Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview

Abstract
:1. Introduction
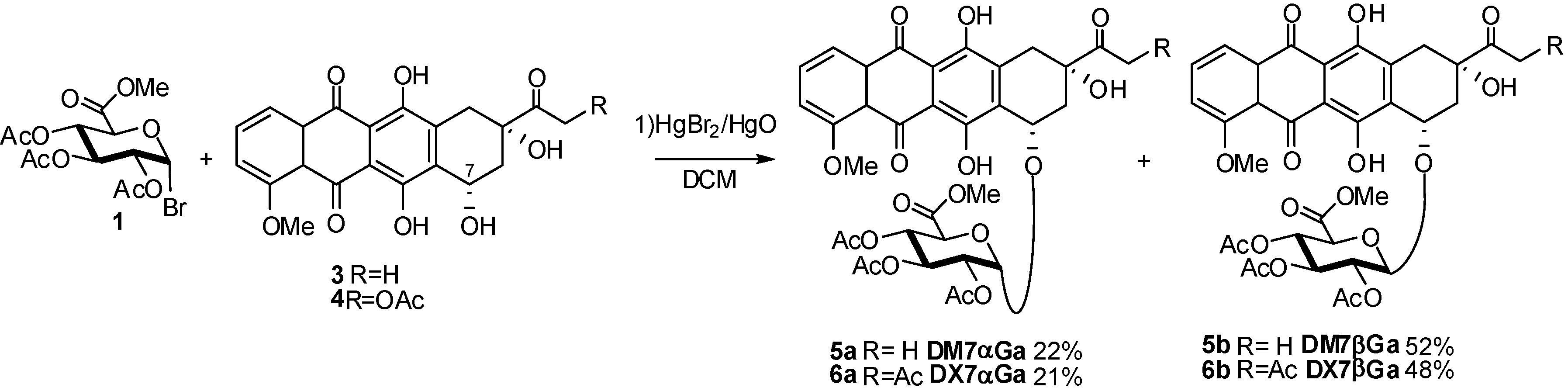
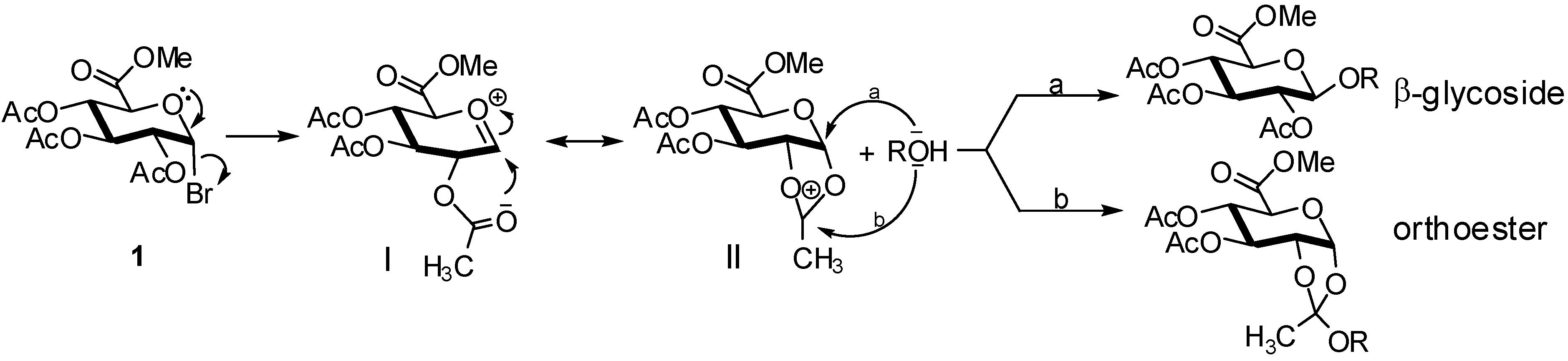
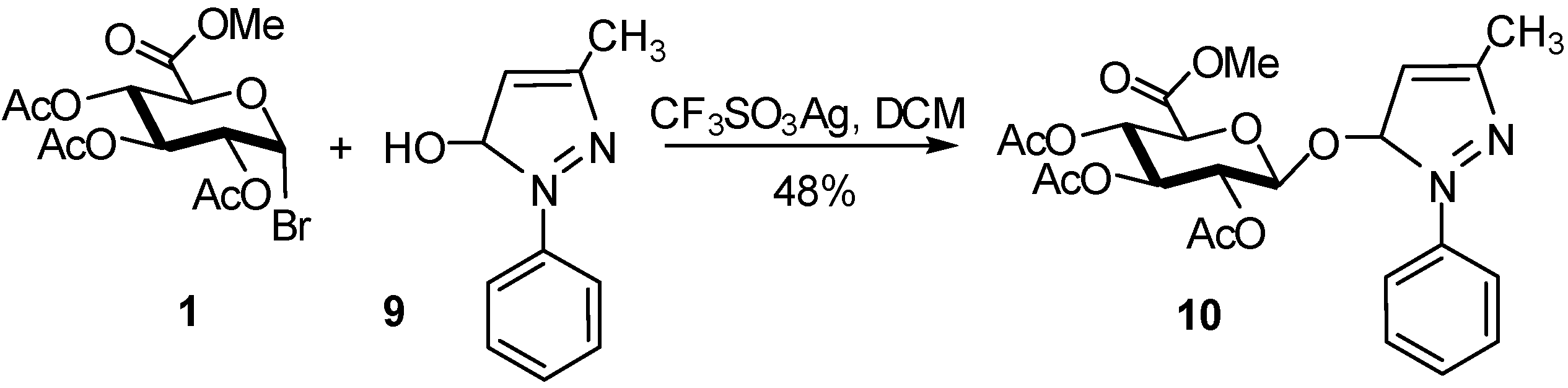
2. Glucuronidation
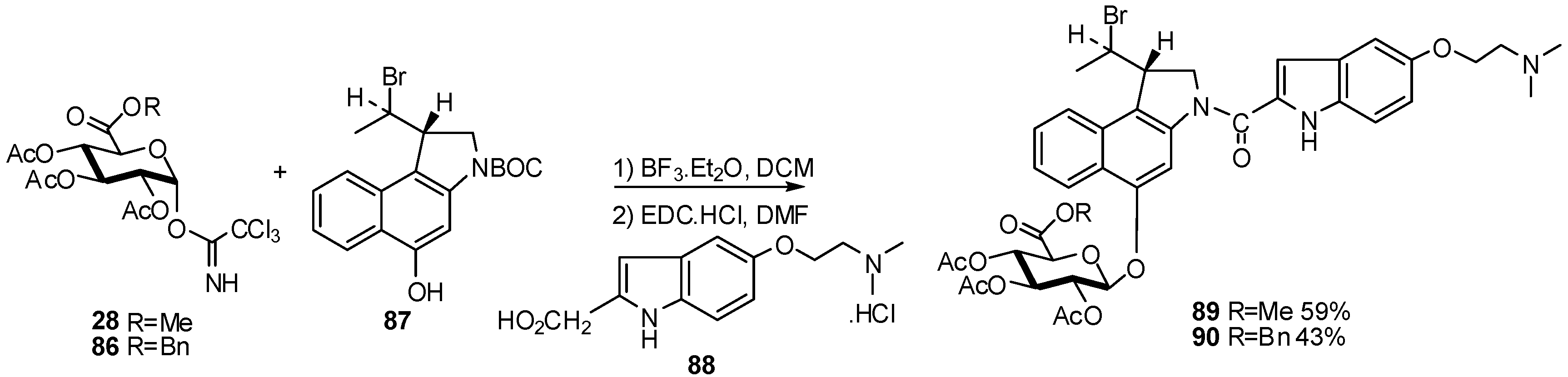
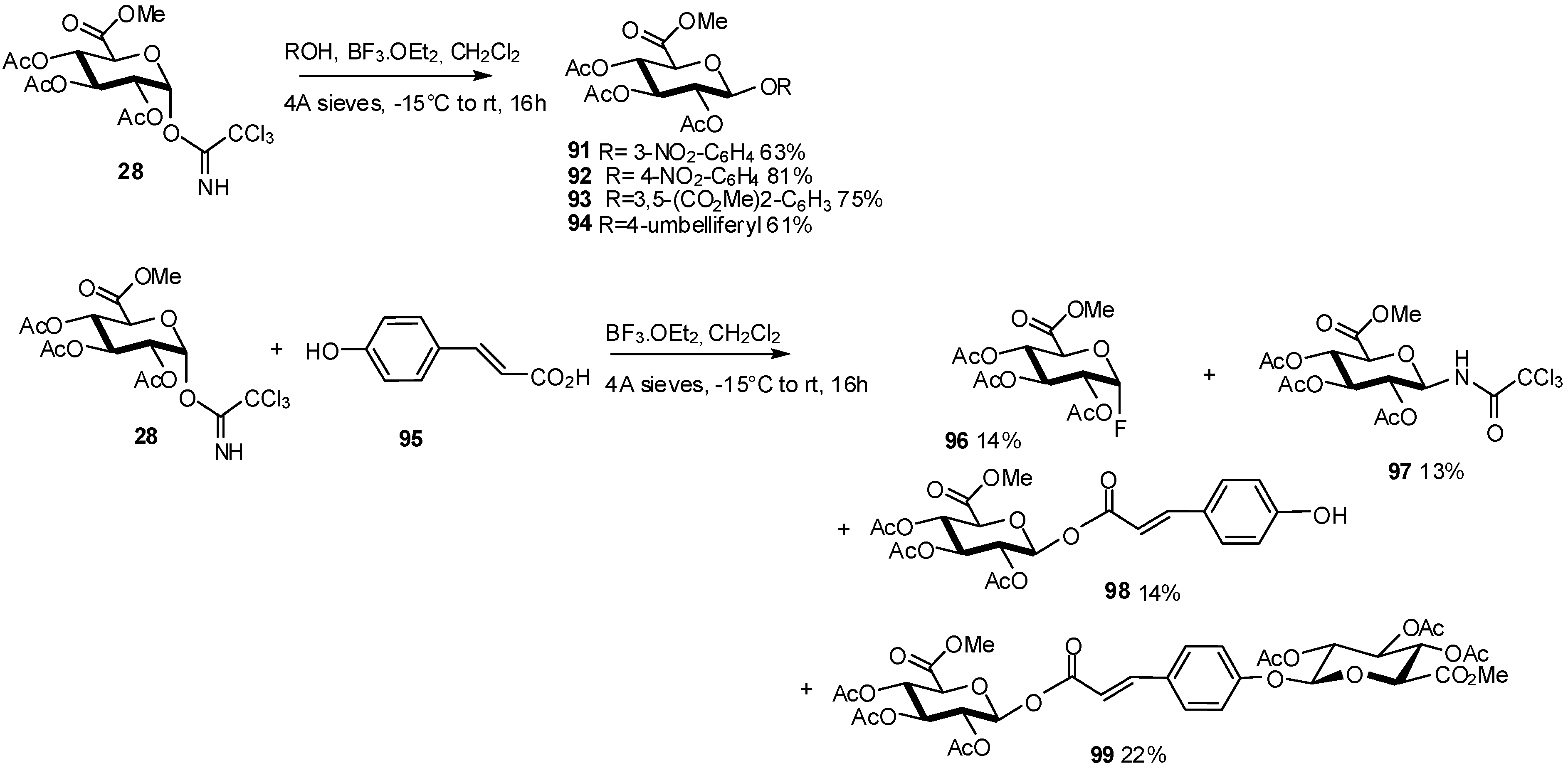
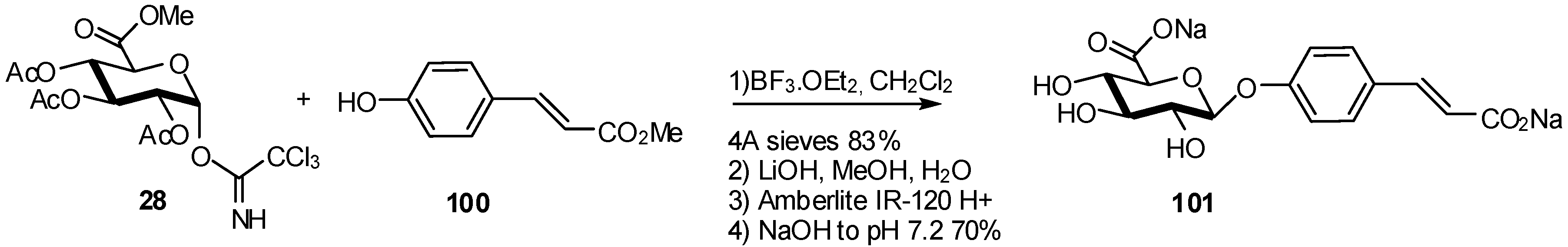
2.1. Metabolites Synthesis



















2.2. Prodrug Therapy



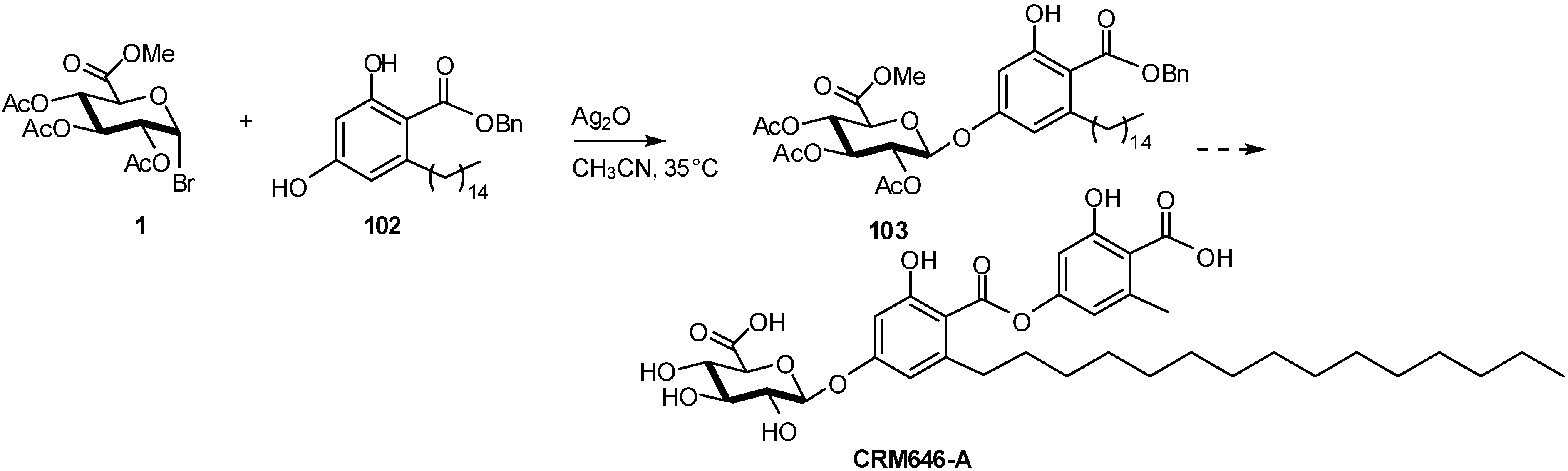
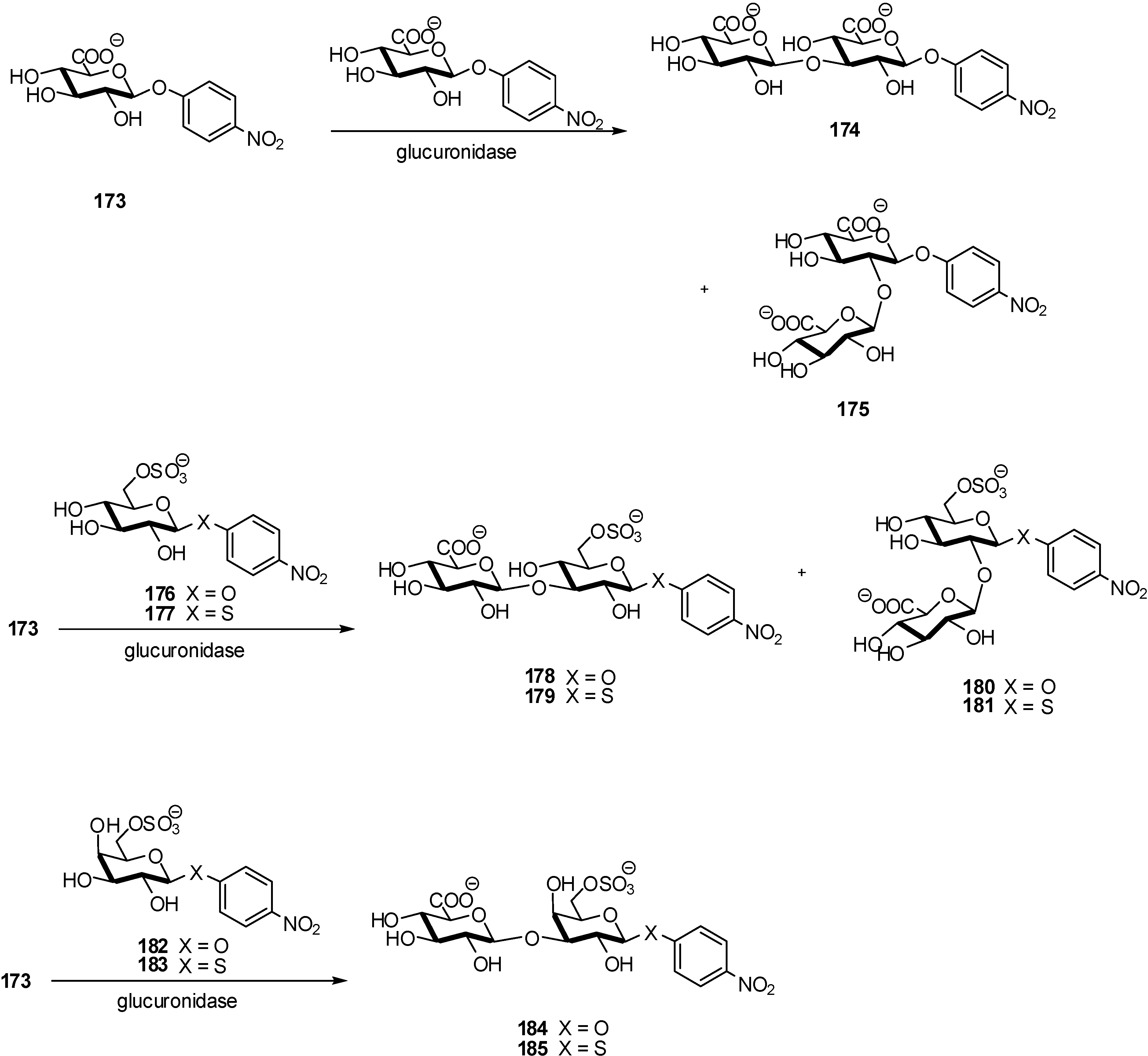
2.3. Antibacterials Inhibitors




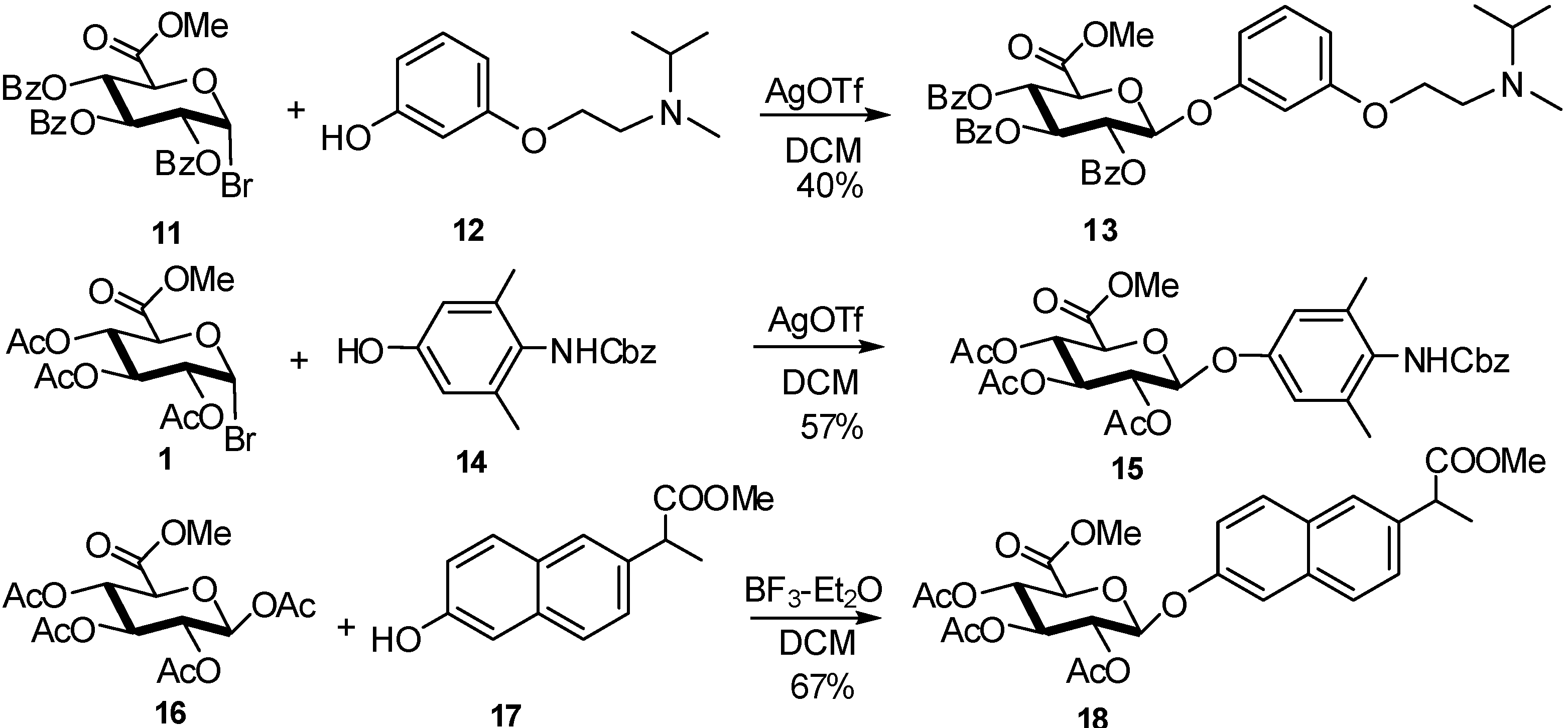
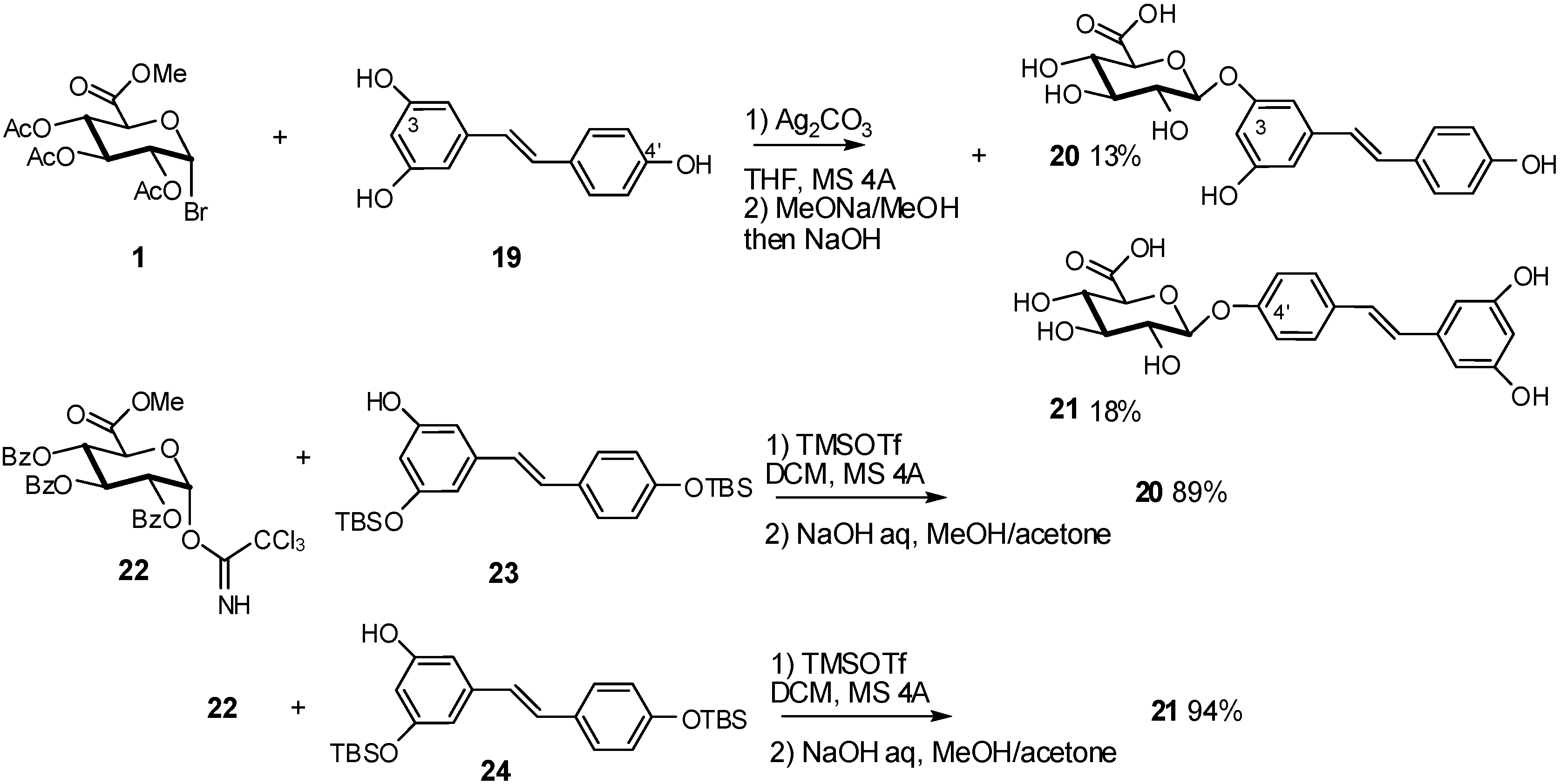
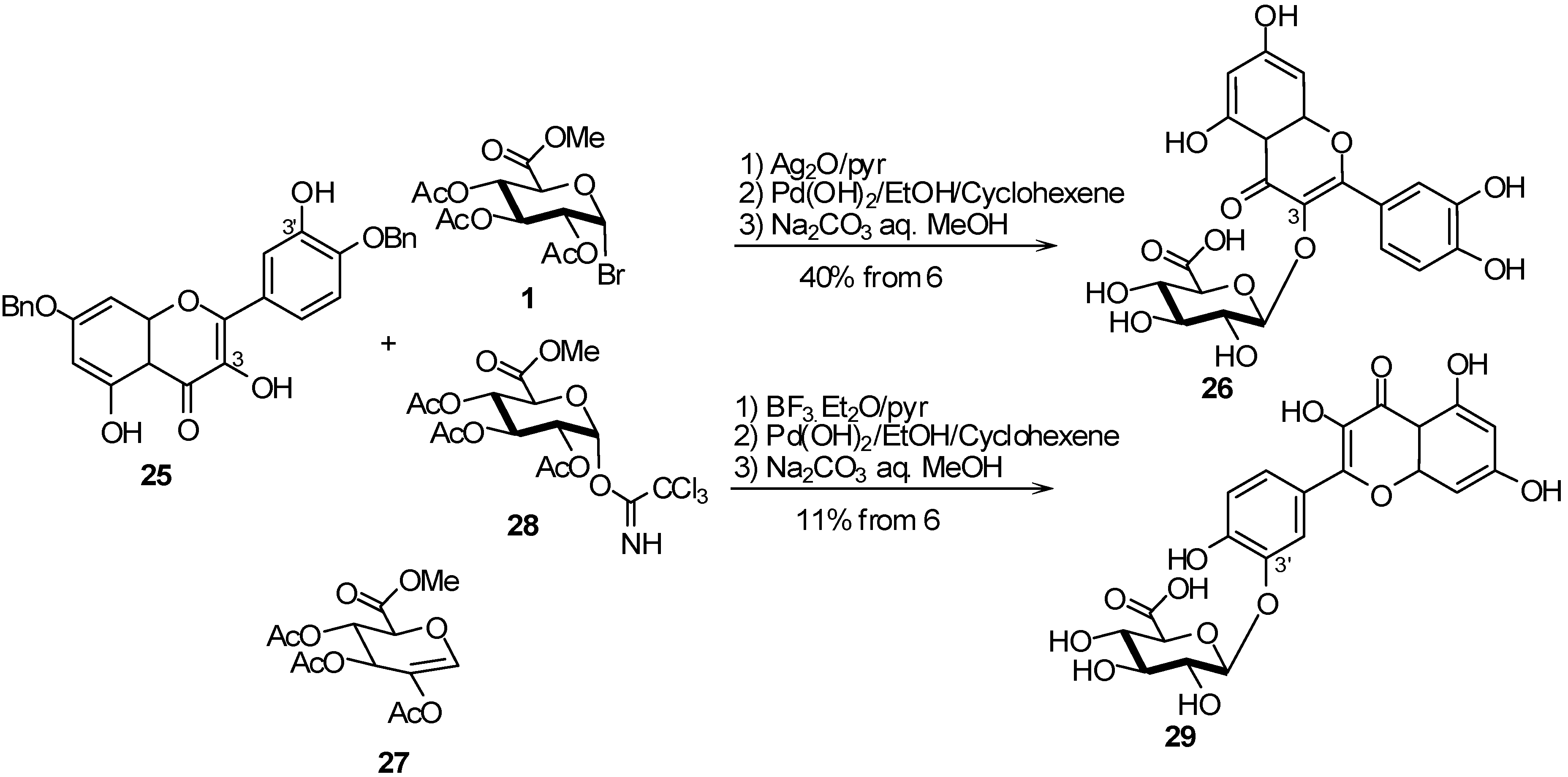
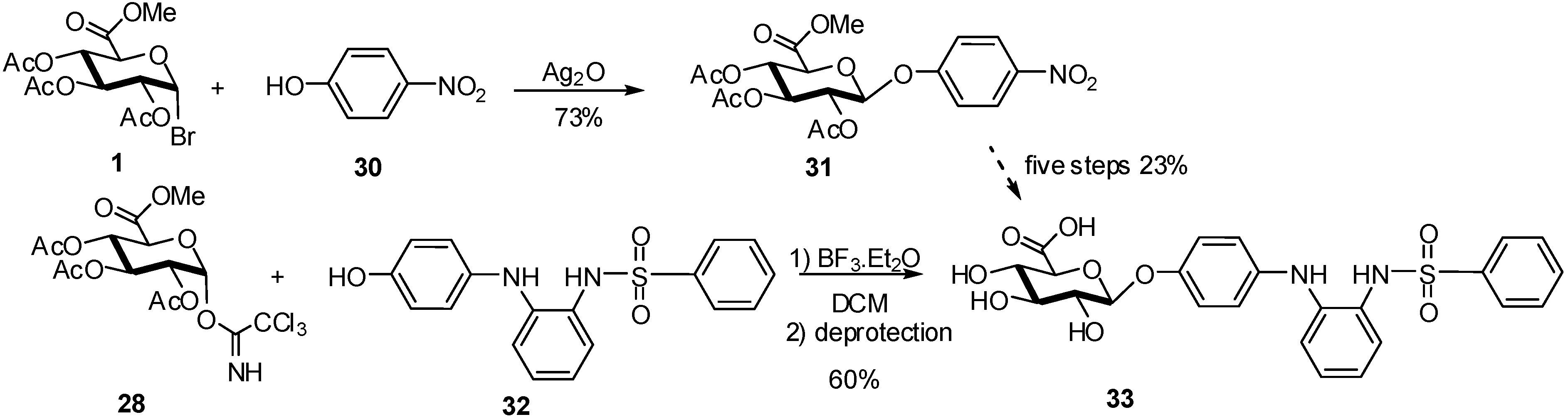
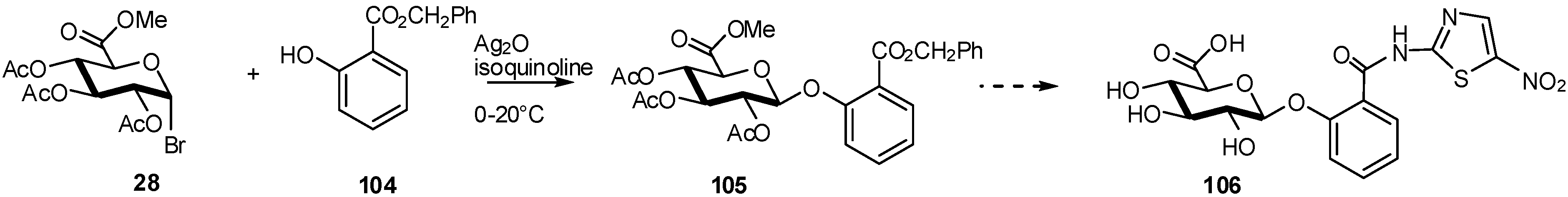
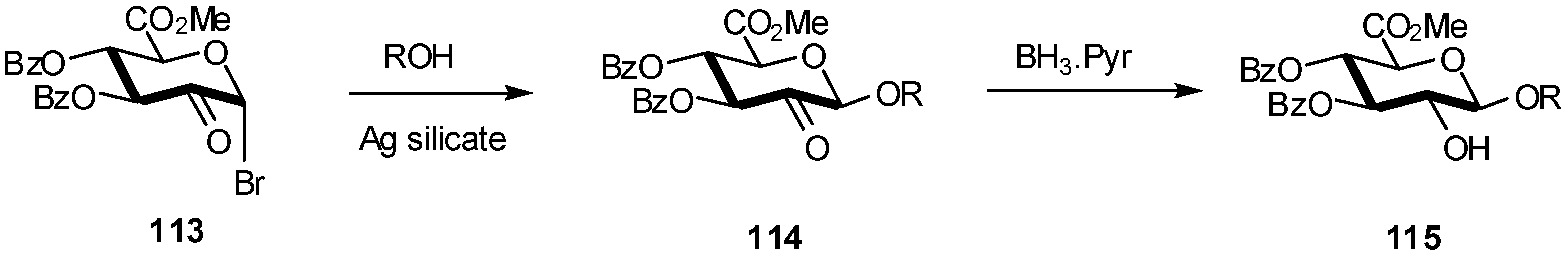
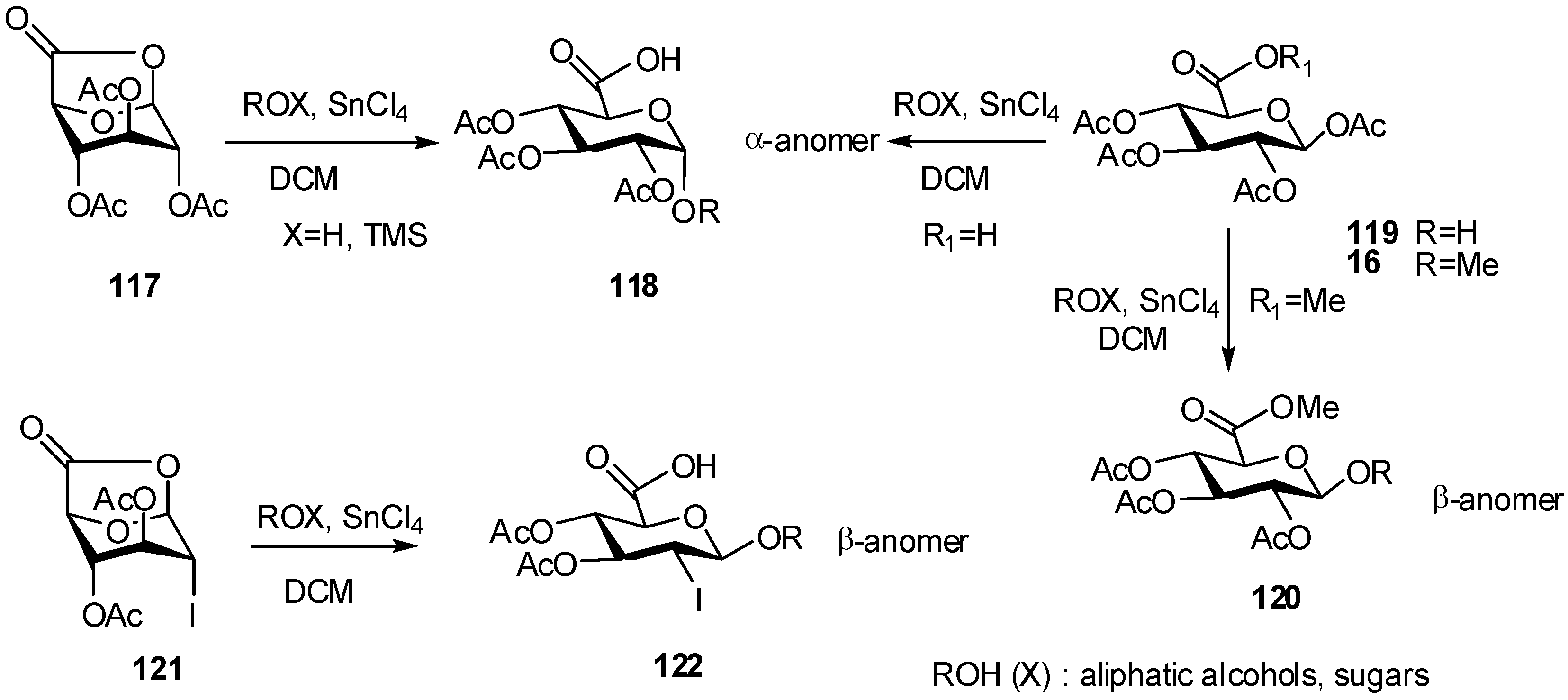
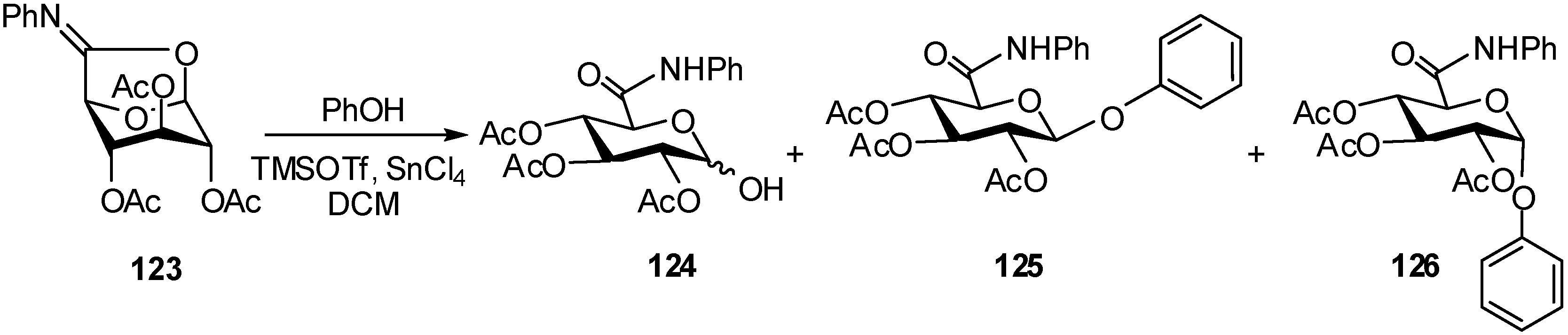
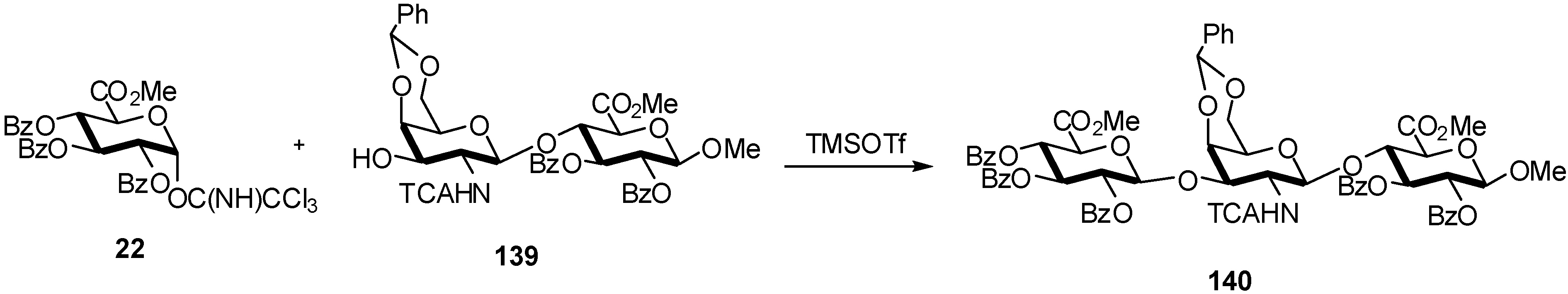
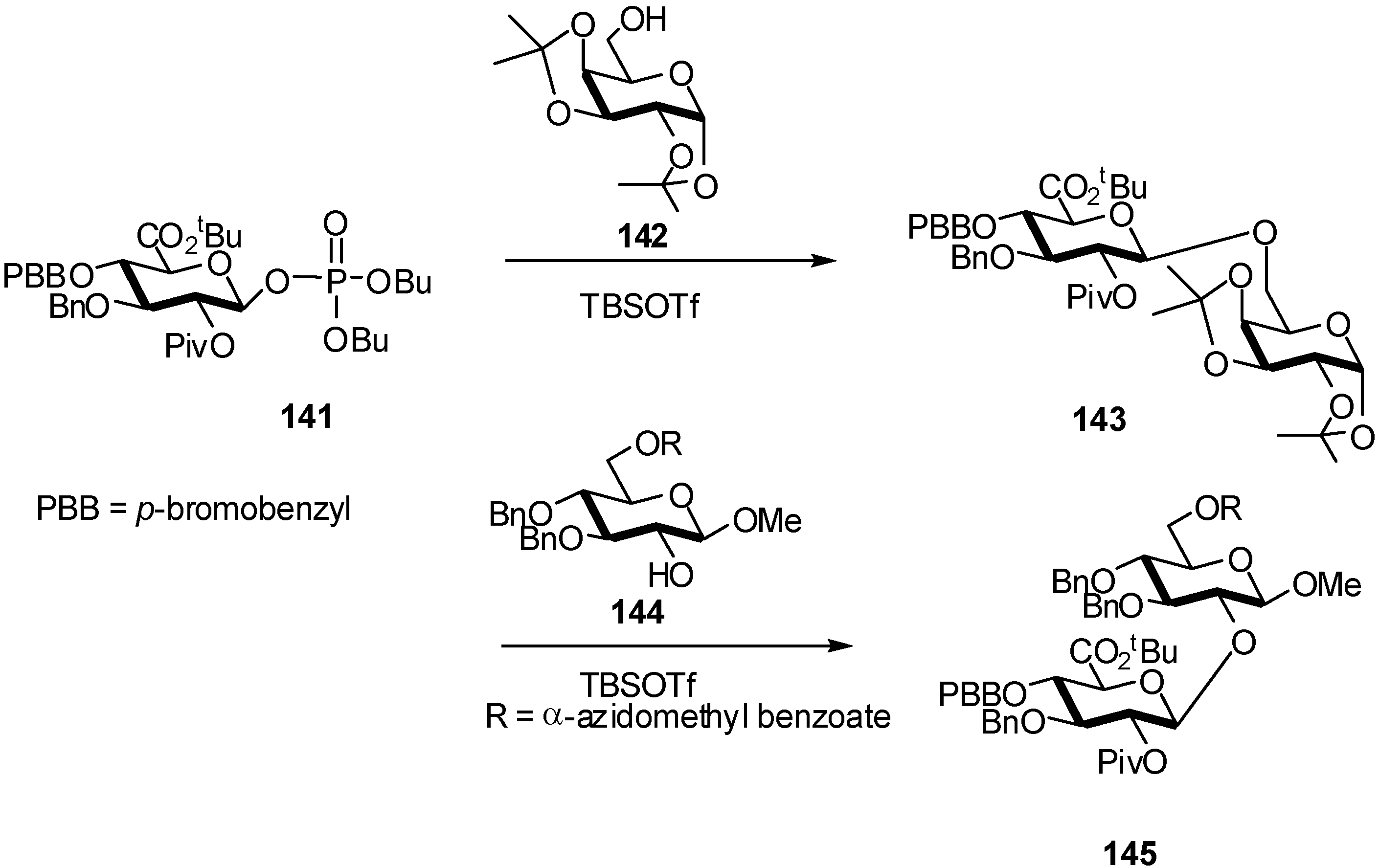
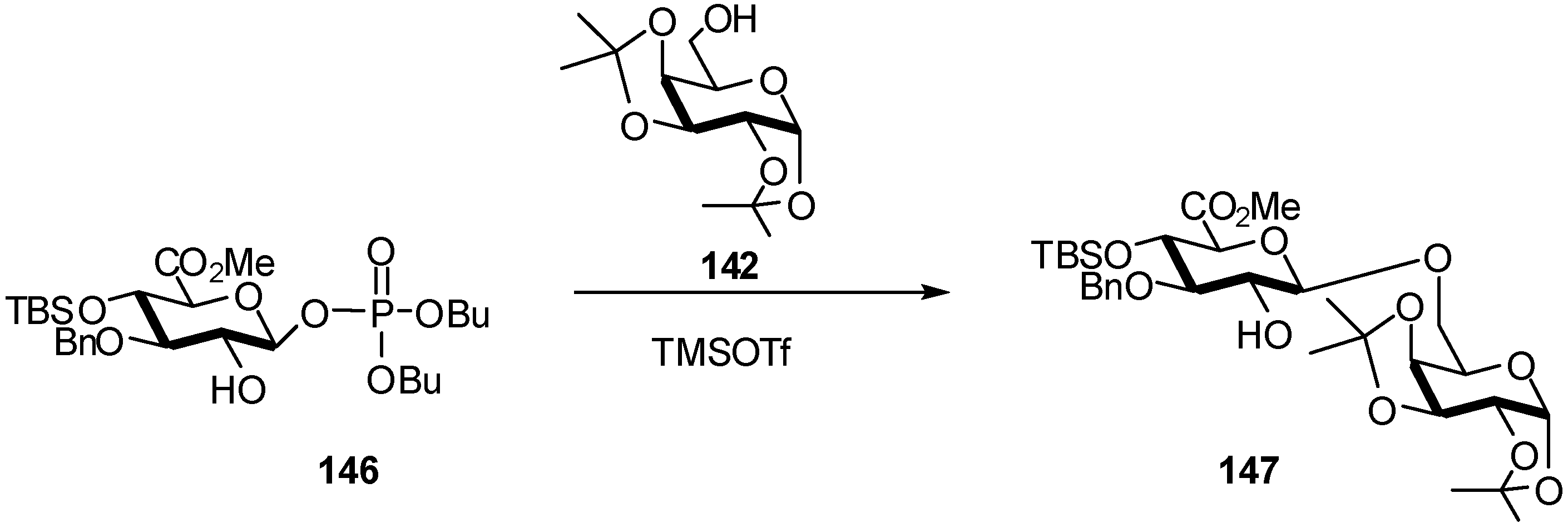
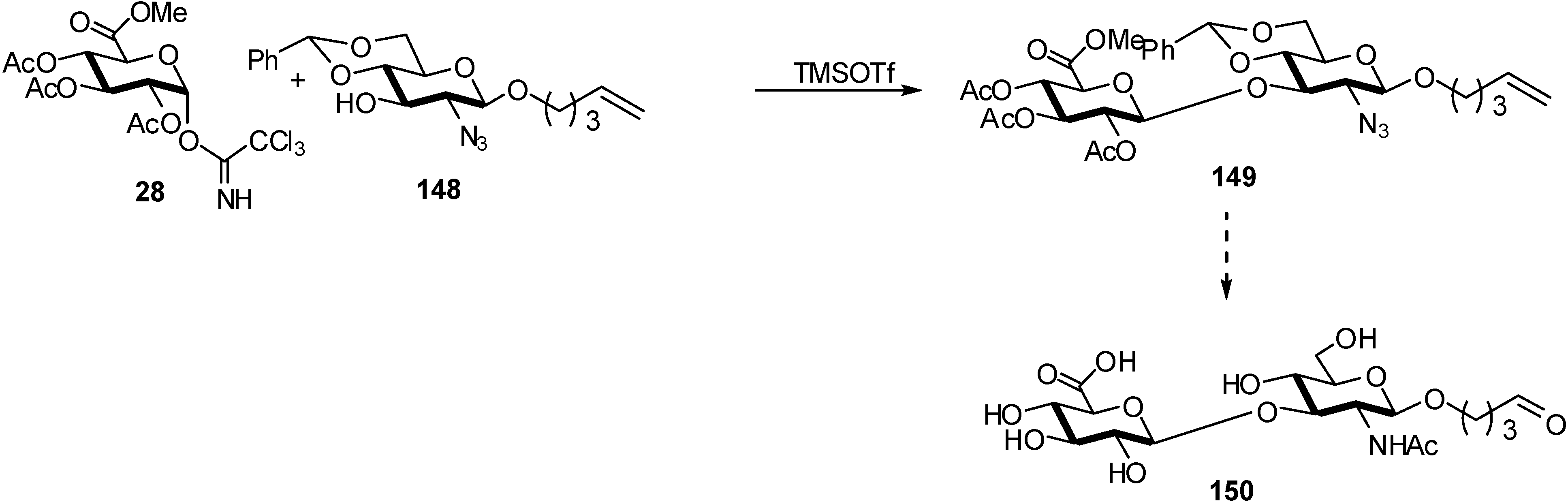
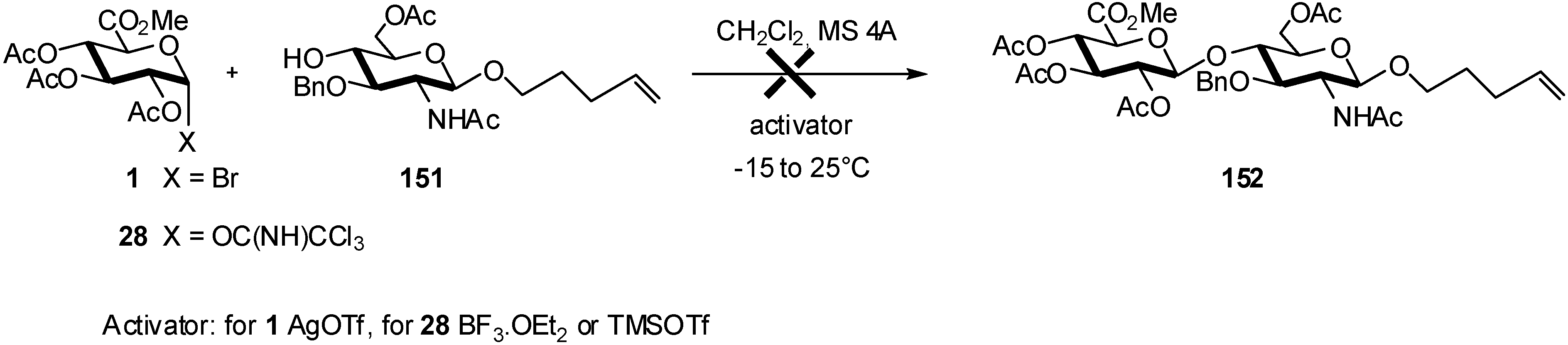
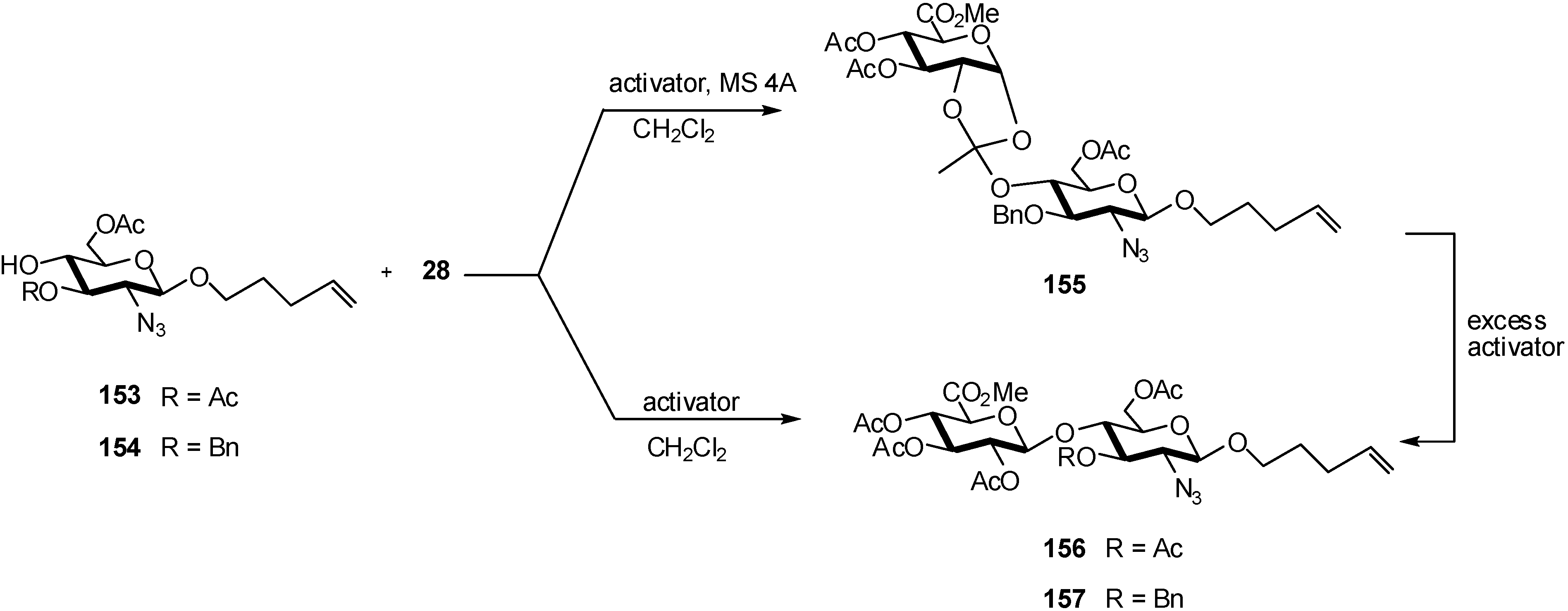
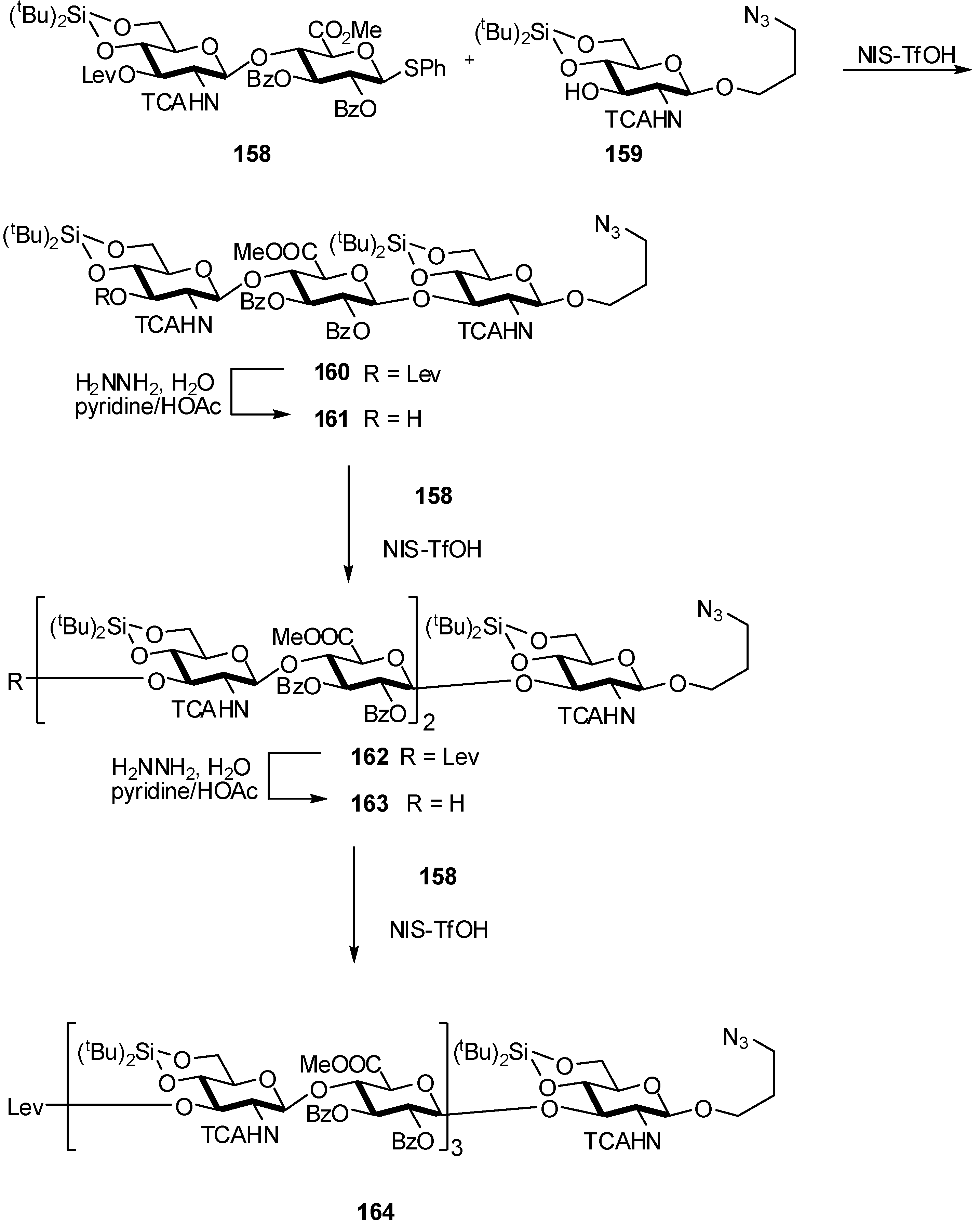
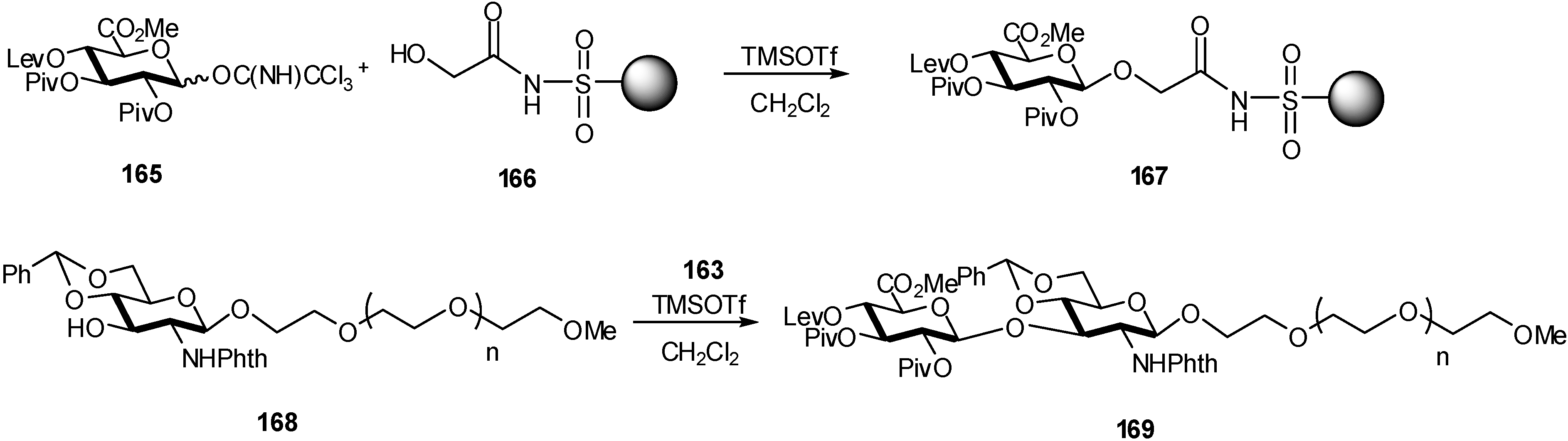
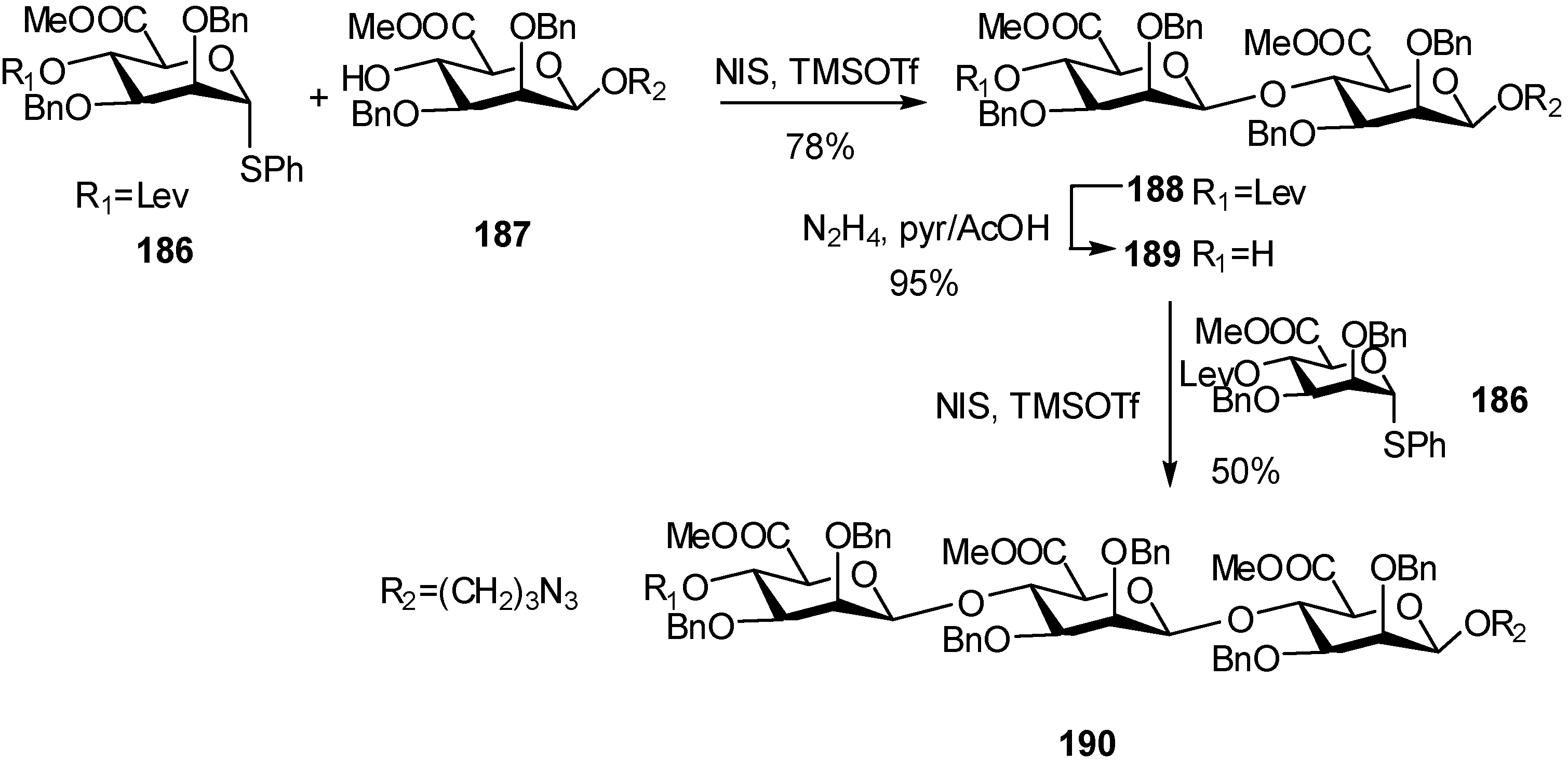
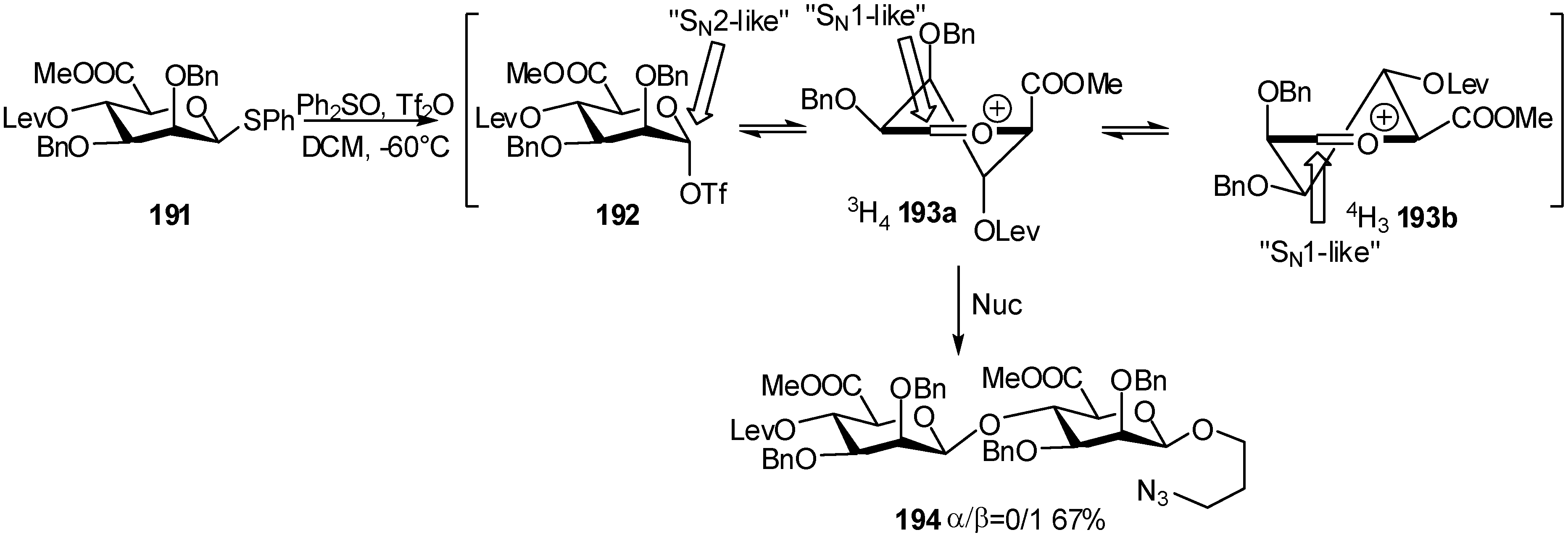
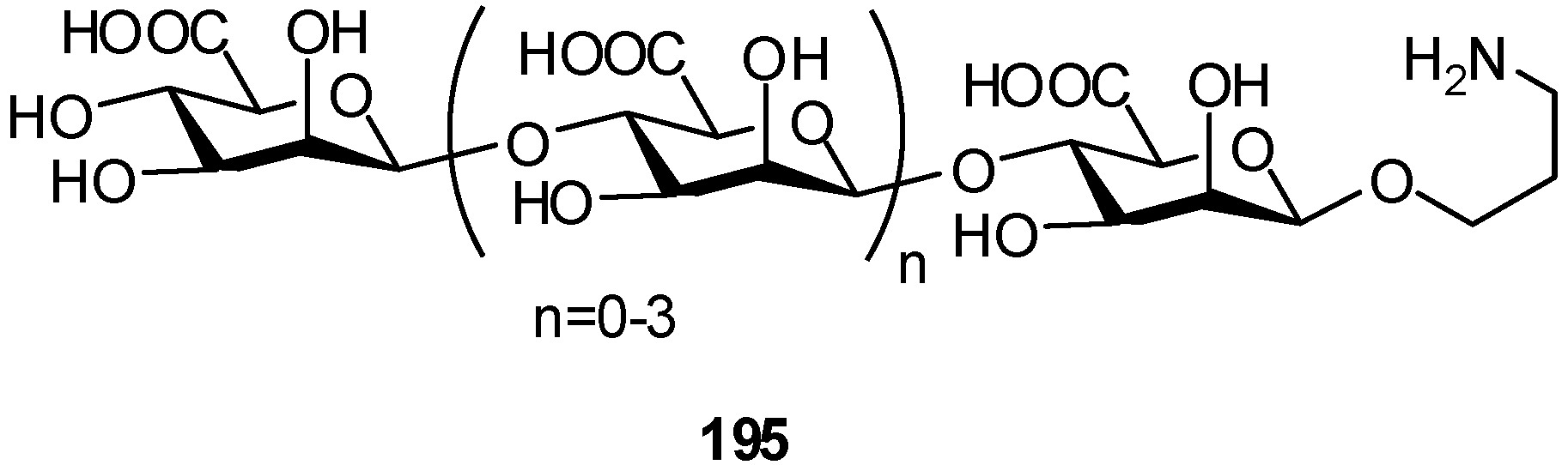
2.4. Methodologies and synthesis of oligosaccharides



















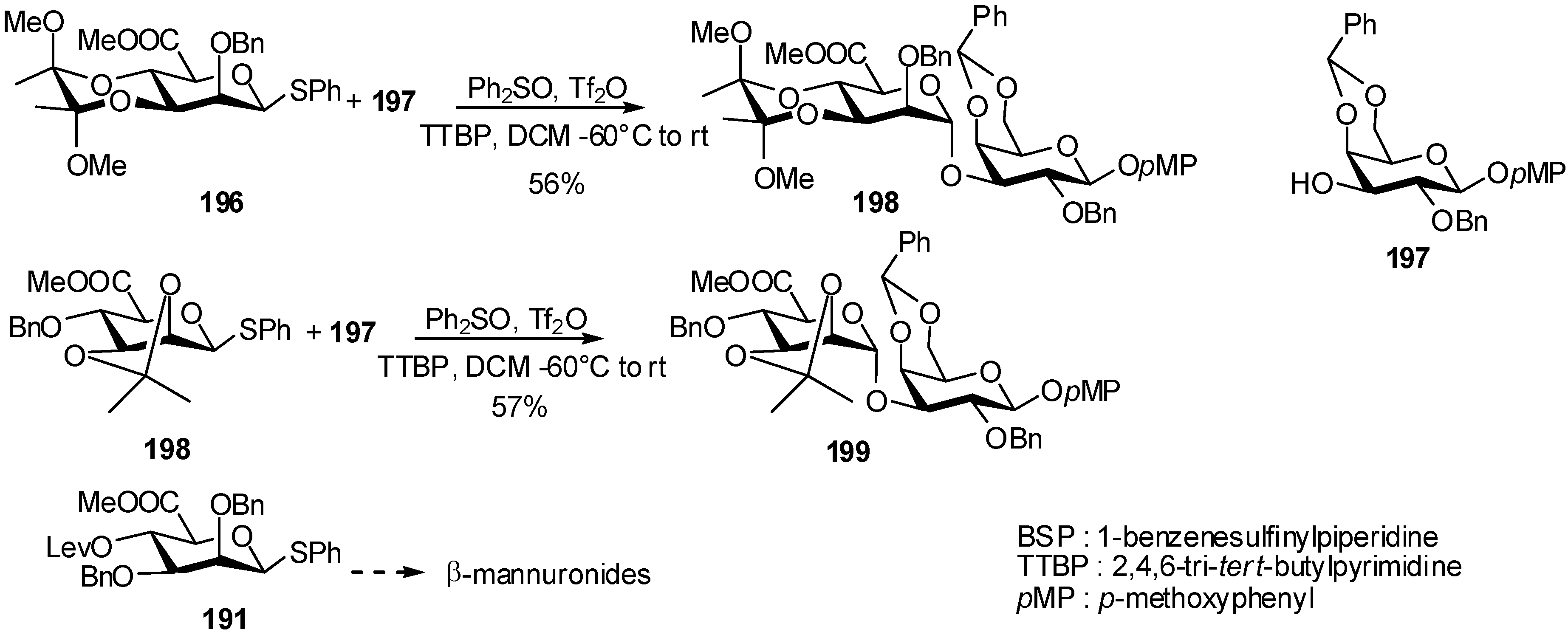
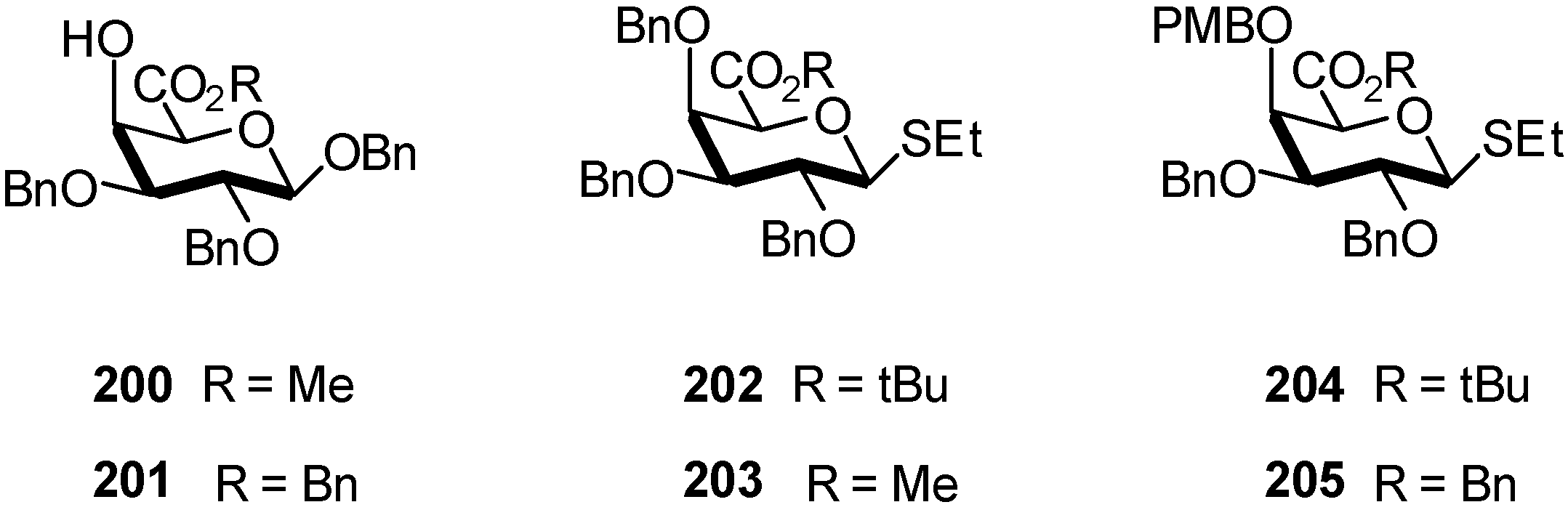
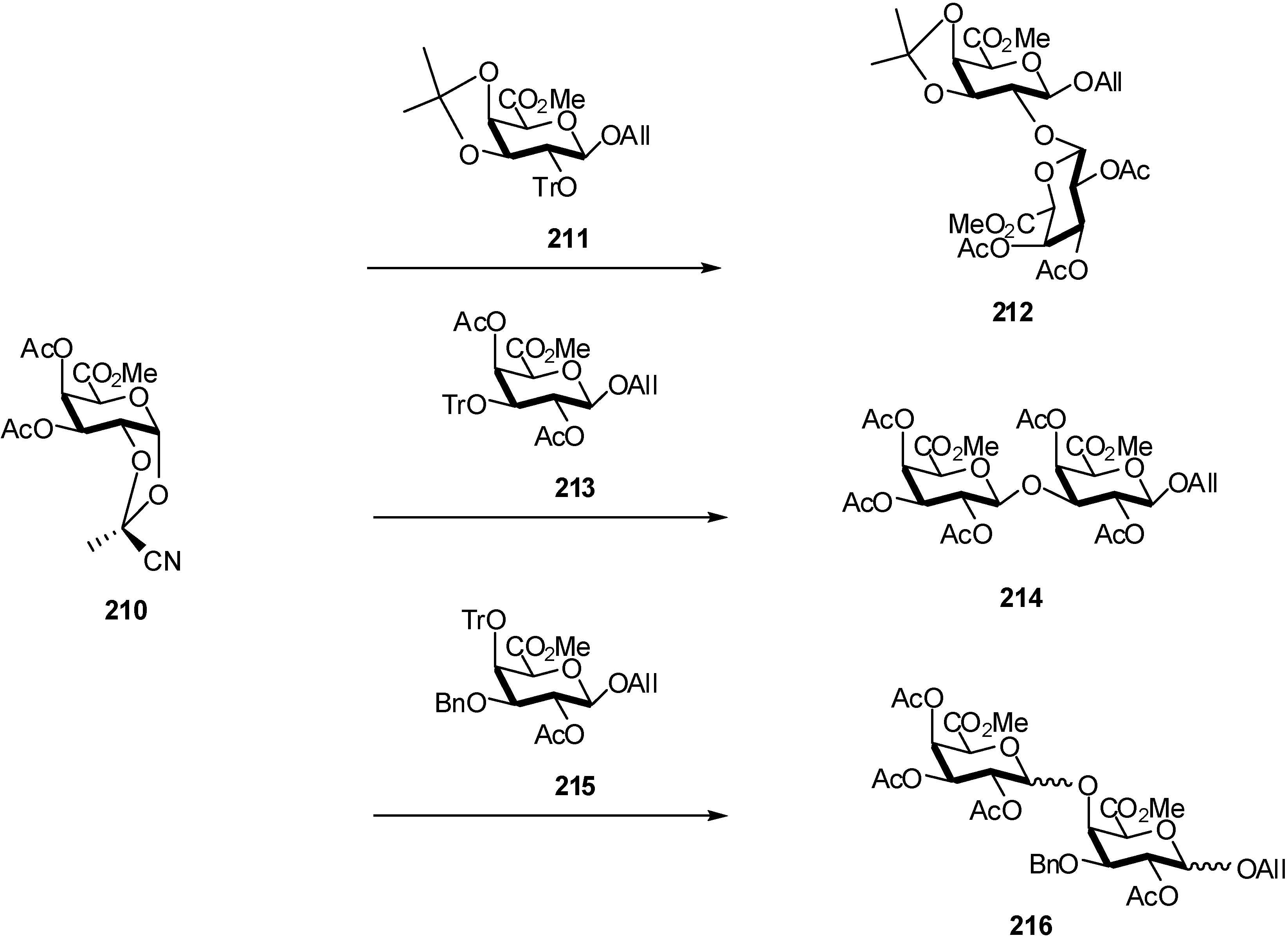
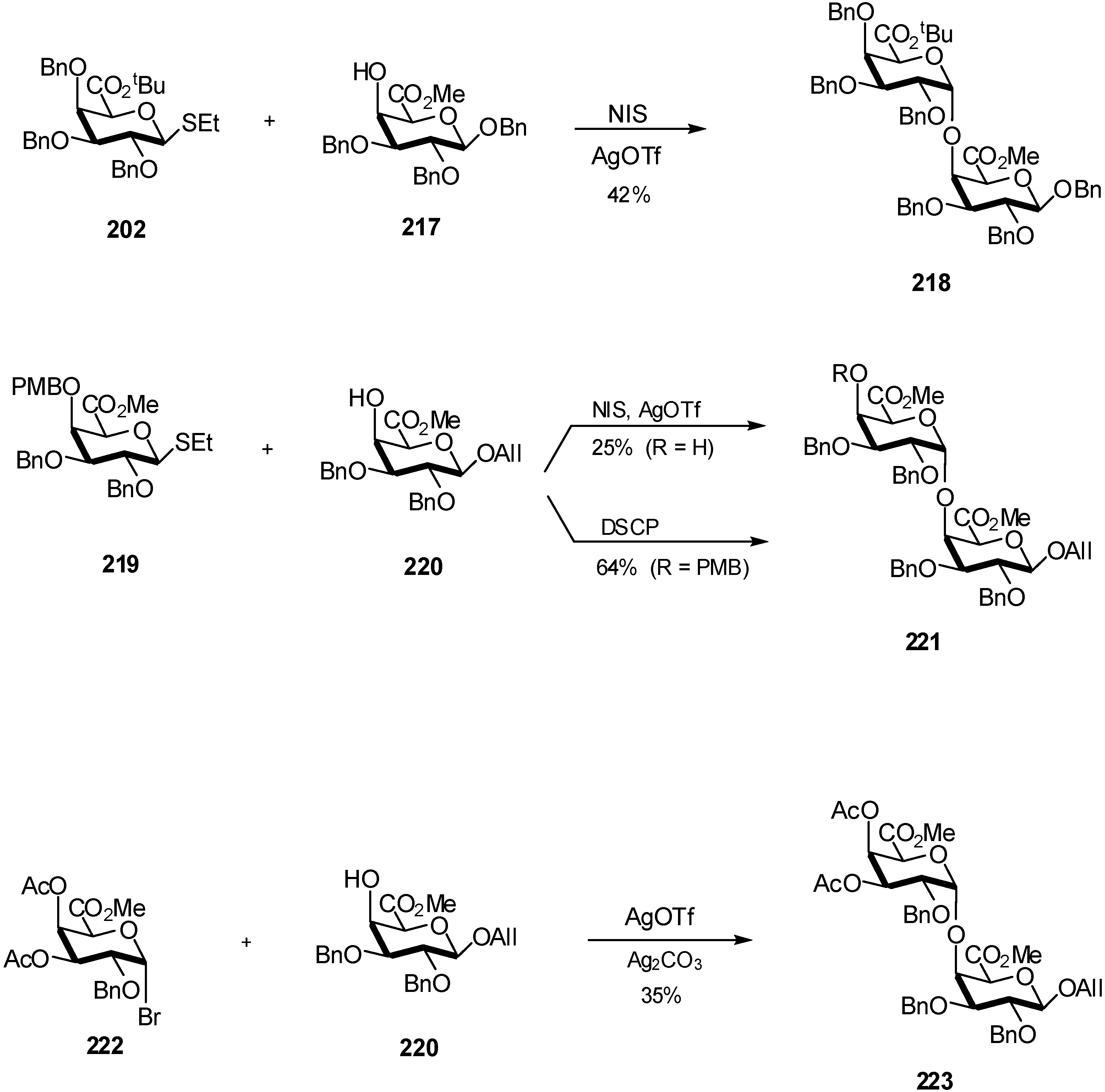
3. Mannuronidation




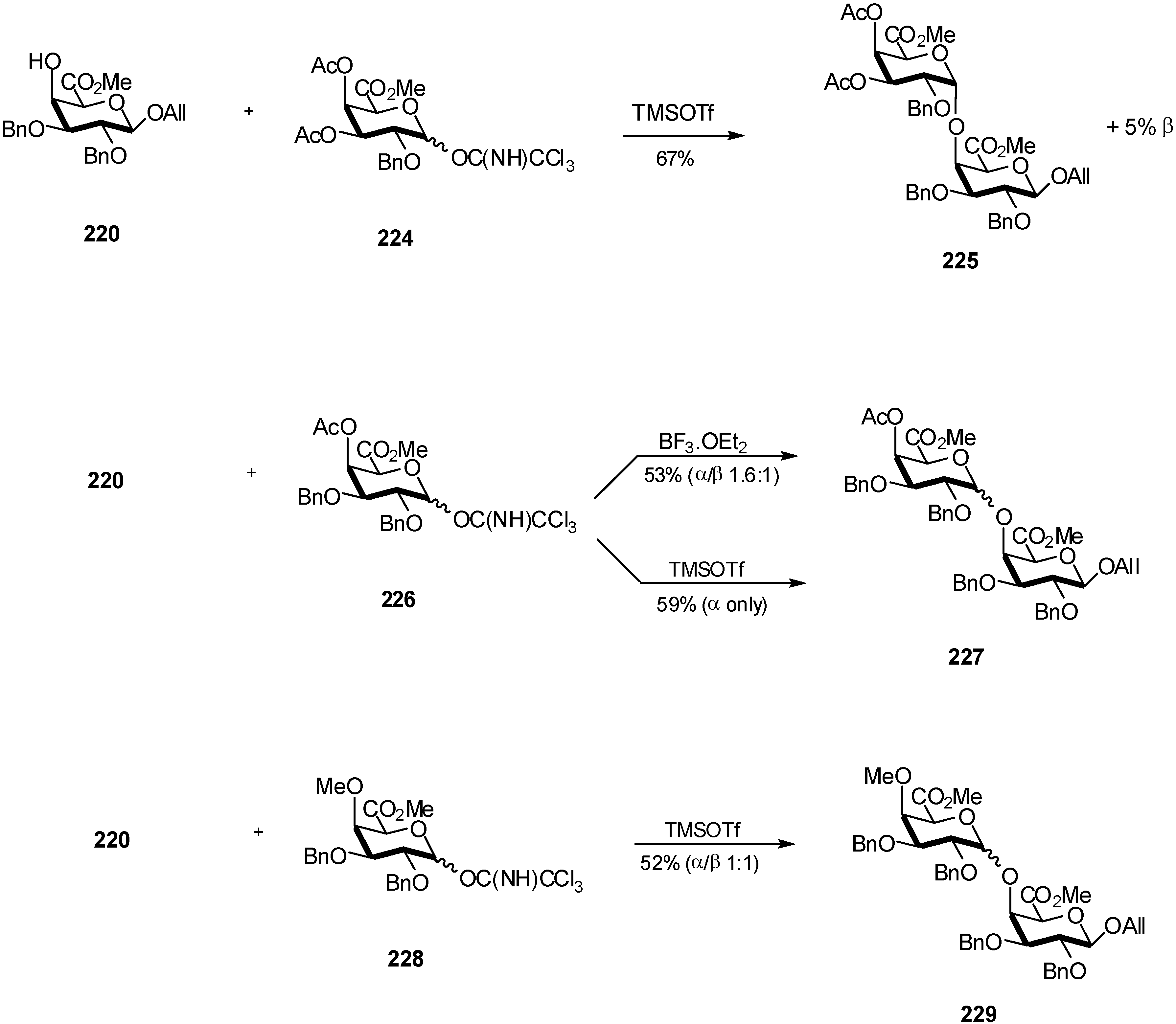
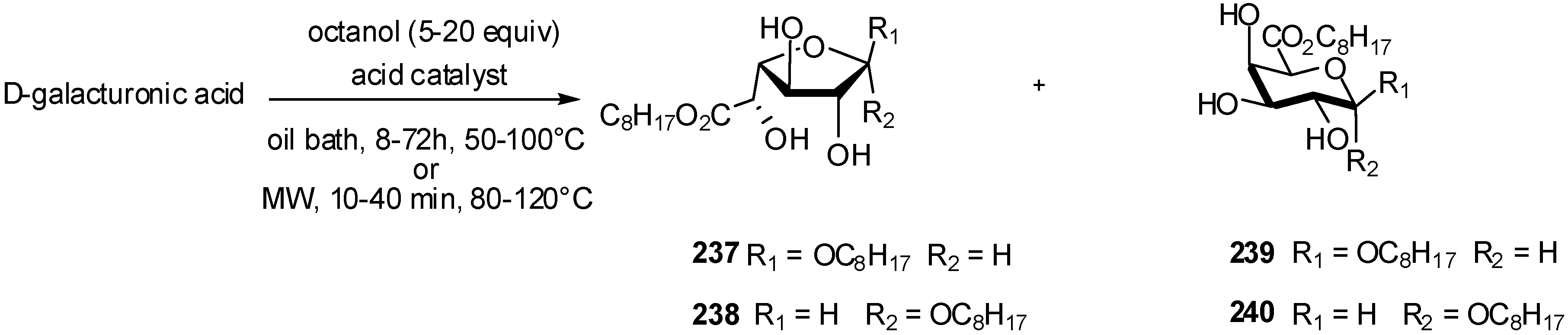
4. Galacturonidation


{kind=link}
{kind=link}
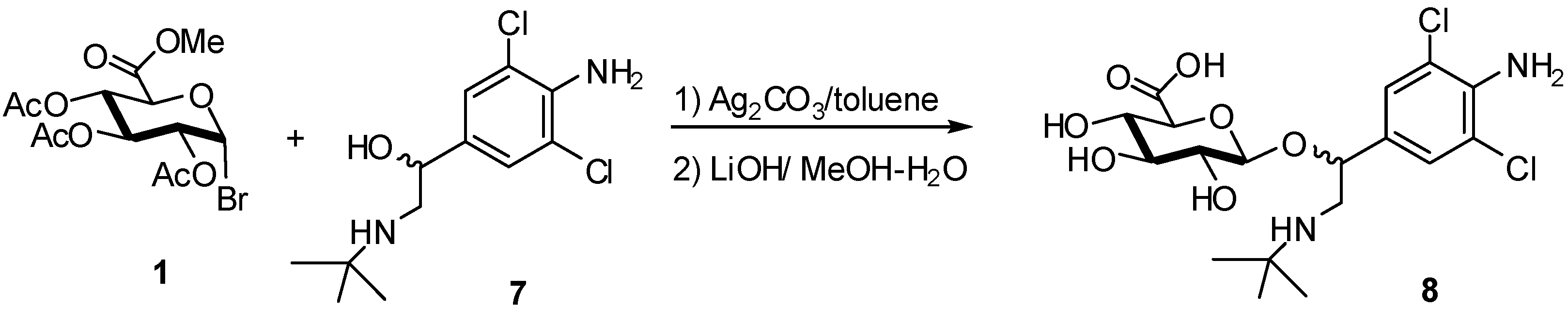{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
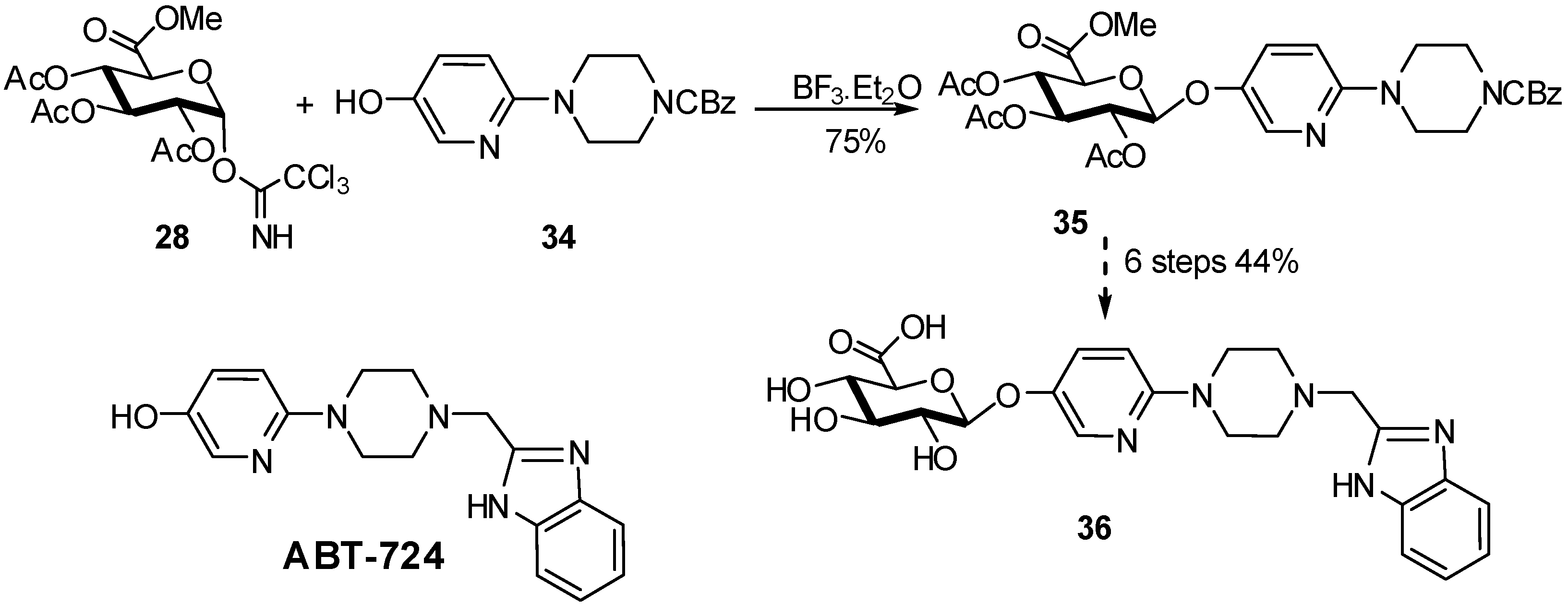{kind=link}
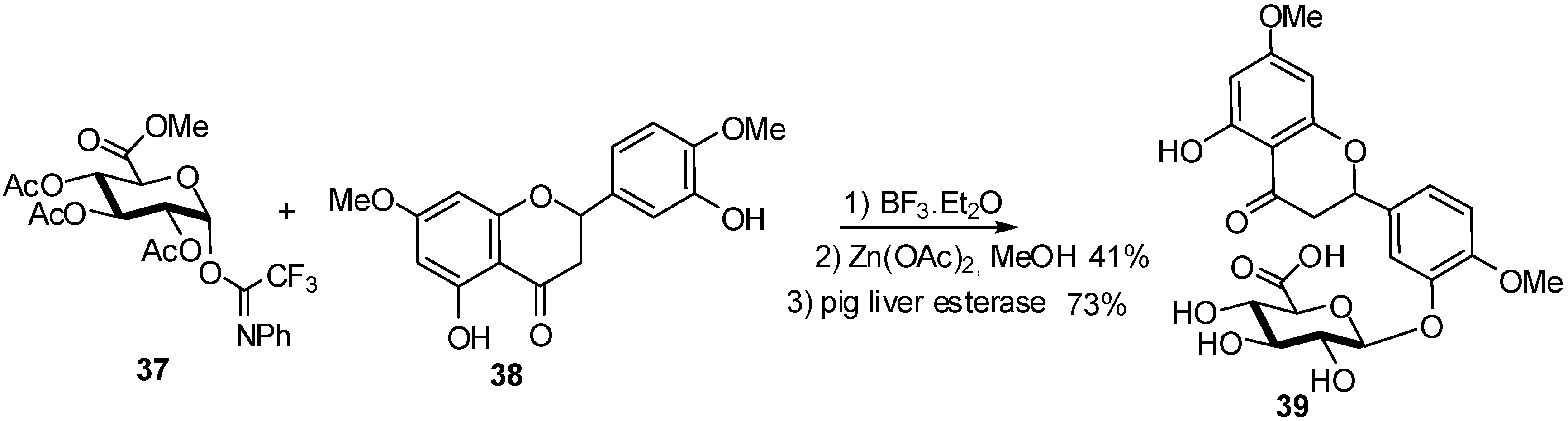{kind=link}
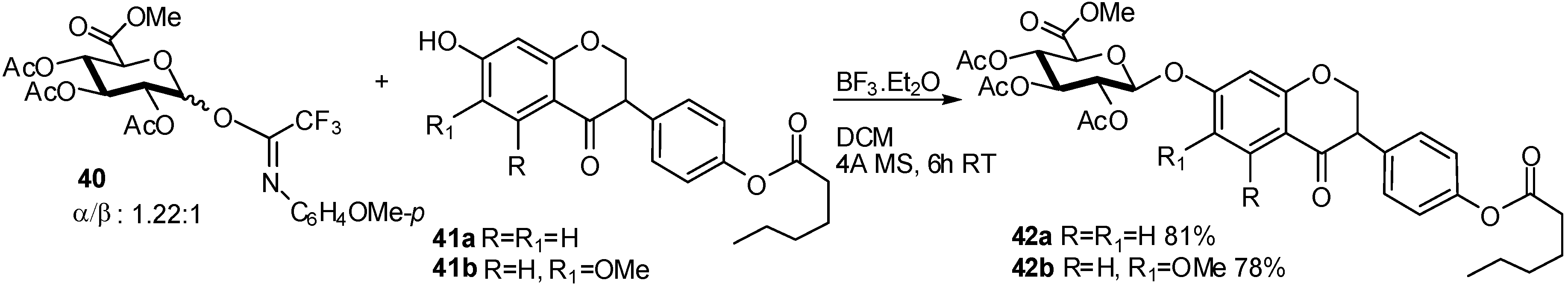{kind=link}
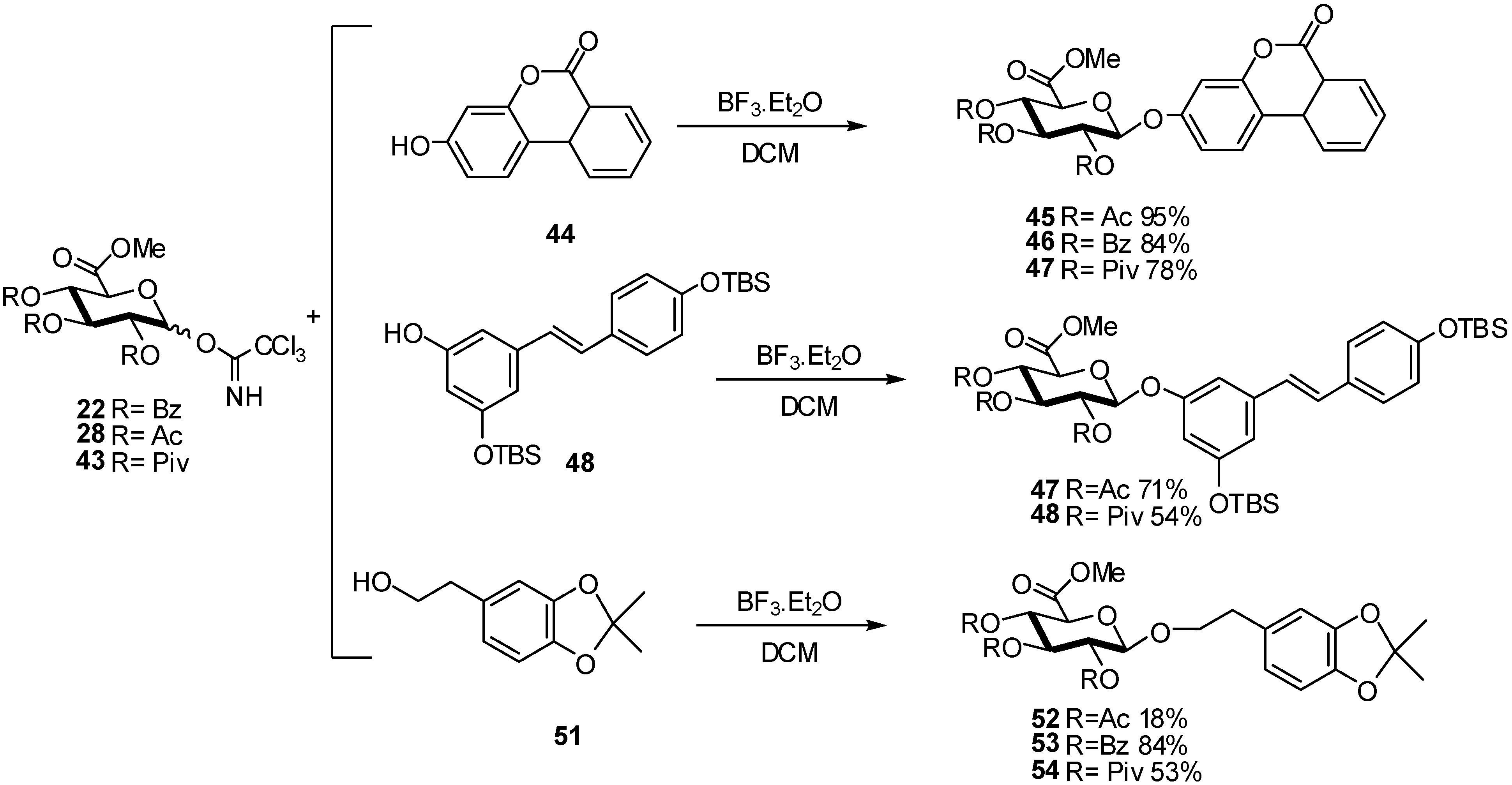{kind=link}
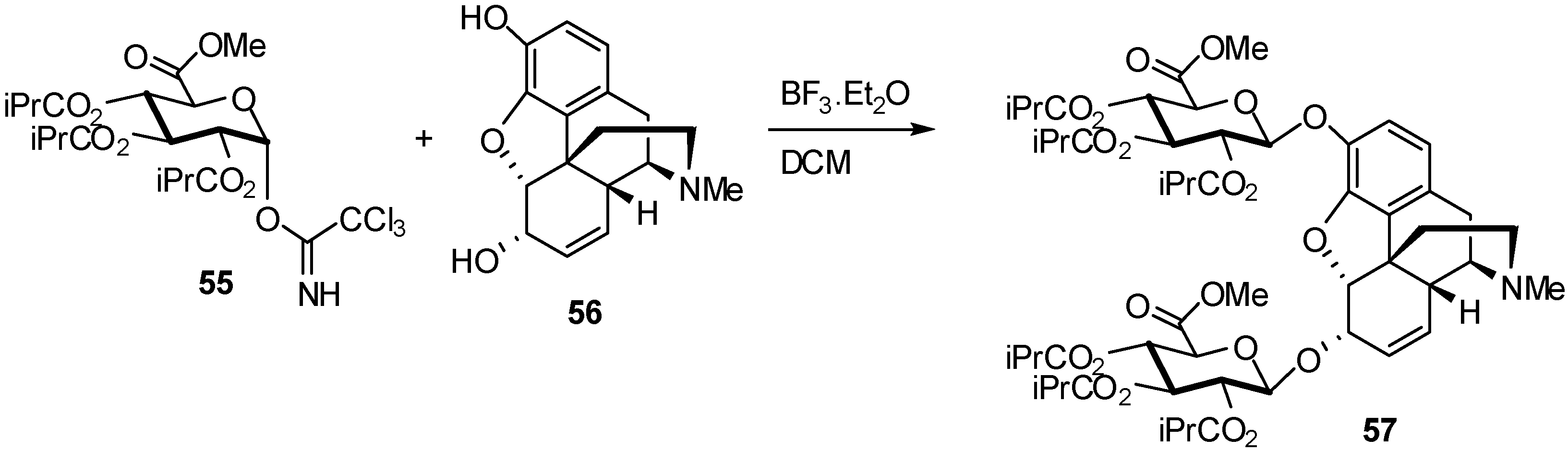{kind=link}
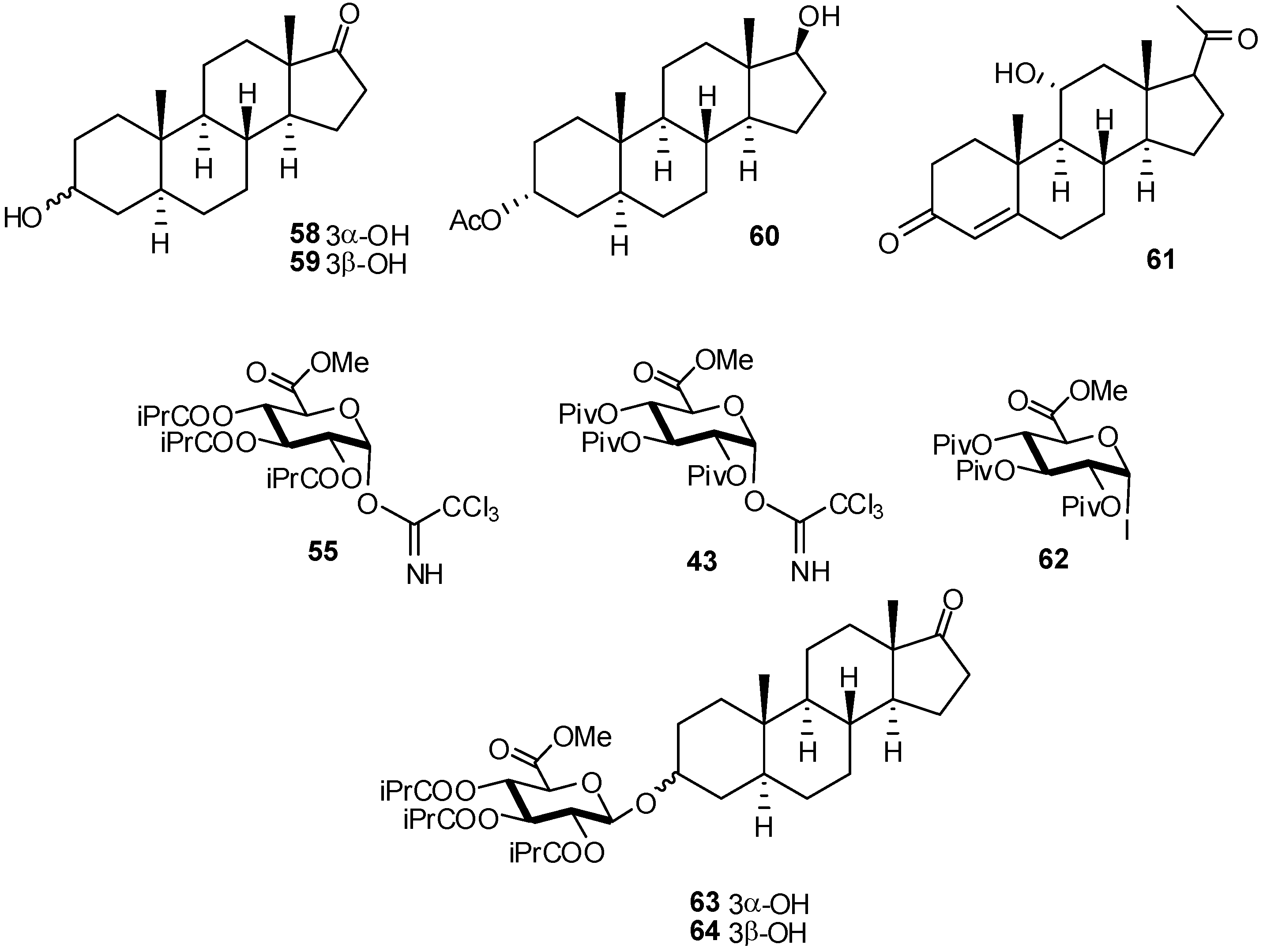{kind=link}
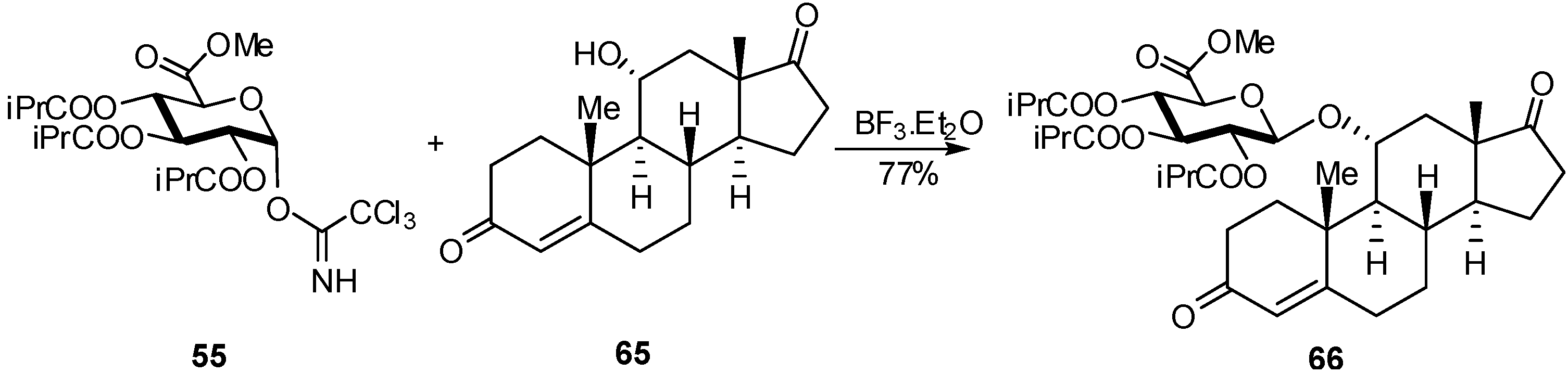{kind=link}
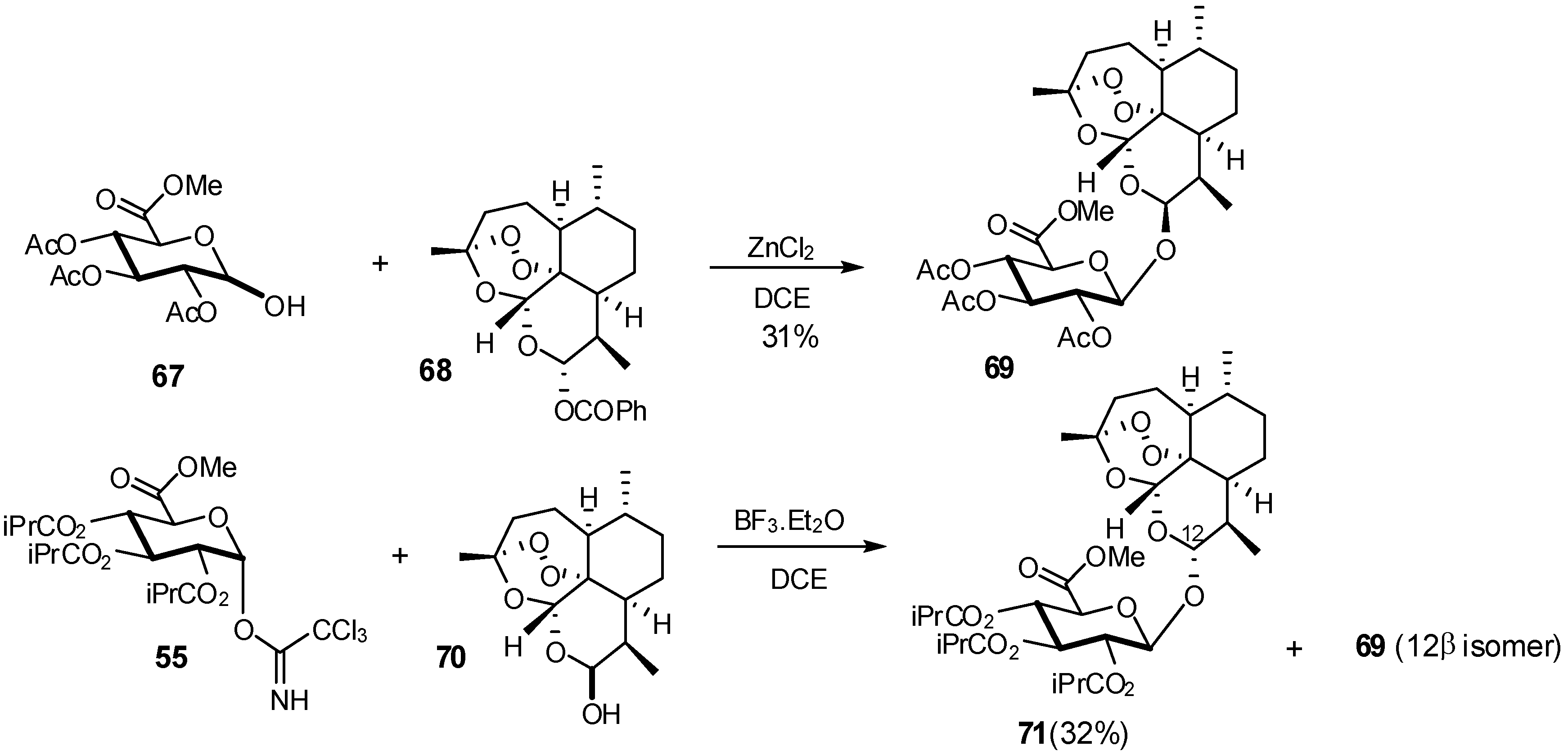{kind=link}
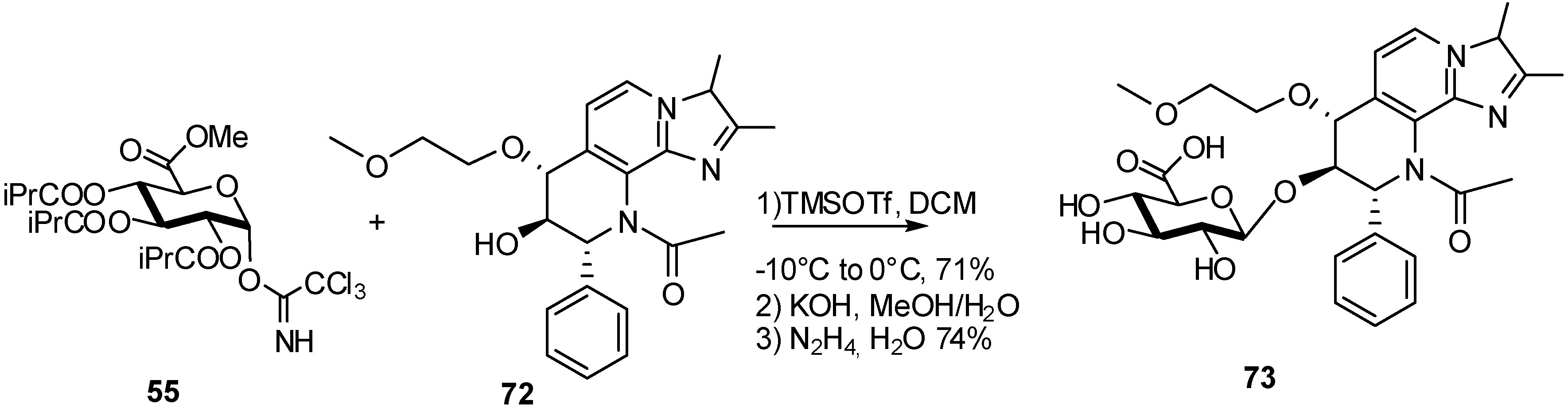{kind=link}
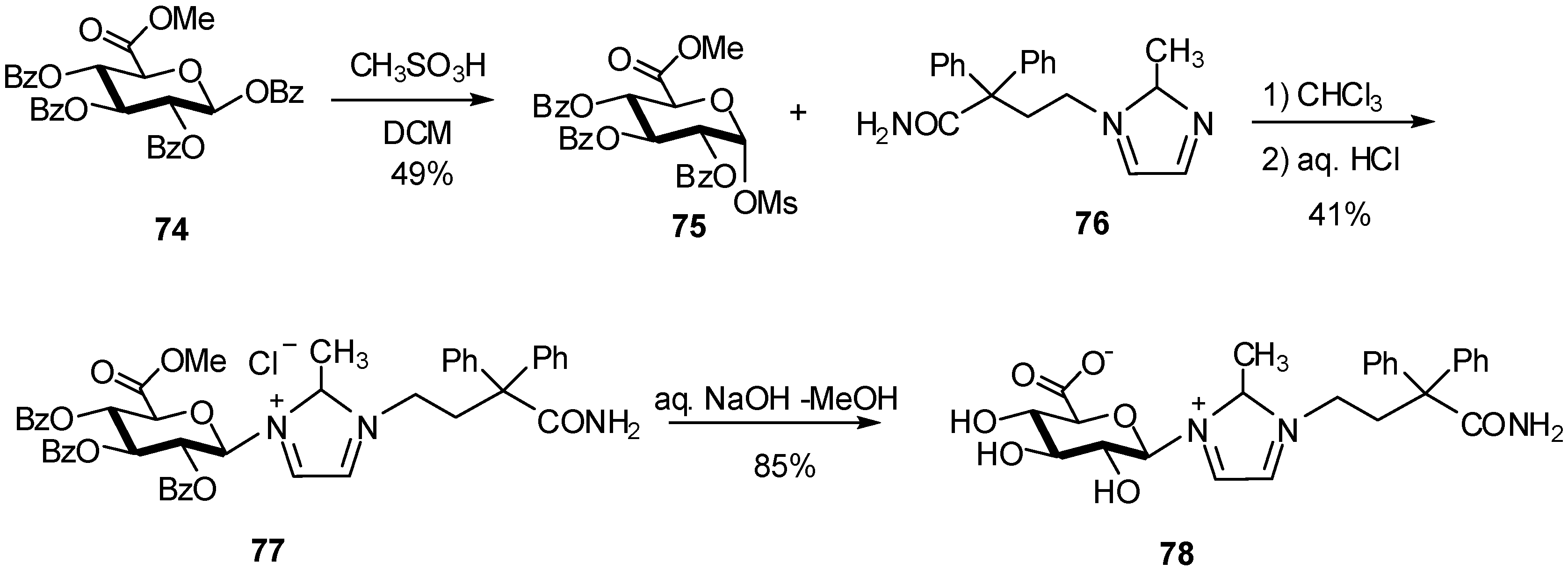{kind=link}
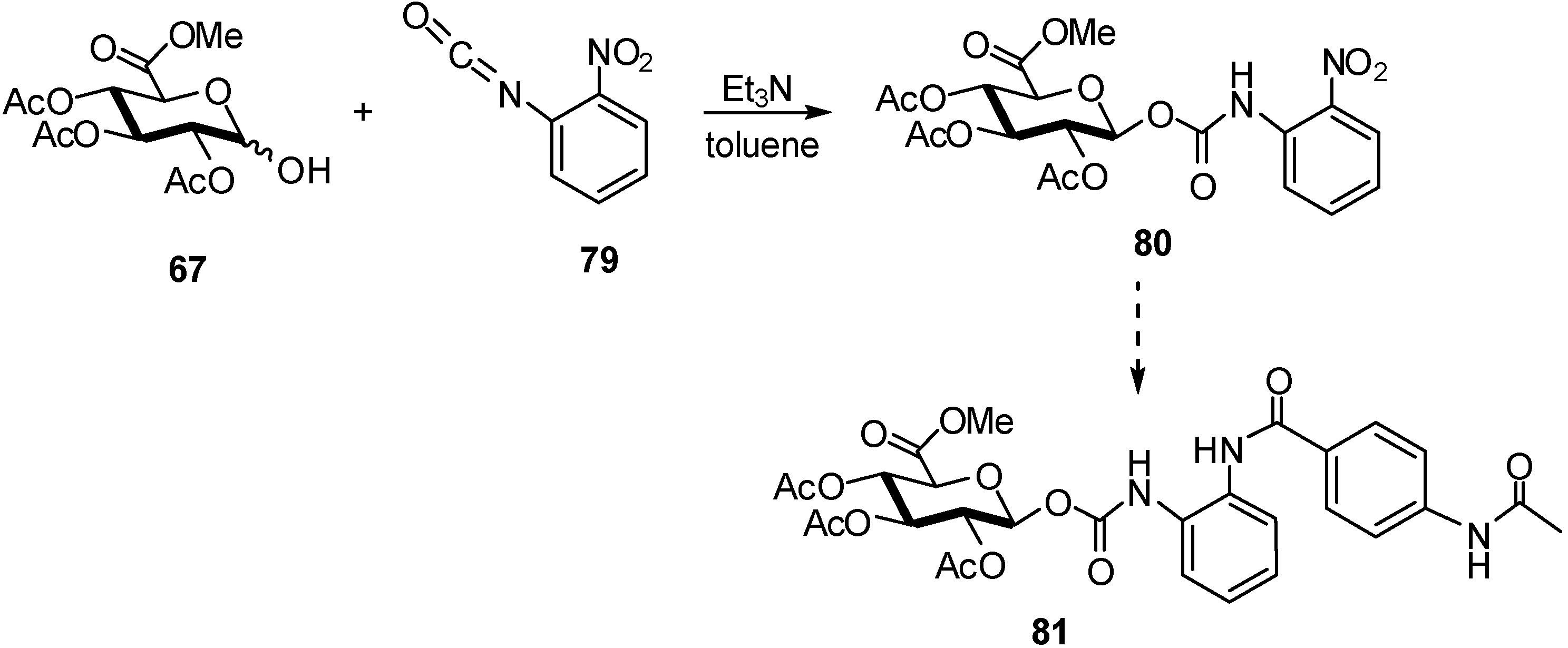{kind=link}
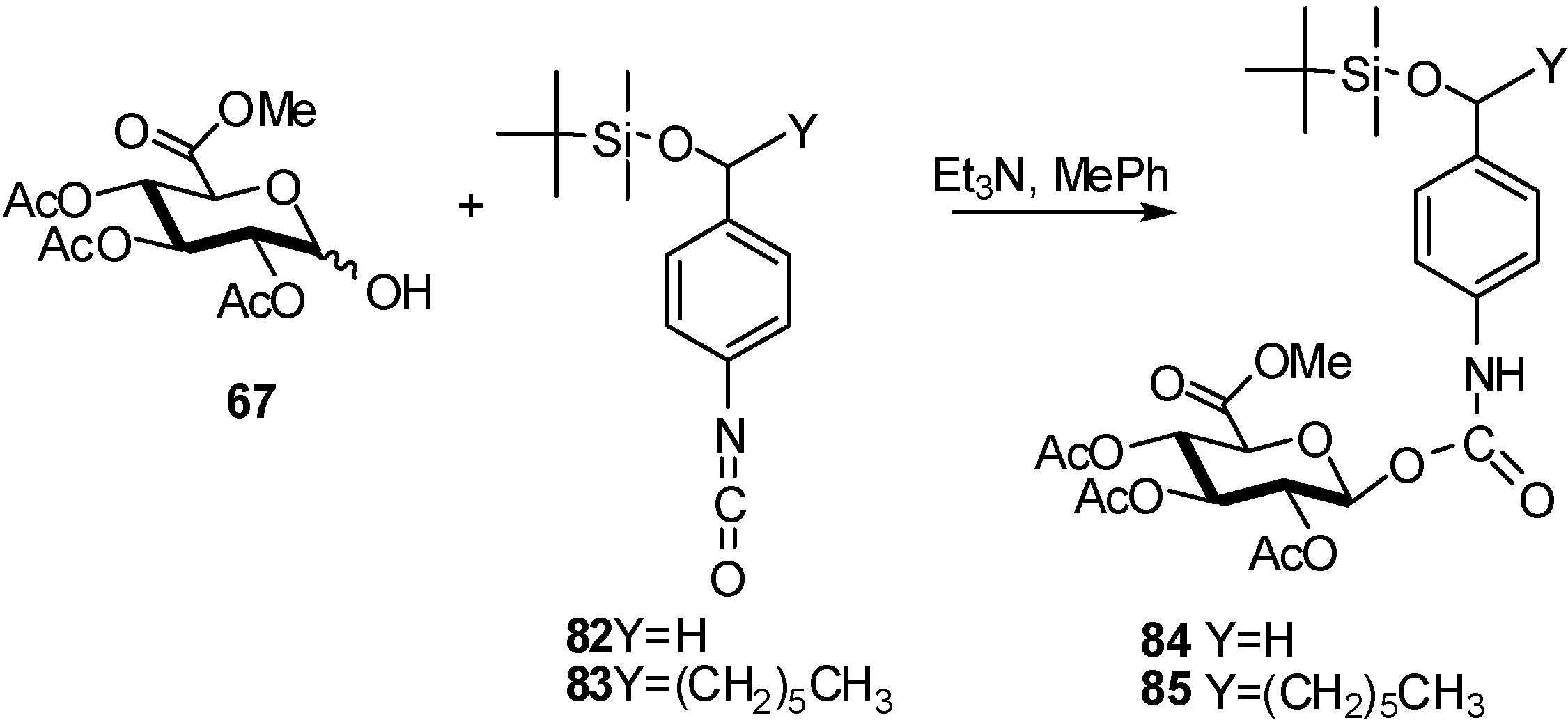{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
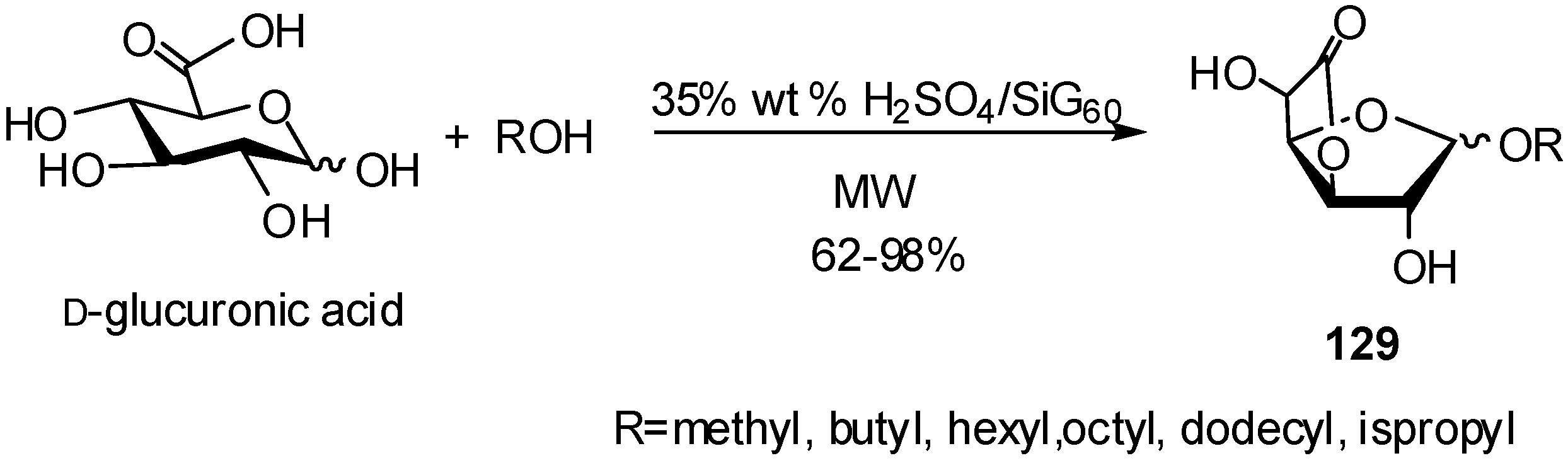{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Donor | Acceptor | Product | α/β ratio | Yield (%) |
