Glycosides, Depression and Suicidal Behaviour: The Role of Glycoside-Linked Proteins



Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleobases | Purine (Adenine, Guanine, Purine analogue), Pyrimidine (Uracil, Thymine, Cytosine, Pyrimidine analogue) |
| Ribonucleosides* | Adenosine, Guanosine, Uridine, Cytidine |
| Deoxyribonucleosides** | Deoxyadenosine, Deoxyguanosine, Thymidine, Deoxyuridine, Deoxycytidine |
| Ribonucleotides† | Monophosphates (AMP, GMP, UMP, CMP), diphosphates (ADP, GDP, UDP,CDP), triphosphates (ATP, GTP, UTP, CTP) |
| Deoxyribonucleotides†† | Monophosphates (dAMP, dGMP, dUMP, TMP, dCMP), diphosphates (dADP, dGDP, TDP, dCDP), triphosphates (dATP, dGTP, dTTP, dCTP) |
| Cyclic∫ | cAMP, cGMP, cGMP, cADPR |
| Deoxyribonucleic acids∫∫ | cDNA, cpDNA, gDNA, msDNA, mtDNA |
| Ribonucleic acids⌠ |
|
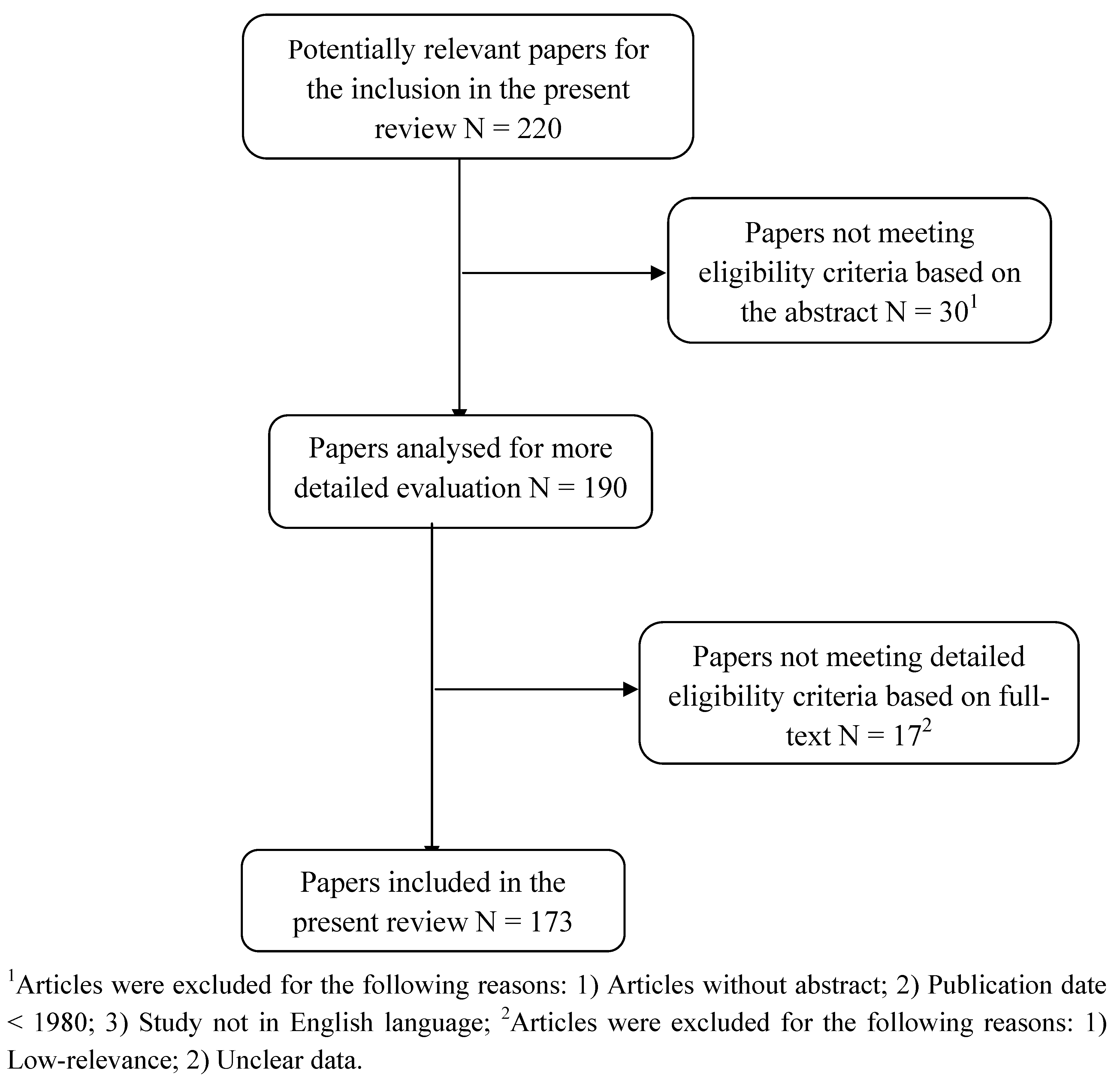
2. Methods

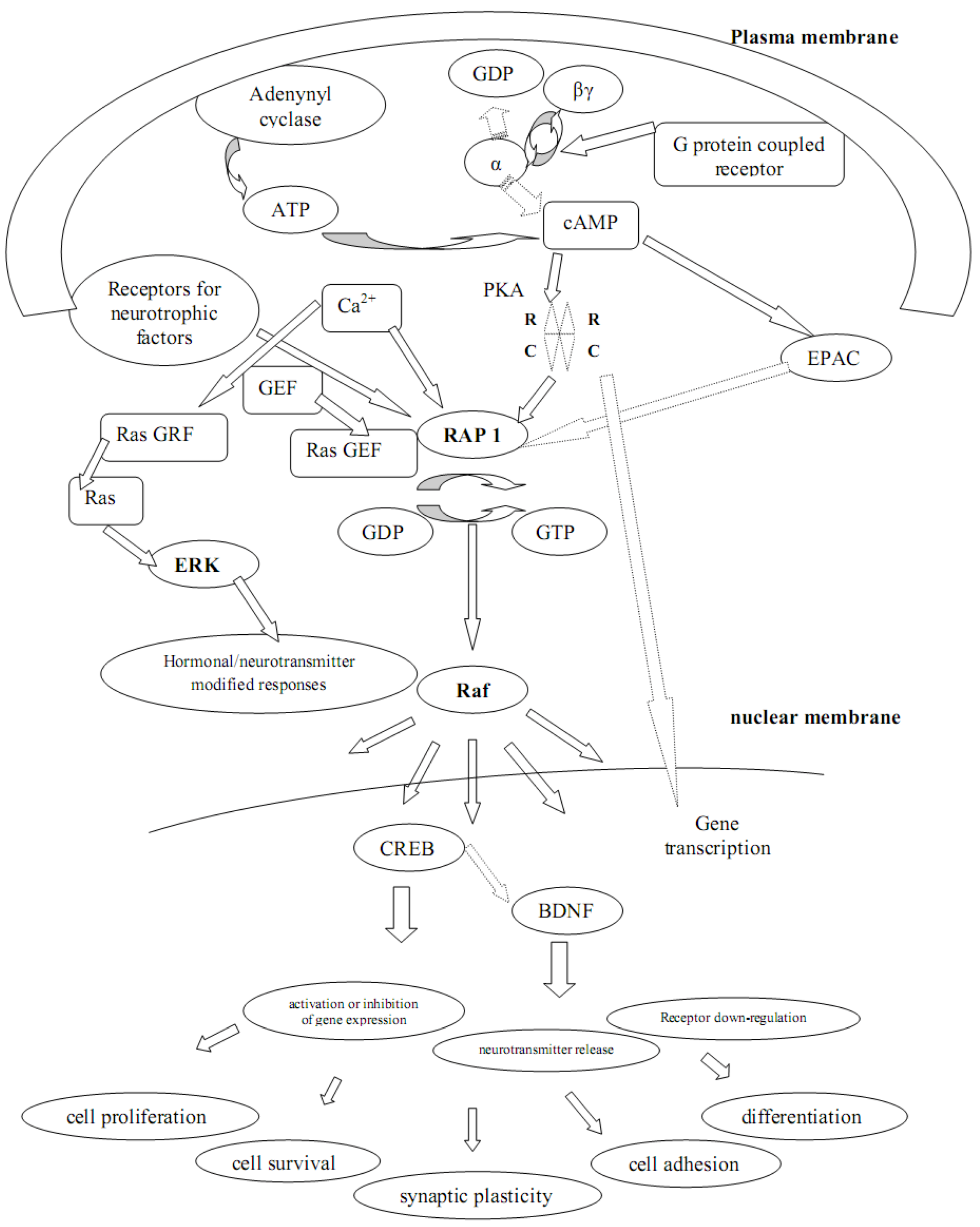
3. G Proteins and Adenylyl Cyclase-cAMP and Their Role in Depression and Suicide

4. PKA, Depression and Suicidal Behaviour
5. Rap-1, Epac-1 and Epac-2 in Depression and Suicide
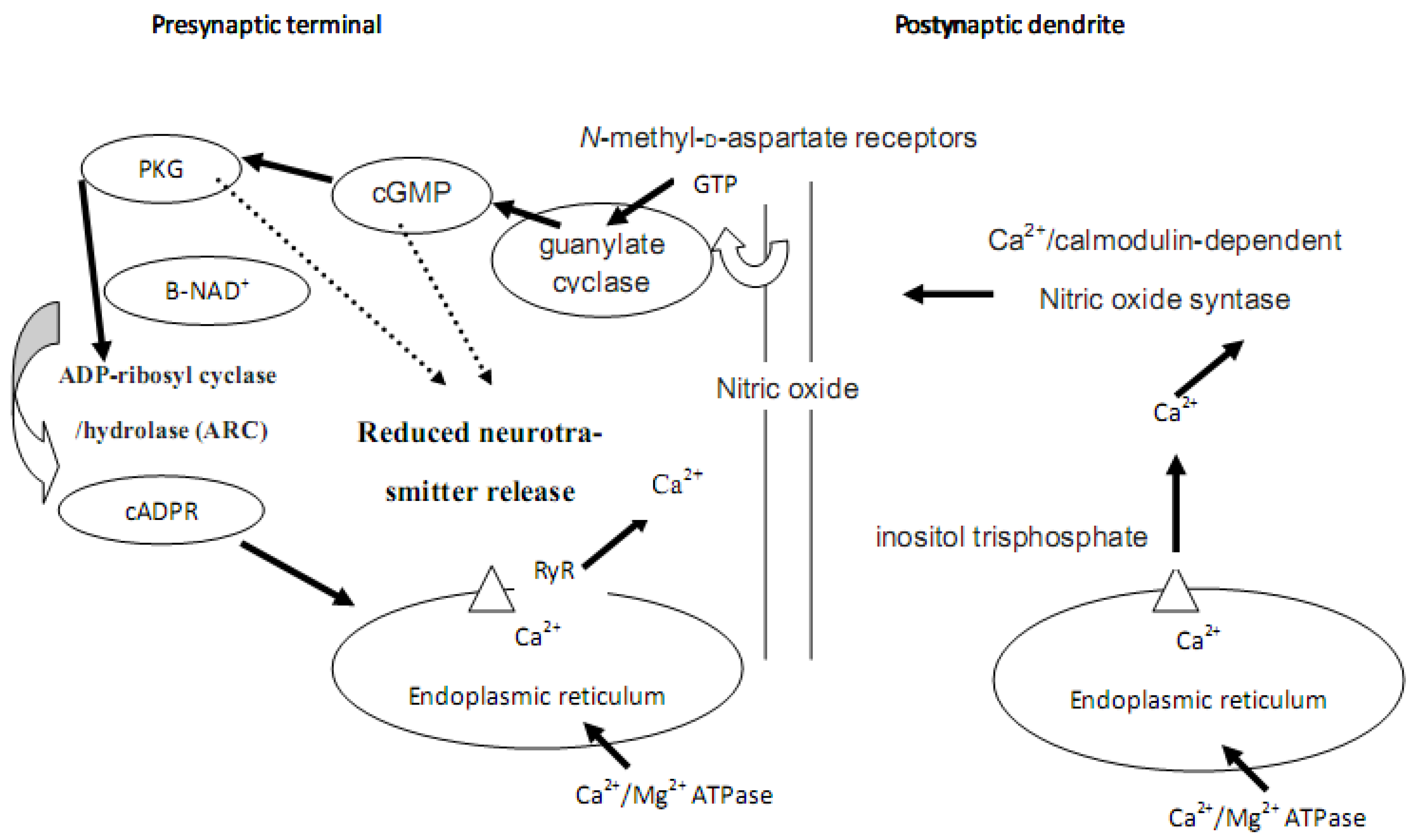
6. Calcium, cADPR, Long-term Depression and Synaptic Plasticity

7. The Pivotal Role of PKA in Synaptic Plasticity and Cell Survival
8. CREB and BDNF in Synaptic Plasticity and Cell Survival
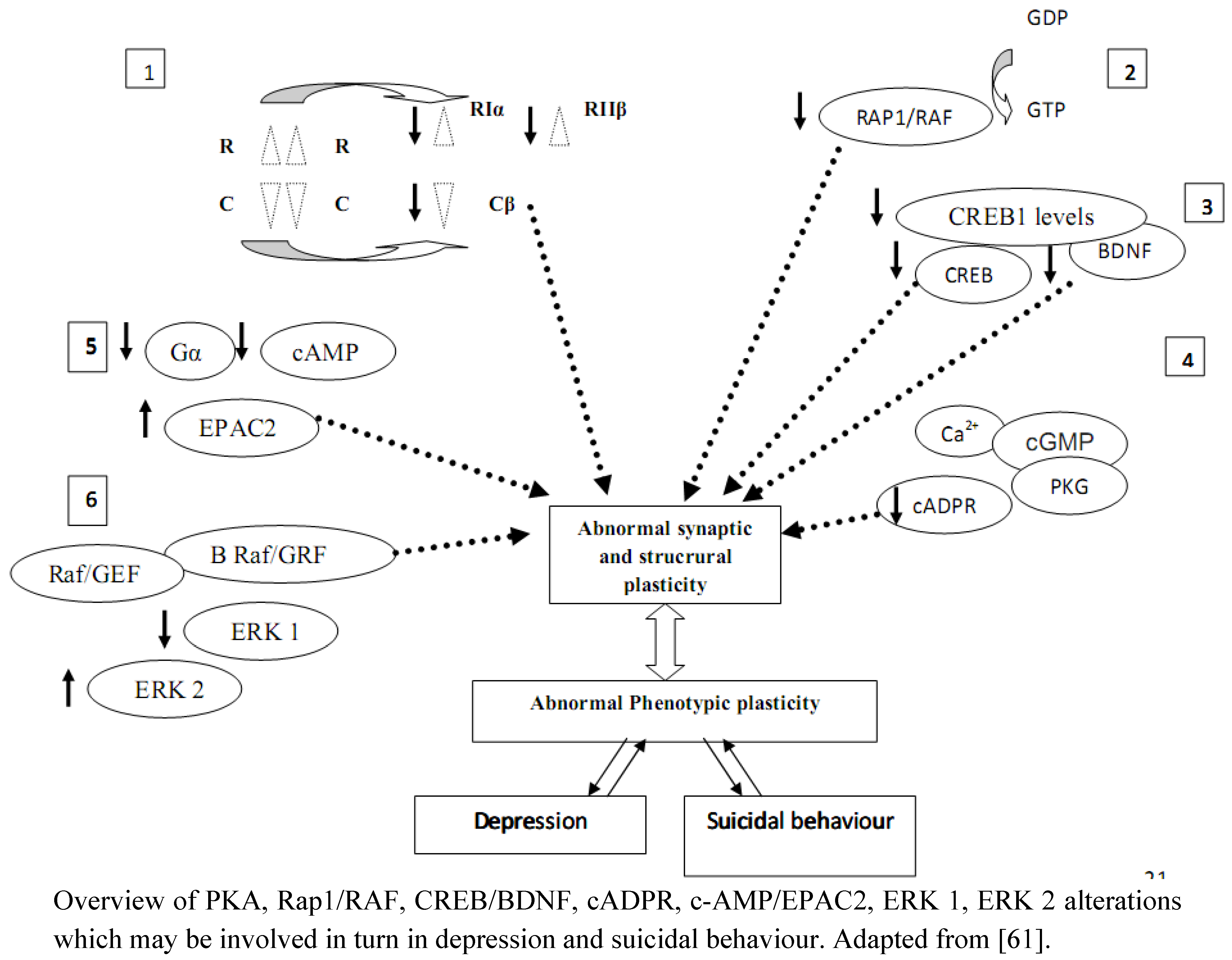
9. Conclusions: Is There Evidence of A Central Role For Glycosides And Glycoside-Linked Signal Transduction Proteins In Depression and Suicide?

Acknowledgements
References
- World Health Organization. The Global Burden of Disease:. 2004. Available online: http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/index.html. (Accessed on 3 December 2010).
- Hirschfeld, R.M.; Weissman, M.M. Risk factors for major depression and bipolar disorder. In Neuropsychopharmacology: the Fifth Generation of Progress; Davis, K.L., Charney, D., Coyle, J.T., Nemeroff, C., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2002; pp. 1017–1025. [Google Scholar]
- Mann, J.J. Neurobiology of suicidal behaviour. Nat. Rev. 2003, 4, 819–828. [Google Scholar]
- Pompili, M.; Rihmer, Z.; Innamorati, M.; Lester, D.; Girardi, P.; Tatarelli, R. Assessment and treatment of suicide risk in bipolar disorders. Expert Rev. Neurother. 2009, 9, 109–136. [Google Scholar] [CrossRef]
- Charney, D.S.; Manji, H.K. Life stress, genes, and depression: Multiple pathways lead to increased risk and new opportunities for intervention. Sci. STKE 2004, 16, 225. [Google Scholar]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef]
- Drevets, W.C. Neuroimaging and neuropathological studies of depression: Implications for the cognitive-emotional features of mood disorders. Curr. Opin. Neurobiol. 2001, 11, 240–249. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Janssens, A.C.; González-Zuloeta Ladd, A.M.; Del-Favero, J.; Claes, S.J.; Oostra, B.A.; van Duijn, C.M. Meta-analyses of genetic studies on major depressive disorder. Mol. Psychiat. 2007, 13, 772–785. [Google Scholar]
- Ressler, K.J.; Mayberg, H.S. Targeting abnormal neural circuits in mood and anxiety disorders: From the laboratory to the clinic. Nat. Neurosci. 2007, 10, 1116–1124. [Google Scholar] [CrossRef]
- Harrison, P.J. The neuropathology of primary mood disorder. Brain 2002, 125, 1428–1449. [Google Scholar] [CrossRef]
- Nemeroff, C. Part IV: Mood Disorders. In Neurobiology of Mental Illness, 2nd; Charney, D.S., Nestler, E.J., Eds.; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Dwivedi, Y.; Rizavi, H.S.; Pandey, G.N. Differential effects of haloperidol and clozapine on [(3)H]cAMP binding, protein kinase A (PKA) activity, and mRNA and protein expression of selective regulatory and catalytic subunit isoforms of PKA in rat brain. J. Pharmacol. Exp. Ther. 2002, 301, 197–209. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Shukla, P.K.; Lyons, J.; Faludi, G.; Palkovits, M.; Sarosi, A.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Protein kinase A in post-mortem brain of depressed suicide victims: Altered expression of specific regulatory and catalytic subunits. Biol. Psychiat. 2004, 55, 234–243. [Google Scholar] [CrossRef]
