar] [CrossRef]
- Engfeldt, T.; Orlova, A.; Tran, T.; Bruskin, A.; Widstrom, C.; Karlstrom, A.E.; Tolmachev, V. Imaging of HER2-expressing tumours using a synthetic Affibody molecule containing the 99mTc-chelating mercaptoacetyl-glycyl-glycyl-glycyl (MAG3) sequence. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 722–733. [Google Scholar] [CrossRef]
- Engfeldt, T.; Tran, T.; Orlova, A.; Widstrom, C.; Feldwisch, J.; Abrahmsen, L.; Wennborg, A.; Karlstrom, A.E.; Tolmachev, V. 99mTc-chelator engineering to improve tumour targeting properties of a HER2-specific Affibody molecule. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1843–1853. [Google Scholar]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.; Nilsson, M.; Larsson, B.; Hoiden-Guthenberg, I.; Widstrom, C.; Carlsson, J.; Tolmachev, V.; Stahl, S.; Nilsson, F.Y. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006, 66, 4339–4348. [Google Scholar]
- Orlova, A.; Tran, T.; Widstrom, C.; Engfeldt, T.; Eriksson Karlstrom, A.; Tolmachev, V. Pre-clinical evaluation of [111In]-benzyl-DOTA-Z(HER2:342), a potential agent for imaging of HER2 expression in malignant tumors. Int. J. Mol. Med. 2007, 20, 397–404. [Google Scholar]
- Tran, T.; Engfeldt, T.; Orlova, A.; Sandstrom, M.; Feldwisch, J.; Abrahmsen, L.; Wennborg, A.; Tolmachev, V.; Karlstrom, A.E. (99m)Tc-maEEE-Z(HER2:342), an Affibody molecule-based tracer for the detection of HER2 expression in malignant tumors. Bioconjug. Chem. 2007, 18, 1956–1964. [Google Scholar] [CrossRef]
- Baum, R.P.; Prasad, V.; Muller, D.; Schuchardt, C.; Orlova, A.; Wennborg, A.; Tolmachev, V.; Feldwisch, J. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J. Nucl. Med. 2010, 51, 892–897. [Google Scholar] [CrossRef]
- Molina, R.; Barak, V.; van Dalen, A.; Duffy, M.J.; Einarsson, R.; Gion, M.; Goike, H.; Lamerz, R.; Nap, M.; Soletormos, G.; Stieber, P. Tumor markers in breast cancer- European Group on Tumor Markers recommendations. Tumour Biol. 2005, 26, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Zidan, J.; Dashkovsky, I.; Stayerman, C.; Basher, W.; Cozacov, C.; Hadary, A. Comparison of HER-2 overexpression in primary breast cancer and metastatic sites and its effect on biological targeting therapy of metastatic disease. Br. J. Cancer 2005, 93, 552–556. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Kohl, A.; Stumpp, M.T.; Briand, C.; Forrer, P.; Grutter, M.G.; Pluckthun, A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004, 22, 575–582. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Forrer, P.; Binz, H.K.; Pluckthun, A. Designing repeat proteins: Modular leucine-rich repeat protein libraries based on the mammalian ribonuclease inhibitor family. J. Mol. Biol. 2003, 332, 471–487. [Google Scholar]
- Dennis, M.S.; Lazarus, R.A. Kunitz domain inhibitors of tissue factor-factor VIIa. I. Potent inhibitors selected from libraries by phage display. J. Biol. Chem. 1994, 269, 22129–22136. [Google Scholar]
- Garnock-Jones, K.P. Ecallantide: In acute hereditary angioedema. Drugs 2010, 70, 1423–1431. [Google Scholar]
- Lehmann, A. Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin. Biol. Ther. 2008, 8, 1187–1199. [Google Scholar] [CrossRef]
- Williams, A.; Baird, L.G. DX-88 and HAE: A developmental perspective. Transfus. Apher. Sci. 2003, 29, 255–258. [Google Scholar] [CrossRef]
- Attucci, S.; Gauthier, A.; Korkmaz, B.; Delepine, P.; Martino, M.F.; Saudubray, F.; Diot, P.; Gauthier, F. EPI-hNE4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J. Pharmacol. Exp. Ther. 2006, 318, 803–809. [Google Scholar] [CrossRef]
- Lindner, T.; Kolmar, H.; Haberkorn, U.; Mier, W. DNA libraries for the construction of phage libraries: Statistical and structural requirements and synthetic methods. Molecules 2011, 16, 1625–1641. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zoller, F.; Haberkorn, U.; Mier, W. Miniproteins as Phage Display-Scaffolds for Clinical Applications. Molecules 2011, 16, 2467-2485. https://doi.org/10.3390/molecules16032467
Zoller F, Haberkorn U, Mier W. Miniproteins as Phage Display-Scaffolds for Clinical Applications. Molecules. 2011; 16(3):2467-2485. https://doi.org/10.3390/molecules16032467
Chicago/Turabian StyleZoller, Frederic, Uwe Haberkorn, and Walter Mier. 2011. "Miniproteins as Phage Display-Scaffolds for Clinical Applications" Molecules 16, no. 3: 2467-2485. https://doi.org/10.3390/molecules16032467
APA StyleZoller, F., Haberkorn, U., & Mier, W. (2011). Miniproteins as Phage Display-Scaffolds for Clinical Applications. Molecules, 16(3), 2467-2485. https://doi.org/10.3390/molecules16032467