|---|---|---|---|---|---|
| 1 | 203 | 201 | 206 | 95/5 | 91 |
| 2 | 202 | 200 | 207 | > 95/5 | 70 |
| 3 | 204 | 201 | 208 | > 95/5 | 70 |
| 4 | 205 | 201 | 209 | > 95/5 | 78 |




| Entry | Donor | Conditions | Product | α/β | Yield (%) |
|---|---|---|---|---|---|
| 1 | 233 | NIS/TBSOTf | 236 | < 10 | |
| 2 | 233 | NIS/TfOH | 236 | 1.0:1 | 45 |
| 3 | 231 | NIS/TfOH | 235 | 2.0:1 | 49 |
| 4 | 231 | DMTST | 235 | < 10 | |
| 5 | 232 | TMSOTf | 234 | 2.1:1 | 96 |
| 6 | 230 | NIS/TfOH | 234 | 3.7:1 | 85 |
| 7 | 230 | NIS/TfOH | 234 | 4.2:1 | 85 |

5. Conclusions
References and Notes
- Jung, K.H.; Mueller, M.; Schmidt, R.R. Intramolecular O-glycoside bond formation. Chem. Rev. 2000, 100, 4423–4442. [Google Scholar] [CrossRef]
- Zhu, X.; Schmidt, R.R. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef]
- Kaspersen, F.M.; Van Boeckel, C.A.A. A review of the methods of chemical synthesis of sulfate and glucuronide conjugates. Xenobiotica 1987, 17, 1451–1471. [Google Scholar] [CrossRef]
- Stachulski, A.V.; Jenkins, G.N. The synthesis of O-glucuronides. Nat. Prod. Rep. 1998, 15, 173–186. [Google Scholar] [CrossRef]
- Barron, D. Recent advances in the chemical synthesis and biological activity of phenolic metabolites. Recent Adv. Polyphenol Res. 2008, 1, 317–358. [Google Scholar] [CrossRef]
- Arewang, C.J.; Lahmann, M.; Oscarson, S.; Tiden, A.K. Synthesis of urine drug metabolites: glucuronic acid glycosides of phenol intermediates. Carbohydr. Res. 2007, 342, 970–974. [Google Scholar] [CrossRef]
- Koga, T.; Meydani, M. Effect of plasma metabolites of (+)-catechin and quercetin on monocyte adhesion to human aortic endothelial cells. Am. J. Clin. Nutr. 2001, 73, 941–948. [Google Scholar]
- Tribolo, S.; Lodi, F.; Connor, C.; Suri, S.; Wilson, V.G.; Taylor, M.A.; Needs, P.W.; Kroon, P.A.; Hughes, D.A. ABCA1 expression in humans is associated with physical activity and alcohol consumption. Atherosclerosis 2008, 197, 50–56. [Google Scholar] [CrossRef]
- Rukhman, I.; Gutman, A.L. Synthesis of morphine 6-α-D-glucuronide. Tetrahedron Lett. 2000, 41, 6889–6892. [Google Scholar] [CrossRef]
- Rukhman, I.; Yudovich, L.; Nisnevich, G.; Gutman, A.L. Selective synthesis of both isomers of morphine 6-β-D-glucuronide and their analogs. Tetrahedron 2001, 57, 1083–1092. [Google Scholar] [CrossRef]
- Rho, Y.S.; Kim, S.Y.; Kim, W.J.; Yun, Y.K.; Sin, H.S.; Yoo, D.J. Convenient syntheses of daunomycinone-7-D-glucuronides and doxorubicinone-7-D-glucuronides. Synth. Comm. 2004, 34, 3497–3511. [Google Scholar] [CrossRef]