- Dwivedi, Y. The concept of dysregulated signal transduction and gene expression in the pathophysiology of mood disorders. Curr. Psychiat. Rev. 1, 227–257. [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Pandey, G.N. ERK MAP kinase signaling in post-mortem brain of suicide subjects: Differential regulation of upstream Raf kinases Raf-1 and B-Raf. Mol. Psychiat. 2006b, 11, 86–98. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Mondal, A.C.; Rizavi, H.S.; Faludi, G.; Palkovits, M.; Sarosi, A.; Conley, R.R.; Roberts, R.C.; Pandey, G.N. Differential and Brain region-specific regulation of Rap-1 and Epac in depressed suicide victims. Arch. Gen. Psychiat. 2006c, 63, 639–648. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Pandey, S.C.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A. Protein kinase C in the postmortem brain of teenage suicide victims. Neurosci. Lett. 1997, 228, 111–114. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Kumari, R.; Janicak, P.G. Protein Kinase C in Platelets of Depressed Patients. Biol. Psychiat. 1998, 44, 909–911. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Ren, X.; Rizavi, H.S.; Roberts, R.C.; Conley, R.R. Cyclic AMP response element-binding protein in postmortem brain of teenage suicide victims: Specific decrease in the prefrontal cortex but not the hippocampus. Int. J. Neuropsychopharmacol. 2006, 18, 1–9. [Google Scholar]
- Shelton, R.C.; Manier, D.H.; Sulser, F. cAMP-dependent protein kinase activity in major depression. Am. J. Psychiat. 1996, 153, 1037–1042. [Google Scholar]
- Shelton, R.C.; Manier, D.H.; Peterson, C.S.; Ellis, T.C.; Sulser, F. Cyclic AMP–dependent protein kinase in subtypes of major depression and normal volunteers. Int. J. Neuropsychopharmacol. 1999, 2, 187–192. [Google Scholar] [CrossRef]
- Manier, D.H.; Shelton, R.C.; Ellis, T.C.; Peterson, C.S.; Eiring, A.; Sulser, F. Human fibroblasts as a relevant model to study signal transduction in affective disorders. J. Affect. Disord. 2000, 61, 51–58. [Google Scholar] [CrossRef]
- Akin, D.; Manier, D.H.; Sanders-Bush, E.; Shelton, R.C. Signal transduction abnormalities in melancholic depression. Int. J. Neuropsychopharmacol. 2005, 8, 5–16. [Google Scholar] [CrossRef]
- Lindhorst, T.K. Essentials of Carbohydrate Chemistry and Biochemistry; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Neer, E.J. Heterotrimeric G proteins: Organizers of transmembrane signals. Cell 1995, 80, 249–257. [Google Scholar] [CrossRef]
- Clapham, D.E.; Neer, E.J. G protein beta gamma subunits. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 167–203. [Google Scholar] [CrossRef]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef]
- Freissmuth, M.; Waldhoer, M.; Bofill-Cardoba, E.; Nanoff, C. G protein antagonists. Trends Pharmacol. Sci. 1999, 20, 237–245. [Google Scholar] [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Borrelli, E.; Montmayeur, J.P.; Foulkes, N.S.; Sassone-Corsi, P. Signal transduction and gene control: The cAMP pathway. Crit. Rev. Oncol. Hematol. 1992, 3, 321–338. [Google Scholar]
- Nestler, E.J.; Greengard, P. Protein phosphorylation and the regulation of neuronal function. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Siegel, G.J., Albers, R.W., Agranoff, B.W., Molinoff, P., Eds.; Little Brown Press: Boston, MA, USA, 1994; pp. 449–474. [Google Scholar]
- Cowburn, R.F.; Marcusson, J.O.; Eriksson, A.; Wiehager, B.; O'Neill, C. Adenylyl cyclase activity and G-protein subunit levels in postmortem frontal cortex of suicide victims. Brain Res. 1994, 633, 297–304. [Google Scholar] [CrossRef]
- Ozawa, H.; Gsell, W.; Frolich, L.; Pantucek, F.; Beckmann, H.; Riederer, P. Imbalance of the Gs and Gi/o function in postmortem human brain of depressed patients. J. Neural. Transm. Gen. Sect. 1993, 94, 63–69. [Google Scholar] [CrossRef]
- Avissar, S.; Nechamkin, Y.; Roitman, G.; Schreiber, G. Reduced G protein functions and immunoreactive levels in mononuclear leukocytes of patients with depression. Am. J. Psychiat. 154, 211–217.
- Karege, F.; Bovier, P.; Stepanian, R.; Malafosse, A. The effect of clinical outcome on platelet G proteins of major depressed patients. Eur. Neuropsychopharmacol. 1998, 8, 89–94. [Google Scholar] [CrossRef]
- Spleiss, O.; van Calker, D.; Scharer, L.; Adamovic, K.; Berger, M.; Gebicke-Haerter, P.J. Abnormal G protein alpha(s)- and alpha (i2)-subunit mRNA expression in bipolar affective disorder. Mol. Psychiat. 1998, 3, 512–520. [Google Scholar]
- Young, L.T.; Li, P.P.; Kish, S.J.; Siu, K.P.; Kamble, A.; Hornykiewicz, O.; Warsh, J.J. Cerebral cortex Gs alpha protein levels and forskolinstimulated cyclic AMP formation are increased in bipolar affective disorder. J. Neurochem. 1993, 61, 890–898. [Google Scholar] [CrossRef]
- Pacheco, M.A.; Stockmeier, C.; Meltzer, H.Y.; Overholser, J.C.; Dilley, G.E.; Jope, R.S. Alterations in phosphoinositide signaling and G-protein levels in depressed suicide brain. Brain Res. 1996, 723, 37–45. [Google Scholar] [CrossRef]
- Schreiber, G.; Avissar, S.; Danon, A.; Belmaker, R.H. Hyperfunctional Gproteins inmononuclear leukocytes of patients with mania. Biol. Psychiat. 1991, 92, 273–280. [Google Scholar]
- Young, L.T.; Li, P.P.; Kish, S.J.; Siu, K.P.; Warsh, J.J. Postmortem cerebral cortex Gsasubunit levels are elevated in bipolar affective disorder. Brain Res. 1991, 553, 323–326. [Google Scholar] [CrossRef]
- Young, L.T.; Bezchlibnyk, Y.B.; Chen, B.; Wang., J.-F.; MacQueen, G.M. Amygdala cyclic adenosine monophosphate response element-binding protein phosphorylation in patients with mood disorders: Effects of diagnosis, suicide, and drug treatment. Biol. Psychiat. 2004, 55, 570–577. [Google Scholar] [CrossRef]
- Avissar, S.; Barki-Harrington, L.; Nechamkin, Y.; Roitman, G.; Schreiber, G. Reduced b-adrenergic receptor-coupled Gs protein function and Gsa immunoreactivity in mononuclear leukocytes of patients with depression. Biol. Psychiat. 1996, 39, 755–760. [Google Scholar] [CrossRef]
- Avissar, A.; Nechamkin, Y.; Barki-Harrington, L.; Roitman, G.; Schreiber, G. Differential G protein measures in mononuclear leukocytes of patients with bipolar mood disorder are state dependent. J. Affect. Disord. 1997b, 43, 85–93. [Google Scholar] [CrossRef]
- Mitchell, P.B.; Manji, H.K.; Chen, G.; Jolkovsky, L.; Smith-Jackson, E.; Denicoff, K.; Schmidt, M.; Potter, W.Z. High levels of Gsa in platelets of euthymic patients with bipolar affective disorder. Am. J. Psychiat. 1997, 154, 218–223. [Google Scholar]
- Lara, J.; Kusano, K.; House, S.; Gainer, H. Interactions of cyclic adenosine monophosphate, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor treatment on the survival and growth of postnatal mesencephalic dopamine neurons in vitro. Exp. Neurol. 2003, 180, 32–45. [Google Scholar] [CrossRef]
- Abel, T.; Nguyen, P.V. Regulation of hippocampus-dependent memory by cyclic AMPdependent protein kinase. Prog. Brain. Res. 2008, 169, 97–115. [Google Scholar] [CrossRef]
- Uhler, M.D.; Chrivia, J.C.; McKnight, G.S. Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1986, 261, 15360–15363. [Google Scholar]
- Odagaki, Y.; Garcia-Sevilla, J.A.; Huguelet, P.; La Harpe, R.; Koyama, T.; Guimon, J. Cyclic AMPmediated signaling components are upregulated in the prefrontal cortex of depressed suicide victims. Brain Res. 2001, 898, 224–231. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Ren, X.; Rizavi, H.S.; Mondal, A.C.; Shukla, P.K.; Conley, R.R. Brain region specific alterations in the protein and mRNA levels of protein kinase A subunits in the postmortem brain of teenage suicide victims. Neuropsychopharmacology 2005, 30, 1548–1556. [Google Scholar] [CrossRef]
- Perez, J.; Tardito, D.; Mori, S.; Racagni, G.; Smeraldi, E.; Zanardi, R. Abnormalities of cyclic adenosine monophosphate signaling in platelets from untreated patients with bipolar disorder. Arch. Gen. Psychiat. 1999, 56, 248–253. [Google Scholar] [CrossRef]
- Brandon, E.P.; Logue, S.F.; Adams, M.R.; Qi, M.; Sullivan, S.P.; Matsumoto, A.M.; Dorsa, D.M.; Wehner, J.M.; McKnight, G.S.; Idzerda, R.L. Defective motor behavior and neural gene expression in RIIβ-protein kinase A mutant mice. J. Neurosci. 1998, 18, 3639–3649. [Google Scholar]
- Thiele, T.E.; Willis, B.; Stadler, J.; Reynolds, J.G.; Bernstein, I.L.; McKnight, G.S. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase Amutant mice. J. Neurosci. 2000, 20, RC75. [Google Scholar]
- Qi, M.; Zhuo, M.; Skalhegg, B.S.; Brandon, E.P.; Kandel, E.R.; McKnight, G.S.; Idzerda, R.L. Impaired hippocampal plasticity in mice lacking the Cβ catalytic subunit of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 1571–1576. [Google Scholar]
- Shelton, R.C.; Hal Manier, D.; Lewis, D.A. Protein kinases A and C in post-mortem prefrontal cortex from persons with major depression and normal controls. Int. J. Neuropsychopharmacol. 2009a, 12, 1223–1232. [Google Scholar] [CrossRef]
- Shelton, R.C.; Sanders-Bush, E.; Manier, D.H.; Lewis, D.A. Elevated 5-HT 2A receptors in postmortem prefrontal cortex in major depression is associated with reduced activity of protein kinase A. Neuroscience 2009b, 158, 1406–1415. [Google Scholar] [CrossRef]
- McEwen, B.S. Effects of adverse experiences for brain structure and function. Biol. Psychiat. 2000, 48, 713–714. [Google Scholar] [CrossRef]
- Manier, D.H.; Eiring, A.; Shelton, R.C.; Sulser, F. Adrenoceptor-linked protein kinase A (PKA) activity in human fibroblasts from normal subjects and from patients with major depression. Neuropsychopharmacology 1996, 15, 555–561. [Google Scholar] [CrossRef]