- Alonen, A.; Gartman, M.; Aitio, O.; Finel, M.; Yli-Kauhaluoma, J.; Kostiainen, R. Synthesis, structure characterization, and enzyme screening of clenbuterol glucuronides. Eur. J. Pharm. Sci. 2009, 37, 581–587. [Google Scholar] [CrossRef]
- Zhu, X.R.; Zheng, Y.; Wang, P.; Yang, S.B.; Chen, R.; Zhu, Y.Q. Synthesis of two metabolites of edaravone. J. Chin. Pharm. Sci. 2010, 19, 307–311. [Google Scholar]
- Arewang, C.J.; Lahmann, M.; Oscarson, S.; Tiden, A.K. Synthesis of urine drug metabolites: glucuronic acid glycosides of phenol intermediate. Carbohydr. Res. 2007, 342, 970–974. [Google Scholar] [CrossRef]
- Wang, L.X.; Heredia, A.; Song, H.; Zhang, Z.; Yu, B.; Davis, C.; Redfield, R. Resveratrol glucuronides as the metabolites of resveratrol in humans: Characterization, synthesis, and anti-HIV activity. J. Pharm. Sci. 2004, 93, 2448–2457. [Google Scholar] [CrossRef]
- Wagner, H.; Danninger, H.; Seligmann, O.; Farkas, L. Synthesis of glucuronides in the flavonoid series. I. First synthesis of a naturally occurring flavonoid glucuronide (quercetin 3-β-D-glucuronide). Chem. Ber. 1970, 103, 3674–3677. [Google Scholar] [CrossRef]
- Needs, P.W.; Kroon, P.A. Convenient syntheses of metabolically important quercetin glucuronides and sulfates. Tetrahedron 2006, 62, 6862–6868. [Google Scholar] [CrossRef]
- Engstrom, K.M.; Henry, R.F.; Marsden, I. Synthesis of the glucuronidemetabolite of ABT-751. Tetrahedron Lett. 2007, 48, 1359–1362. [Google Scholar] [CrossRef]
- Engstrom, K.M.; Daanen, J.F.; Wagaw, S.; Stewart, A.O. Gram scale synthesis of the glucuronidemetabolite of ABT-724. J. Org. Chem. 2006, 71, 8378–8383. [Google Scholar] [CrossRef]
- Boumendjel, A.; Blanc, M.; Williamson, G.; Barron, D. Efficient synthesis of flavanone glucuronides. J. Agr. Food Chem. 2009, 57, 7264–7267. [Google Scholar] [CrossRef]
- Al-Maharik, N.; Botting, N.P. A facile synthesis of isoflavone 7-O-glucuronides. Tetrahedron Lett. 2006, 47, 8703–8706. [Google Scholar] [CrossRef]
- Al-Maharik, N.; Botting, N.P. An efficient method for the glycosylation of isoflavones. Eur. J. Org. Chem. 2008, 33, 5622–5629. [Google Scholar] [CrossRef]
- Lucas, R.; Alcantara, D.; Morales, J.C. A concise synthesis of glucuronide metabolites of urolithin-B, resveratrol, and hydroxytyrosol. Carbohydr. Res. 2009, 344, 1340–1346. [Google Scholar] [CrossRef]
- Brown, R.T.; Carter, N.E.; Mayalarp, S.P.; Scheinmann, F. A Simple synthesis of morphine-3,6-di-β-D-glucuronide. Tetrahedron 2000, 56, 7591–7594. [Google Scholar] [CrossRef]