- Seligman, M.E.; Maier, S.F. Failure to escape traumatic shock. J. Exp. Psychol. 1967, 74, 1–9. [Google Scholar] [CrossRef]
- Petty, F.; Sherman, A.D. Reversal of learned helplessness by imipramine. Commun. Psychopharmacol. 1979, 3, 371–373. [Google Scholar]
- Sherman, A.D.; Sacquitne, J.L.; Petty, F. Specificity of the learned helplessness model of depression. Pharmacol. Biochem. Behav. 1982, 16, 449–454. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Pandey, G.N. Elucidating biological risk factors in suicide: Role of protein kinase A. Prog. Neuro-Psychopharmacol. Biol. Psychiat. 2010, in press. [Google Scholar]
- Altschuler, D.L.; Peterson, S.N.; Ostrowski, M.C.; Lapetina, E.G. Cyclic AMP-dependent activation of Rap1b. J. Biol. Chem. 1995, 270, 373–376. [Google Scholar]
- Zwartkruis, F.J.T.; Bos, J.L. Ras and Rap1: Two highly related small GTPases with distinct function. Exp. Cell Res. 1999, 253, 157–165. [Google Scholar] [CrossRef]
- Bos, J.L.; de Rooijm, J.; Reedquist, K.A. Rap1 signaling: Adhering to new models. Nat. Rev. Mol. Cell Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef]
- Baldassa, S.; Zippel, R.; Sturani, E. Depolarization-induced signaling to Ras, Rap1 and MAPKs in cortical neurons. Mol. Brain Res. 2003, 119, 111–122. [Google Scholar] [CrossRef]
- Hattori, M.; Minato, N. Rap1 GTPase: Functions, regulation, and malignancy. J. Biochem. (Tokyo) 2003, 134, 479–484. [Google Scholar] [CrossRef]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity andmemory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef]
- Kolkova, K.; Novitskaya, V.; Pedersen, N.; Berzin, V.; Bock, E. Neural cell adhesion molecule- stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J. Neurosci. 2000, 20, 2238–2246. [Google Scholar]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef]
- Marshall, R.; Dragunow, M. Is CREB a key to neuronal survival? Trends Neurosci. 2000, 23, 48–53. [Google Scholar] [CrossRef]
- Yao, H.; York, R.D.; Misra-Press, A.; Carr, D.W.; Stork, P.J.S. The cyclic adenosine monophosphate-dependent protein kinase (PKA) is required for the sustained activation of mitogen-activated kinases and gene expression by nerve growth factor. J. Biol. Chem. 1998, 273, 8240–8247. [Google Scholar] [CrossRef]
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.J.S. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 1998, 392, 622–626. [Google Scholar] [CrossRef]
- Qiu, W.; Zhuang, S.; von Lintig, F.C.; Boss, G.R.; Pilz, R.B. Cell type-specific regulation of B-Raf kinase by cAMP and 14-3-3 proteins. J. Biol. Chem. 2000, 275, 31921–31929. [Google Scholar]
- Grewal, S.S.; Fass, D.M.; Yao, H.; Ellig, C.L.; Goodman, R.H.; Stork, P.J. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J. Biol. Chem. 2000, 275, 34433–34441. [Google Scholar]
- Morozov, A.; Muzzio, I.A.; Bourtcholadze, R.; Van-Strien, N.; Lapidus, K.; Yin, D.Q.; Winder, D.G.; Adams, J.P.; Sweatt, J.D.; Kandel, E.R. Rap1 couples cAMP signaling to a distinct pool of p42/44 MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 2003, 39, 309–325. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Roberts, R.C.; Conley, R.C.; Tamminga, C.A.; Pandey, G.N. Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J. Neurochem. 2001, 77, 916–928. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar]
- Perez, J.; Zanardi, R.; Mori, S.; Gasperini, M.; Smeraldi, E.; Racagni, G. Abnormalities of cAMP-dependent endogenous phosphorylation in platelets from patients with bipolar disorder. Am. J. Psychiat. 1995, 152, 1204–1206. [Google Scholar]
- Zanardi, R.; Racagni, G.; Smeraldi, E.; Perez, J. Differential effects of lithium on platelet protein phosphorylation in bipolar patients and healthy subjects. Psychopharmacology 1997, 129, 44–47. [Google Scholar] [CrossRef]
- Perez, J.; Tardito, D.; Mori, S.; Racagni, G.; Smeraldi, E.; Zanardi, R. Altered Rap1 endogenous phosphorylation and levels in platelets from patients with bipolar disorder. J. Psychiatr. Res. 2000a, 34, 99–104. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Schmitt, J.M.; Stork, P.J. PKA phosphorylation of Src mediates cAMP's inhibition of cell growth via Rap1. Mol. Cell 2002, 9, 85–94. [Google Scholar] [CrossRef]
- Hoshijima, M.; Kikuchi, A.; Kawata, M.; Ohmori, T.; Hashimoto, E.; Yamamura, H.; Takai, Y. Phosphorylation by cyclic AMP-dependent protein kinase of a human platelet Mr 22,000 GTP-binding protein (smg p21) having the same putative effector domain as the ras gene products. Biochem. Biophys. Res. Commun. 1988, 157, 851–860. [Google Scholar] [CrossRef]
- Bokoch, G.M. Biology of the Rap proteins, members of the ras superfamily of GTP-binding proteins. Biochem. J. 1993, 289(Pt 1), 17–24. [Google Scholar]
- Vossler, M.R.; Yao, H.; York, R.D.; Pan, M.G.; Rim, C.S.; Stork, P.J. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 1997, 89, 73–82. [Google Scholar] [CrossRef]
- Dugan, L.L.; Kim, J.S.; Zhang, Y.; Bart, R.D.; Sun, Y.; Holtzman, D.M.; Gutmann, D.H. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J. Biol. Chem. 1999, 274, 25842–25848. [Google Scholar]
- Zanassi, P.; Paolillo, M.; Feliciello, A.; Avvedimento, E.V.; Gallo, V.; Schinelli, S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 2001, 276, 11487–11495. [Google Scholar]
- Qiao, J.; Mei, F.C.; Popov, V.L.; Vergara, L.A.; Cheng, X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J. Biol. Chem. 2002, 277, 26581–26586. [Google Scholar]
- Rehmann, H.; Prakash, B.; Wolf, E.; Rueppel, A.; de Rooij, J.; Bos, J.L.; Wittinghofer, A. Structure and regulation of the cAMP-binding domains of Epac2. Nat. Struct. Biol. 2003, 10, 26–32. [Google Scholar] [CrossRef]
- Kaneko, M.; Takahashi, T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J. Neurosci. 2004, 24, 5202–5208. [Google Scholar] [CrossRef]
- Zhu, J.J.; Qin, Y.; Zhao, M.; Van Aelst, L.; Malinow, R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 2002, 110, 443–455. [Google Scholar] [CrossRef]
- Ceni, C.; Pochon, N.; Villaz, M.; Muller-Steffner, H.; Schuber, F.; Baratier, J.; De Waard, M.; Ronjat, M.; Moutin, M.-J. The CD38-independent ADP-ribosyl cyclase from mouse brain synaptosomes: A comparative study of neonate and adult brain. Biochem. J. 2006, 395, 417–426. [Google Scholar] [CrossRef]
- Guse, A.H. Cyclic ADP-ribose: A novel Ca2+-mobilising second messenger. Cell. Signal. 1999, 11, 309–316. [Google Scholar] [CrossRef]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R. Kinet, J.P.; Scharenberg, A.M. ADP-ribose gating of the calciumpermeable LTRPC2 channel revealed by Nudix motif homology. Nature (London) 2001, 411, 595–599. [Google Scholar]
- Hua, S.Y.; Tokimasa, T.; Takasawa, S.; Furuya, Y.; Nohmi, M.; Okamoto, H.; Kuba, K. Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron 1994, 12, 1073–1079. [Google Scholar] [CrossRef]
- Empson, R.M.; Galione, A. Cyclic ADP-ribose enhances coupling between voltage-gated Ca2+ entry and intracellular Ca2+ release. J. Biol. Chem. 1997, 272, 20967–20970. [Google Scholar] [CrossRef]
- Hashii, M.; Minabe, Y.; Higashida, H. cADP-ribose potentiates cytosolic Ca2+ elevation and Ca2+ entry via L-type voltage-activated Ca2+ channels in NG108-NG115 neuronal cells. Biochem. J. 2000, 345, 207–215. [Google Scholar] [CrossRef]
- Budde, T.; Sieg, F.; Braunewell, K.H.; Gundelfinger, E.D.; Pape, H.C. Ca2+-induced Ca2+ release supports the relay mode of activity in thalamocortical cells. Neuron 2000, 26, 483–492. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef]
- Higashida, H.; Yokoyama, S.; Hashii, M.; Taketo, M.; Higashida, M.; Takayasu, T.; Ohshima, T.; Takasawa, S.; Okamoto, H.; Noda, M. Muscarinic receptormediated dual regulation of ADP-ribosyl cyclase in NG108-NG115 neuronal cell membranes. J. Biol. Chem. 1997, 272, 31272–31277. [Google Scholar]
- Morita, K.; Kitayama, S.; Dohi, T. Stimulation of cyclic ADP-ribose synthesis by acetylcholine and its role in catecholamine release in bovine adrenal chromaffin cells. J. Biol. Chem. 1997, 272, 21002–21009. [Google Scholar] [CrossRef]
- Pollock, J.; Crawford, J.H.; Wootton, J.F.; Seabrook, G.R.; Scott, R.H. Metabotropic glutamate receptor activation and intracellular cyclic ADP-ribose release Ca2+ from the same store in cultured DRG neurones. Cell Calcium 1999, 26, 139–148. [Google Scholar] [CrossRef]
- Hotta, T.; Asai, K.; Fujita, K.; Kato, T.; Higashida, H. Membrane-bound form of ADP-ribosyl cyclase in rat cortical astrocytes in culture. J. Neurochem. 2000, 74, 669–675. [Google Scholar]
- Noda, M.; Yasuda, S.; Okada, M.; Higashida, H.; Shimada, A.; Iwata, N.; Ozaki, N.; Nishikawa, K.; Shirasawa, S.; Uchida, M.; Aoki, S.; Wada, K. Recombinant human serotonin 5A receptors stably expressed in C6 glioma cells couple to multiple signal transduction pathways. J. Neurochem. 2003, 84, 222–232. [Google Scholar] [CrossRef]
- Morikawa, H.; Khodakhah, K.; Williams, J.T. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J. Neurosci. 2003, 23, 149–157. [Google Scholar]