- Harding, J.R.; King, C.D.; Perrie, J.A.; Sinnott, D.; Stachulski, A.V. Glucuronidation of steroidal alcohols using iodo-sugar and imidate donors. Org. Biomol. Chem. 2005, 3, 1501–1507. [Google Scholar] [CrossRef]
- Ferguson, J.R.; Harding, J.R.; Lumbard, K.W.; Scheinmann, F.; Stachulski, A.V. Glucuronide and sulfate conjugates of ICI 182,780, a pure anti-estrogenic steroid. Order of addition, catalysis and substitution effects in glucuronidation. Tetrahedron Lett. 2000, 41, 389–392. [Google Scholar] [CrossRef]
- O'Neill, P.M.; Scheinmann, F.; Stachulski, A.V.; Maggs, J.L.; Park, B.K. Efficient preparations of the β-glucuronides of dihydroartemisinin and structural confirmation of the human glucuronide metabolite. J. Med. Chem. 2001, 44, 1467–1470. [Google Scholar] [CrossRef]
- Senn-Bilfinger, J.; Ferguson, J.R.; Holmes, M.A.; Lumbard, K.W.; Huber, R.; Zech, K.; Hummel, R.P.; Zimmermann, P.J. Glucuronide conjugates of Soraprazan (BY359), a new potassium-competitive acid blocker (P-CAB) for the treatment of acid-related diseases. Tetrahedron Lett. 2006, 47, 3321–3323. [Google Scholar]
- Araya, I.; Tsubuki, T.; Saito, T.; Numata, M.; Akita, H. Synthesis of the metabolites of 4-(2-methyl-1H-imidazol-1-yl)-2,2-diphenylbutanamide (KRP-197/ONO-8025). Chem. Pharm. Bull. 2007, 55, 1039–1043. [Google Scholar] [CrossRef]
- van Dorp, E.L.A.; Morariu, A.; Dahan, A. Morphine-6-glucuronide: Potency and safety compared with morphine. Exp. Opin. Pharmacother. 2008, 9, 1955–1961. [Google Scholar] [CrossRef]
- Bosslet, K.; Straub, R.; Blumrich, M.; Czech, J.; Gerken, M.; Sperker, B.; Kroemer, H.K.; Gesson, J.P.; Koch, M.; Monneret, C. Elucidation of the mechanism enabling tumor selective prodrug monotherapy. Cancer Res. 1998, 58, 1195–1201. [Google Scholar]
- Kratz, F.; Mueller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef]
- Bagshawe, K.D.; Sharma, S.K.; Begent, R.H.J. Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Exp. Opin. Biol. Ther. 2004, 4, 1777–1789. [Google Scholar] [CrossRef]
- Niculescu-Duvaz, I.; Springer, C.J. Introduction to the background, principles, and state of the art in suicide gene therapy. Mol. Biotechnol. 2005, 30, 71–88. [Google Scholar] [CrossRef]
- De Graaf, M.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. Beta-glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–403. [Google Scholar] [CrossRef]
- Thomas, M.; Clarhaut, J.; Tranoy-Opalinski, I.; Gesson, J.P.; Roche, J.; Papot, S. Synthesis and biological evaluation of glucuronide prodrugs of the histone deacetylase inhibitor CI-994 for application in selective cancer chemotherapy. Bioorg. Med. Chem. 2008, 16, 8109–8116. [Google Scholar] [CrossRef]