- Mothet, J.P.; Fossier, P.; Meunier, F.M.; Stinnakre, J.; Tauc, L.; Baux, G. Cyclic ADP-ribose and calcium-induced calcium release regulate neurotransmitter release at a cholinergic synapse of Aplysia. J. Physiol. 1998, 507, 405–414. [Google Scholar] [CrossRef]
- Reyes-Harde, M.; Empson, R.; Potter, B.V.; Galione, A.; Stanton, P.K. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc. Natl. Acad. Sci. USA 1999, 96, 4061–4066. [Google Scholar] [CrossRef]
- Brailoiu, E.; Miyamoto, M.D. Inositol trisphosphate and cyclic adenosine diphosphate-ribose increase quantal transmitter release at frog motor nerve terminals: Possible involvement of smooth endoplasmic reticulum. Neuroscience 2000, 95, 927–931. [Google Scholar] [CrossRef]
- Oliet, S.H.; Malenka, R.C.; Nicoll, R.A. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 1997, 18, 969–982. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong Calcium Entry Activates Mitochondrial Superoxide Generation, Upregulating Kinase Signaling in Hippocampal Neurons. J. Neurosci. 2004, 24, 19878–10887. [Google Scholar]
- Tokuda, M.; Hatase, O. Regulation of neuronal plasticity in the central nervous system by phosphorylation and dephosphorylation. Mol. Neurobiol. 1998, 17, 137–156. [Google Scholar] [CrossRef]
- Soderling, T.R.; Derkach, V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000, 23, 75–80. [Google Scholar] [CrossRef]
- Martin, K.C.; Michael, D.; Rose, J.C.; Barad, M.; Casadio, A.; Zhu., H.; Kandel, E.R. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 1997, 18, 899–912. [Google Scholar] [CrossRef]
- English, J.D.; Sweatt, J.D. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J. Biol. Chem. 1997, 272, 19103–19106. [Google Scholar] [CrossRef]
- Impey, S.; Obrietan, K.; Wong, S.T.; Poser, S.; Yano, S.; Wayman, G.; Deloulme, J.C.; Chan, G.; Storm, D.R. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 1998, 21, 869–883. [Google Scholar] [CrossRef]
- Bailey, C.H.; Kaang, B.K.; Chen, M.; Martin, K.C.; Lim, C.S.; Casadio, A.; Kandel, E.R. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron 1997, 18, 913–924. [Google Scholar] [CrossRef]
- Anderson, A.E.; Adams, J.P.; Qian, Y.; Cook, R.G.; Pfaffinger, P.J.; Sweatt, J.D. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J. Biol. Chem. 2000, 275, 5337–5346. [Google Scholar]
- Lin, T.A.; Kong, X.; Haystead, T.A.; Pause, A.; Belsham, G.; Sonenberg, N.; Lawrence, J.C. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 1994, 266, 653–656. [Google Scholar]
- Waskiewicz, A.J.; Johnson, J.C.; Penn, B.; Mahalingam, M.; Kimball, S.R.; Cooper, J.A. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 1999, 19, 1871–1880. [Google Scholar]
- Jovanovic, J.N.; Benfenati, F.; Siow, Y.L.; Sihra, T.S.; Sanghera, J.S.; Pelech, S.L.; Greengard, P.; Czernik, A.J. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 3679–3683. [Google Scholar]
- Hardingham, G.E.; Arnold, F.J.; Bading, H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 2001, 4, 261–267. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. The yin and yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Grewal, S.S.; York, R.D.; Stork, P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 1999, 9, 544–553. [Google Scholar] [CrossRef]
- Villalba, M.; Bockaert, J.; Journot, L. Pituitary adenylate cyclaseactivating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J. Neurosci. 1997, 17, 83–90. [Google Scholar]
- Perez, J.; Tardito, D.; Racagni, G.; Smeraldi, E.; Zanardi, R. cAMP signaling pathway in depressed patients with psychotic features. Mol. Psychiat. 2002, 7, 208–212. [Google Scholar] [CrossRef]
- Perez, J.; Tardito, D.; Racagni, G.; Smeraldi, E.; Zanardi, R. Protein kinase A and Rap1 levels in platelets of untreated patients with major depression. Mol. Psychiat. 2001, 6, 44–49. [Google Scholar] [CrossRef]
- Dowlatshahi, D.; MacQueen, G.M.; Wang, J.F.; Young, L.T. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 1998, 352, 1754–1755. [Google Scholar]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Ann. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef]
- Viola, H.; Furman, M.; Izquierdo, L.A.; Alonso, M.; Barros, D.M.; de Souza, M.M.; Izquierdo, I.; Medina, J.H. Phosphorylated cAMP response element-binding protein as a molecular marker of memory processing in rat hippocampus: Effect of novelty. J. Neurosci. 2000, 20, RC112. [Google Scholar]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar]