- Leenders, R.G.G.; Ruytenbeek, R.; Damen, E.W.P.; Scheeren, H.W. Highly diastereoselective synthesis of anomeric β-O-glycopyranosyl carbamates from isocyanates. Synthesis 1996, 1309–1312. [Google Scholar]
- Leenders, R.G.G.; Damen, E.W.P.; Bijsterveld, E.J.A.; Scheeren, H.W.; Houba, P.H.J.; Van der Meulen-Muileman, I.H.; Boven, E.; Haisma, H.J. Novel anthracycline-spacer-β-glucuronide, -β-glucoside, and -β-galactoside prodrugs for application in selective chemotherapy. Bioorg. Med. Chem. 1999, 7, 1597–1610. [Google Scholar] [CrossRef]
- Tietze, L.F.; Schuster, H.J.; Schmuck, K.; Schuberth, I.; Alves, F. Duocarmycin-based prodrugs for cancer prodrug monotherapy. Bioorg. Med. Chem. 2008, 16, 6312–6318. [Google Scholar] [CrossRef]
- Pearson, A.G.; Kiefel, M.J.; Ferro, V.; von Itzstein, M. Towards the synthesis of aryl glucuronides as potential heparanase probes. An interesting outcome in the glycosidation of glucuronic acid with 4-hydroxycinnamic acid. Carbohydr. Res. 2005, 340, 2077–2085. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Z.; Yu, B. Total Synthesis of CRM646-A and -B, Two Fungal Glucuronides with Potent Heparinase Inhibition Activities. J. Org. Chem. 2005, 70, 8884–8889. [Google Scholar] [CrossRef]
- Rossignol, J.F.; Stachulski, A.V. Syntheses and antibacterial activities of tizoxanide, an N-(nitrothiazolyl)salicylamide, and its O-aryl glucuronide. J. Chem. Res. Synop. 1999, 1, 44–45. [Google Scholar]
- Pews-Davtyan, A.; Pirojan, A.; Shaljyan, I.; Awetissjan, A.A.; Reinke, H.; Vogel, C. Comparison of Several Glucuronate Glycosyl Donors. J. Carbohydr. Chem. 2003, 22, 939–962. [Google Scholar]
- Lergenmüller, M.; Lichtenthaler, F.W. Glucuronic acid-based ulosyl donors for introducing α-D-GlcA and β-D-ManA units. Carbohydr. Res. 2007, 342, 2132–2137. [Google Scholar] [CrossRef]
- Perrie, J.A.; Harding, J.R.; King, C.; Sinnott, D.; Stachulski, A.V. Glycosidation with a disarmed glycosyl iodide: promotion and scope. Org. Lett. 2003, 5, 4545–4548. [Google Scholar]
- Polakova, M.; Pitt, N.; Tosin, M.; Murphy, P.V. Stereoselective glycoside synthesis: Glycosidation reactions of silyl ethers with conformationally inverted donors derived from glucuronic acid: stereoselective synthesis of glycosides and 2-deoxy-glycosides. Angew. Chem. Int. Ed. 2004, 43, 2518–2521. [Google Scholar] [CrossRef]
- Cronin, L.; Tosin, M.; Mueller-Bunz, H.; Murphy, P.V. The synthesis of cyclic imidates from amides of glucuronic acid and investigation of glycosidation reactions. Carbohydr. Res. 2007, 342, 111–118. [Google Scholar] [CrossRef]
- O'Brien, C.; Polakova, M.; Pitt, N.; Tosin, M.; Murphy, P.V. Glycosidation-anomerization reactions of 6,1-anhydroglucopyranuronic acid and anomerization of β-D-glucopyranosiduronic acids promoted by SnCl4. Chem. Eur. J. 2007, 13, 902–909. [Google Scholar] [CrossRef]
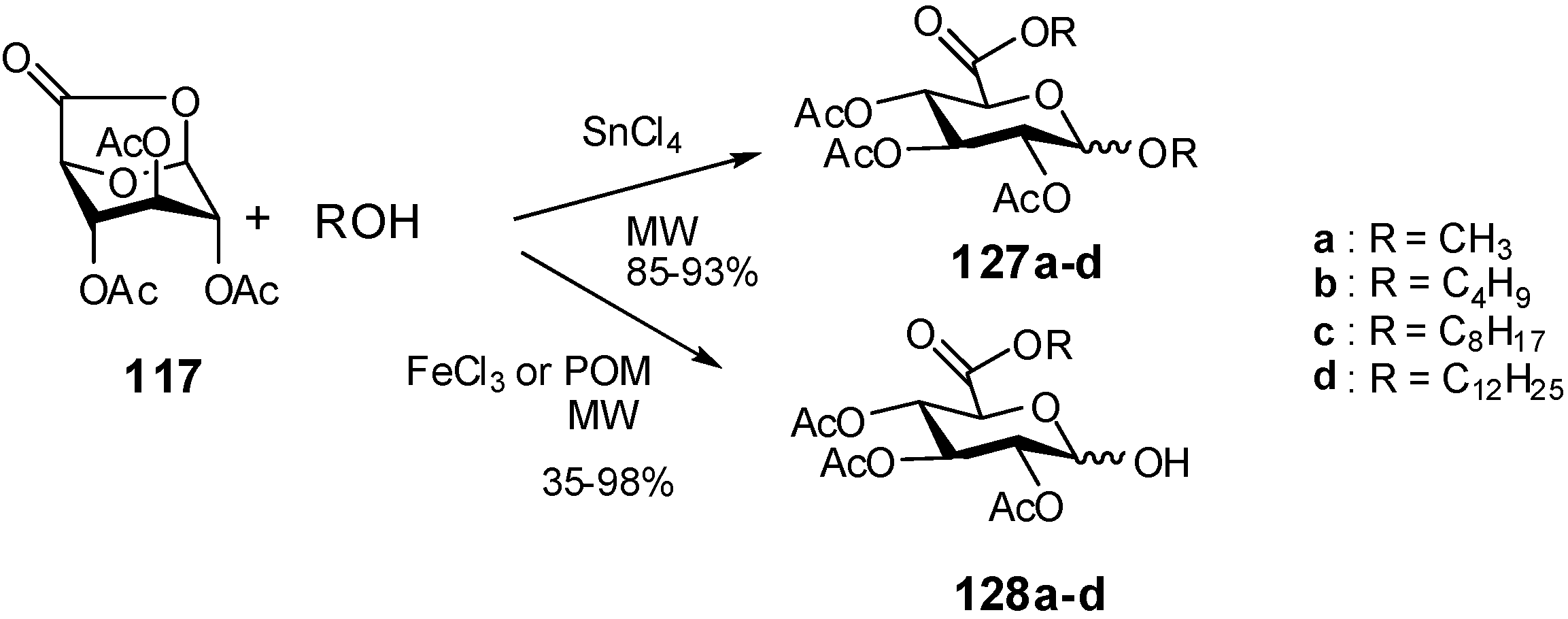
- Rat, S.; Mathiron, D.; Michaud, P.; Kovensky, J.; Wadouachi, A. Efficient glycosylation and/or esterification of D-glucuronic acid and its 6,1-lactone under solvent-free microwave irradiation. Tetrahedron 2007, 63, 12424–12428. [Google Scholar] [CrossRef]
- Bosco, M.; Rat, S.; Dupre, N.; Hasenknopf, B.; Lacote, E.; Malacria, M.; Remy, P.; Kovensky, J.; Thorimbert, S.; Wadouachi, A. Lewis-Acidic polyoxometalates as reusable catalysts for the synthesis of glucuronic acid esters under microwave irradiation. ChemSusChem 2010, 3, 1249–1252. [Google Scholar] [CrossRef]
- Richel, A.; Laurent, P.; Whatelet, B.; Whatelet, J.P.; Paquot, M. Microwave-assisted synthesis of D-glucuronic acid derivatives using cost-effective solid acid catalyst. Tetrahedron Lett. 2010, 51, 1356–1360. [Google Scholar]
- Ferrieres, V.; Bertho, J.N.; Plusquellec, D. A convenient synthesis of alkyl D-glycofuranosiduronic acids and alkyl D-glycofuranosides from unprotected carbohydrates. Carbohydr. Res. 1998, 311, 25–35. [Google Scholar] [CrossRef]
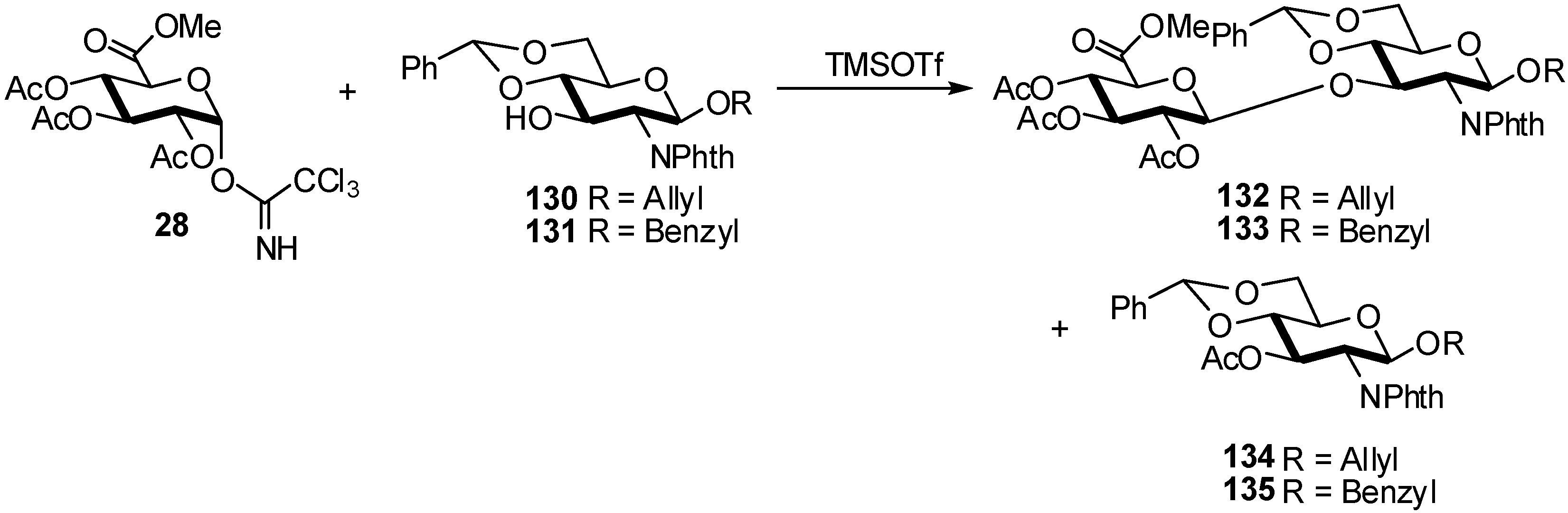
- Soliman, S.E.; Bassily, R.W.; El-Sokkary, R.I.; Nashed, M.A. Acetylated methyl glucopyranuronate trichloroacetimidate as a glycosyl donor for efficient synthesis of disaccharides. Carbohydr. Res. 2003, 338, 2337–2340. [Google Scholar] [CrossRef]
- Nilsson, M.; Svahn, C.M.; Westman, J. Synthesis of the methyl glycosides of a tri- and a tetra-saccharide related to heparin and heparan sulfate. Carbohydr. Res. 1993, 246, 161–72. [Google Scholar] [CrossRef]
- Belot, F.; Jacquinet, J.C. Unexpected stereochemical outcome of activated 4,6-O-benzylidene derivatives of the 2-deoxy-2-trichloroacetamido-D-galacto series in glycosylation reactions during the synthesis of a chondroitin 6-sulfate trisaccharide methyl glycoside. Carbohydr. Res. 2000, 325, 93–106. [Google Scholar] [CrossRef]
- Plante, O.J.; Palmacci, E.R.; Andrade, R.B.; Seeberger, P.H. Oligosaccharide synthesis with glycosyl phosphate and dithiophosphate triesters as glycosylating agents. J. Am. Chem. Soc. 2001, 123, 9545–9554. [Google Scholar] [CrossRef]