- Carlezon, W.A., Jr.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Yamada, S.; Yamamoto, M.; Ozawa, H.; Piederer, P.; Saito, T. Reduced phosphorylation of cyclic AMP responsive element-binding protein in the postmortem orbitofrontal cortex of patients with major depressive disorder. J. Neural. Transm. 2003, 110, 671–680. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rao, J.S.; Rizavi, H.S.; Kotowski, J.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Abnormal expression and functional characteristics of cyclic adenosine monophosphate response element-binding protein in postmortem brain of suicide subjects. Arch. Gen. Psychiat. 2003, 60, 273–282. [Google Scholar] [CrossRef]
- Nibuya, M.; Nestler, E.J.; Duman, R.S. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 1996, 16, 2365–2372. [Google Scholar]
- Zubenko, G.S.; Hughes, H.B., III; Maher, B.S.; Stiffler, J.S.; Zubenko, W.N.; Marazita, M.L. Genetic linkage of region containing the CREB1 gene to depressive disorders in women from families with recurrent, early-onset, major depression. Am. J. Med. Genet. 2002, 114, 980–987. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kim, J.E.; Lee, R.; Malberg, J.E.; Chen, J.; Steffen, C.; Zhang, Y.J.; Nestler, E.J.; Duman, R.S. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 2002, 22, 3673–3682. [Google Scholar]
- Arnould, T.; Vankoningsloo, S.; Renard, P.; Houbion, A.; Ninane, N.; Demazy, C.; Remacle, J.; Raes, M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002, 21, 53–63. [Google Scholar] [CrossRef]
- Cherukuri, D.P.; Chen, X.B.; Goulet, A.C.; Young, R.N.; Han, Y.; Heimark, R.L.; Regan, J.W.; Meuillet, E.; Nelson, M.A. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp. Cell Res. 2007, 313, 2969–2979. [Google Scholar] [CrossRef]
- Gur, T.L.; Conti, A.C.; Holden, J.; Bechtholt, A.J.; Hill, T.E.; Lucki, I.; Malberg, J.E.; Blendy, J.A. cAMP Response Element-Binding Protein Deficiency Allows for Increased Neurogenesis and a Rapid Onset of Antidepressant Response. J. Neurosci. 2007, 27, 7860–7868. [Google Scholar] [CrossRef]
- Groves, J.O. Is it time to reassess the BNDF hypothesis of depression? Mol. Psychiat. 2007, 12, 1079–1088. [Google Scholar] [CrossRef]
- Lowther, S.; De Paermentier, F.; Cheetham, S.C.; Crompton, M.R.; Katona, C.L.; Horton, R.W. 5-HT1A receptor binding sites in post-mortem brain samples from depressed suicides and controls. J. Affect. Disord. 1997, 42, 199–207. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Thoenen, H. Neurotrophins and neuronal plasticity. Science 1995, 270, 593–598. [Google Scholar]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar]
- Bartrup, J.T.; Moorman, J.M.; Newberry, N.R. BDNF enhances neuronal growth and synaptic activity in hippocampal cell cultures. NeuroReport 1997, 8, 3791–3794. [Google Scholar] [CrossRef]
- Conti, A.C.; Cryan, J.F.; Dalvi, A.; Lucki, I.; Blendy, J.A. cAMP Response Element-Binding Protein Is Essential for the Upregulation of Brain-Derived Neurotrophic Factor Transcription, But Not the Behavioral or Endocrine Responses to Antidepressant Drugs. J. Neurosci. 2002, 22, 3262–3268. [Google Scholar]
- Karege, F.; Vaudan, G.; Schwald, M.; Perroud, N.; La Harpe, R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Mol. Brain Res. 2005, 136, 29–37. [Google Scholar] [CrossRef]
- Karege, F.; Schwald, M.; Cisse, M. Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci. Lett. 2002, 328, 261–264. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lee, H.P.; Won, S.D.; Park, E.Y.; Lee, H.Y.; Lee, B.H. Low plasma BDNF is associated with suicidal behavior in depression. Prog. Neuropsychopharmacol. Biol. Psychiat. 2007, 31, 578–579. [Google Scholar] [CrossRef]
- Sarchiapone, M.; Carli, V.; Roy, A.; Iacoviello, L.; Cuomo, C.; Latella, M.C.; di Giannantonio, M.; Janiri, L.; de Gaetano, M.; Janal, M.N. Association of polymorphism (Val66Met) of brain-derived neurotrophic factor with suicide attempts in depressed patients. Neuropsychobiology 2008, 57, 139–145. [Google Scholar] [CrossRef]
- Kohli, M.A.; Salyakina, D.; Pfennig, A.; Lucae, S.; Horstmann, S.; Menke, A.; Kloiber, S.; Hennings, J.; Bradley, B.B.; Ressler, K.J.; Uhr, M.; Müller-Myhsok, B.; Holsboer, F.; Binder, E.B. Association of genetic variants in the neurotrophic receptor-encoding gene NTRK2 and a lifetime history of suicide attempts in depressed patients. Arch. Gen. Psychiat. 2010, 67, 348–359. [Google Scholar] [CrossRef]
- Deveci, A.; Aydemir, O.; Taskin, O.; Taneli, F.; Esen-Danaci, A. Serum BDNF levels in suicide attempters related to psychosocial stressors: A comparative study with depression. Neuropsychobiology 2007, 56, 93–97. [Google Scholar] [CrossRef]
- Lee, B.H.; Kim, Y.K. Reduced platelet BDNF level in patients with depression. Prog. Neuropsychopharmacol. Biol. Psychiat. 2009, 33, 849–853. [Google Scholar] [CrossRef]
- Dawood, T.; Anderson, J.; Barton, D.; Lambert, E.; Esler, M.; Hotchkin, E. Reduced overflow of BDNF from the brain is linked with suicide risk in depressive illness. Mol. Psychiat. 2007, 12, 981–983. [Google Scholar] [CrossRef]
- Garcia, R. Stress, synaptic plasticity, and psychopathology. Rev. Neurosci. 2002, 13, 195–208. [Google Scholar]
- Duman, R.S.; Malberg, J.; Nakagawa, S.; D'Sa, C. Neuronal plasticity and survival in mood disorders. Biol. Psychiat. 2000, 48, 732–739. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Mondal, A.C.; Rizavi, H.S.; Conley, R.R. Suicide brain is associated with decreased expression of neurotrophins. Biol. Psychiat. 2005, 58, 315–324. [Google Scholar] [CrossRef]
- Dwivedi, Y. BDNF in suicide pathogenesis. Ann. Med. 2010, 42, 87–96. [Google Scholar] [CrossRef]
- Altshuler, L.L.; Casanova, M.F.; Goldberg, T.E.; Kleinman, J.E. The hippocampus and parahippocampus in schizophrenia, suicide, and control brains. Arch. Gen. Psychiat. 1990, 47, 1029–1034. [Google Scholar] [CrossRef]
- Rajkowska, G. Morphometric methods for studying the prefrontal cortex in suicide victims and psychiatric patients. Ann. NY. Acad. Sci. USA 1997, 836, 253–268. [Google Scholar] [CrossRef]
- Rajkowska, G. Cell pathology in mood disorders. Semin. Clin. Neuropsychiatry 2002, 7, 281–292. [Google Scholar]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiat. 2000, 157, 115–118. [Google Scholar]
- Rosoklija, G.; Toomayan, G.; Ellis, S.P.; Keilp, J.; Mann, J.J.; Latov, N. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: Preliminary findings. Arch. Gen. Psychiat. 2000, 57, 349–356. [Google Scholar] [CrossRef]
- Cotter, D.; Mackay., D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef]
- Aganova, E.A.; Uranova, N.A. Morphometric analysis of synaptic contacts in the anterior limbic cortex in the endogenous psychoses. Neurosci. Behav. Physiol. 1992, 22, 59–65. [Google Scholar] [CrossRef]
- Honer, W.G. Assessing the machinery of mind: Synapses in neuropsychiatric disorders. J. Psychiatr. Neurosci. 1999, 24, 16–21. [Google Scholar]
- Hajsza, T.; MacLusky, N.J.; Leranth, C. Short-term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur. J. Neurosci. 2005, 21, 1299–1303. [Google Scholar] [CrossRef]
- Sheline, Y.I. 3D MRI studies of neuroanatomic changes in unipolar depression: The role of stress and medical comorbidity. Biol. Psychiat. 2000, 48, 791–800. [Google Scholar] [CrossRef]
- Sackeim, H.A. Functional brain circuits in depression and remission. Arch. Gen. Psychiat. 2001, 58, 649–650. [Google Scholar] [CrossRef]
- Young, L.T. Postreceptor pathways for signal transduction in depression and bipolar disorder. J. Psychiat. Neurosci. 2001, 26 (Suppl.), S17–S22. [Google Scholar]
- Dwivedi, Y.; Rizavi, H.S.; Zhang, H.; Mondal, A.C.; Roberts, R.C.; Conley, R.R.; Pandey, G.N. Neurotrophin receptor activation and expression in human postmortem brain: Effect of suicide. Biol. Psychiat. 2009, 65, 319–328. [Google Scholar] [CrossRef]
- Joëls, M.; Karst, H.; Alfarez, D.; Heine, V.M.; Qin, Y.; van Riel, E.; Verkuyl, M.; Lucassen, P.J.; Krugers, H.J. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 2004, 7, 221–231. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiat. 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Serafini, G.; Pompili, M.; Innamorati, M.; Giordano, G.; Tatarelli, R.; Lester, D.; Girardi, P.; Dwivedi, Y. Glycosides, Depression and Suicidal Behaviour: The Role of Glycoside-Linked Proteins. Molecules 2011, 16, 2688-2713. https://doi.org/10.3390/molecules16032688
Serafini G, Pompili M, Innamorati M, Giordano G, Tatarelli R, Lester D, Girardi P, Dwivedi Y. Glycosides, Depression and Suicidal Behaviour: The Role of Glycoside-Linked Proteins. Molecules. 2011; 16(3):2688-2713. https://doi.org/10.3390/molecules16032688
Chicago/Turabian StyleSerafini, Gianluca, Maurizio Pompili, Marco Innamorati, Gloria Giordano, Roberto Tatarelli, David Lester, Paolo Girardi, and Yogesh Dwivedi. 2011. "Glycosides, Depression and Suicidal Behaviour: The Role of Glycoside-Linked Proteins" Molecules 16, no. 3: 2688-2713. https://doi.org/10.3390/molecules16032688
APA StyleSerafini, G., Pompili, M., Innamorati, M., Giordano, G., Tatarelli, R., Lester, D., Girardi, P., & Dwivedi, Y. (2011). Glycosides, Depression and Suicidal Behaviour: The Role of Glycoside-Linked Proteins. Molecules, 16(3), 2688-2713. https://doi.org/10.3390/molecules16032688