- Iyer, S.S.; Rele, S.M.; Baskaran, S.; Chaikof, E.L. Design and synthesis of hyaluronan-mimetic gemini disaccharides. Tetrahedron 2003, 59, 631–638. [Google Scholar] [CrossRef]
- Rele, S.M.; Iyer, S.S.; Baskaran, S.; Chaikof, E.L. Design and synthesis of dimeric heparinoid mimetics. J. Org. Chem. 2004, 69, 9159–9170. [Google Scholar] [CrossRef]
- Dinkelaar, J.; Gold, H.; Overkleeft, H.S.; Codee, J.D.C.; van der Marel, G.A. Synthesis of hyaluronic acid oligomers using chemoselective and one-pot strategies. J. Org. Chem. 2009, 74, 4208–4216. [Google Scholar]
- de Paz, J.L.; Mar Kayser, M.; Macchione, G.; Nieto, P.M. Exploration of the use of an acylsulfonamide safety-catch linker for the polymer-supported synthesis of hyaluronic acid oligosaccharides. Carbohydr. Res. 2010, 345, 565–571. [Google Scholar] [CrossRef]
- Daragics, K.; Fügedi, P. Synthesis of glycosaminoglycan oligosaccharides. Part 5: Synthesis of a putative heparan sulfate tetrasaccharide antigen involved in prion diseases. Tetrahedron 2010, 66, 8036–8046. [Google Scholar]
- Nagatsukaa, T.; Uzawab, H.; Asanumaa, H.; Nishidac, Y. Glucuronidase-assisted transglycosylation for the synthesis of highly functional disaccharides: β-D-glucuronyl 6-O-sulfo-β-D-gluco- and -β-D-galactopyranosides. J. Carbohydr. Chem. 2009, 28, 94–106. [Google Scholar]
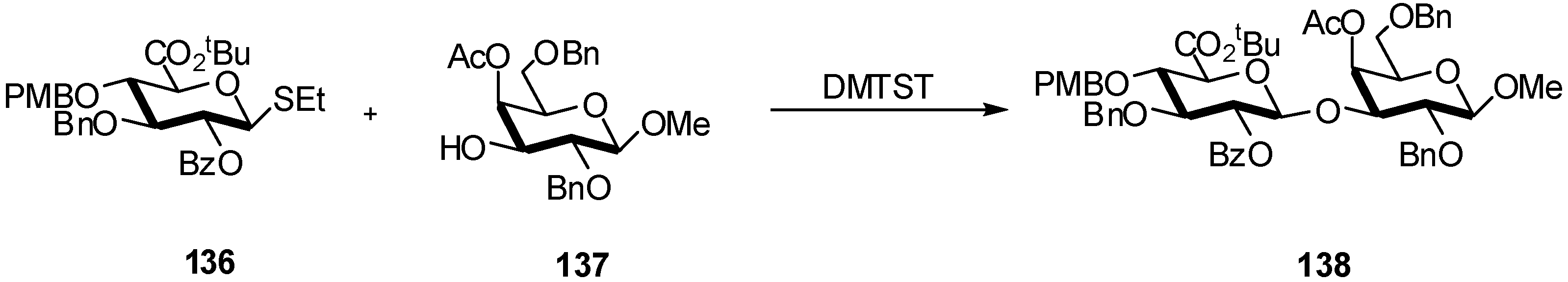
- van den Bos, L.J.; Dinkelaar, J.; Overkleeft, H.S.; van der Marel, G.A. Stereocontrolled synthesis of β-D-mannuronic acid esters: synthesis of an alginate trisaccharide. J. Am. Chem. Soc. 2006, 128, 13066–13067. [Google Scholar]
- Codee, J.D.C.; van den Bos, L.J.; de Jong, A.R.; Dinkelaar, J.; Lodder, G.; Overkleeft, H.S.; van der Marel, G.A. The stereo-directing effect of the glycosyl C5-carboxylate ester: stereoselective synthesis of β-mannuronic acid alginates. J. Org. Chem. 2009, 74, 38–47. [Google Scholar] [CrossRef]
- Dinkelaar, J.; de Jong, A.R.; van Meer, R.; Somers, M.; Lodder, G.; Overkleeft, H.S.; Codee, J.D.C.; van der Marel, G.A. Stereodirecting effect of the pyranosyl C-5 substituent in glycosylation reactions. J. Org. Chem. 2009, 74, 4982–4991. [Google Scholar]
- Walvoort, M.T.C.; Lodder, G.; Mazurek, J.; Overkleeft, H.S.; Codee, J.D.C.; van der Marel, G.A. Equatorial anomeric triflates from mannuronic acid esters. J. Am. Chem. Soc. 2009, 131, 12080–12081. [Google Scholar]
- Codee, J.D.C.; de Jong, A.R.; Dinkelaar, J.; Overkleeft, H.S.; van der Marel, G.A. Stereoselectivity of glycosylation of conformationally restricted mannuronate esters. Tetrahedron 2009, 65, 3780–3788. [Google Scholar] [CrossRef]
- Walvoort, M.T.C.; Dinkelaar, J.; van den Bos, L.J.; Lodder, G.; Overkleeft, H.S.; Codee, Je.D. C.; van der Marel, G.A. The impact of oxacarbenium ion conformers on the stereochemical outcome of glycosylation. Carbohydr. Res. 2010, 345, 1252–1263. [Google Scholar] [CrossRef]
- Magaud, D.; Grandjean, C.; Doutheau, M.; Ankera, D.; Shevehlk, V.; Cotte.Pattat, N.; Robert, J. An Efficient and Highly Stereoselective α(1→4) Glycosylation between two D-galacturonic acid ester Derivatives. Tetrahedron Lett. 1997, 38, 241–244. [Google Scholar]
- Vogel, C.; Steffan, W.; Ott, A.Y.; Betaneli, V.I. Galacturonic acid derivatives. Part IV. D-Galacturonic acid derivatives as acceptors and donors in glycosylation reactions. Carbohydr. Res. 1992, 237, 115–129. [Google Scholar] [CrossRef]
- Kramer, S.; Nolting, B.; Ott, A.J.; Vogel, C. Synthesis of homogalacturonan fragments. J. Carbohydr. Chem. 2000, 19, 891–921. [Google Scholar]
- Nolting, B.; Boye, H.; Vogel, C. Block synthesis with galacturonate trichloroacetimidates. J. Carbohydr. Chem. 2001, 20, 585–610. [Google Scholar]
- Stallforth, P.; Adibekian, A.; Seeberger, P.H. De Novo Synthesis of a D-Galacturonic Acid Thioglycoside as Key to the Total synthesis of a glycosphingolipid from Sphingomonas yanoikuyae. Org. Lett. 2008, 10, 1573–1576. [Google Scholar]
- Allam, A.; Behr, J.B.; Dupont, L.; Nardello-Rataj, V.; Plantier-Royon, R. Synthesis, physico-chemical properties and complexing abilities of new amphiphilic ligands from D-galacturonic acid. Carbohydr. Res. 2010, 345, 731–739. [Google Scholar] [CrossRef]
- Bordoni, A.; Lima, C.; Mariño, K.; de Lederkremer, R.M.; Marino, C. Facile synthesis of methyl α- and β-D-[6-3H]galactofuranosides from D-galacturonic acid. Substrates for the detection of galactofuranosidases. Carbohydr. Res. 2008, 343, 1863–1869. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wadouachi, A.; Kovensky, J. Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview. Molecules 2011, 16, 3933-3968. https://doi.org/10.3390/molecules16053933
Wadouachi A, Kovensky J. Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview. Molecules. 2011; 16(5):3933-3968. https://doi.org/10.3390/molecules16053933
Chicago/Turabian StyleWadouachi, Anne, and José Kovensky. 2011. "Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview" Molecules 16, no. 5: 3933-3968. https://doi.org/10.3390/molecules16053933
APA StyleWadouachi, A., & Kovensky, J. (2011). Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview. Molecules, 16(5), 3933-3968. https://doi.org/10.3390/molecules16053933