Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents

Abstract
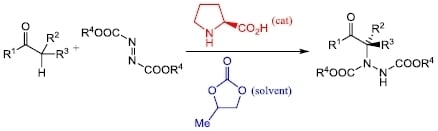
:
1. Introduction
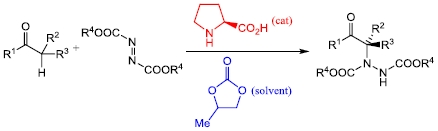
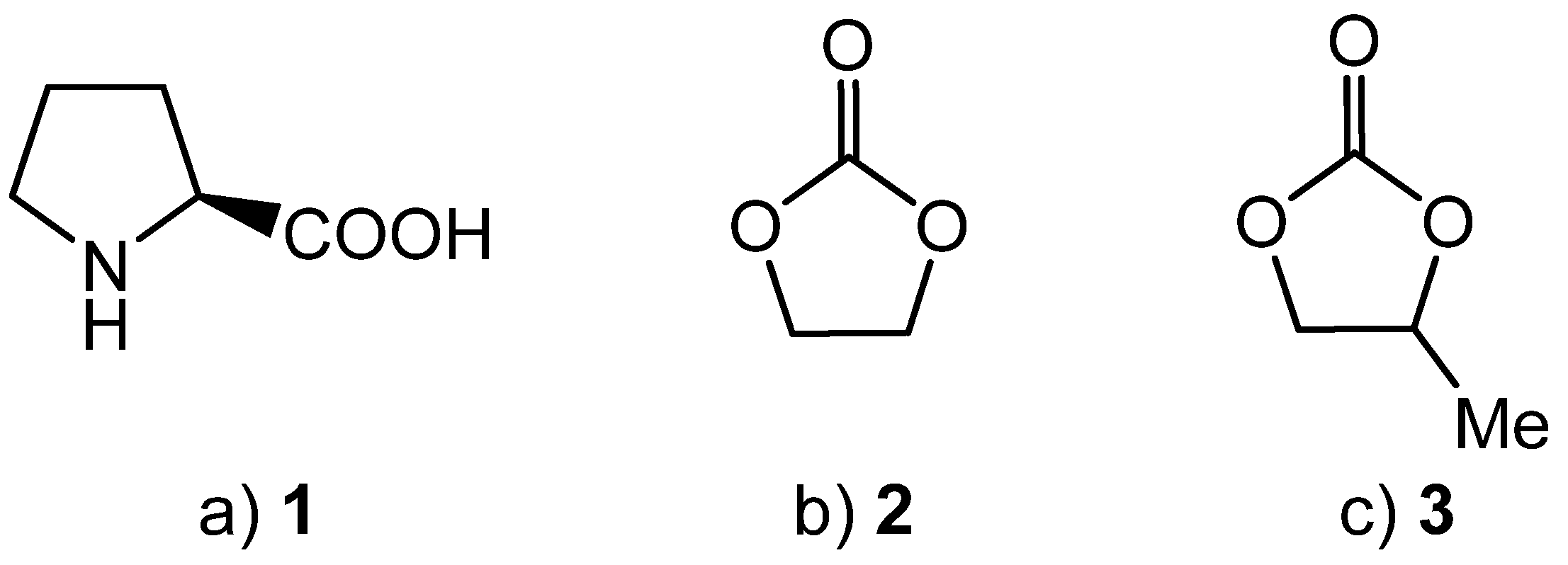
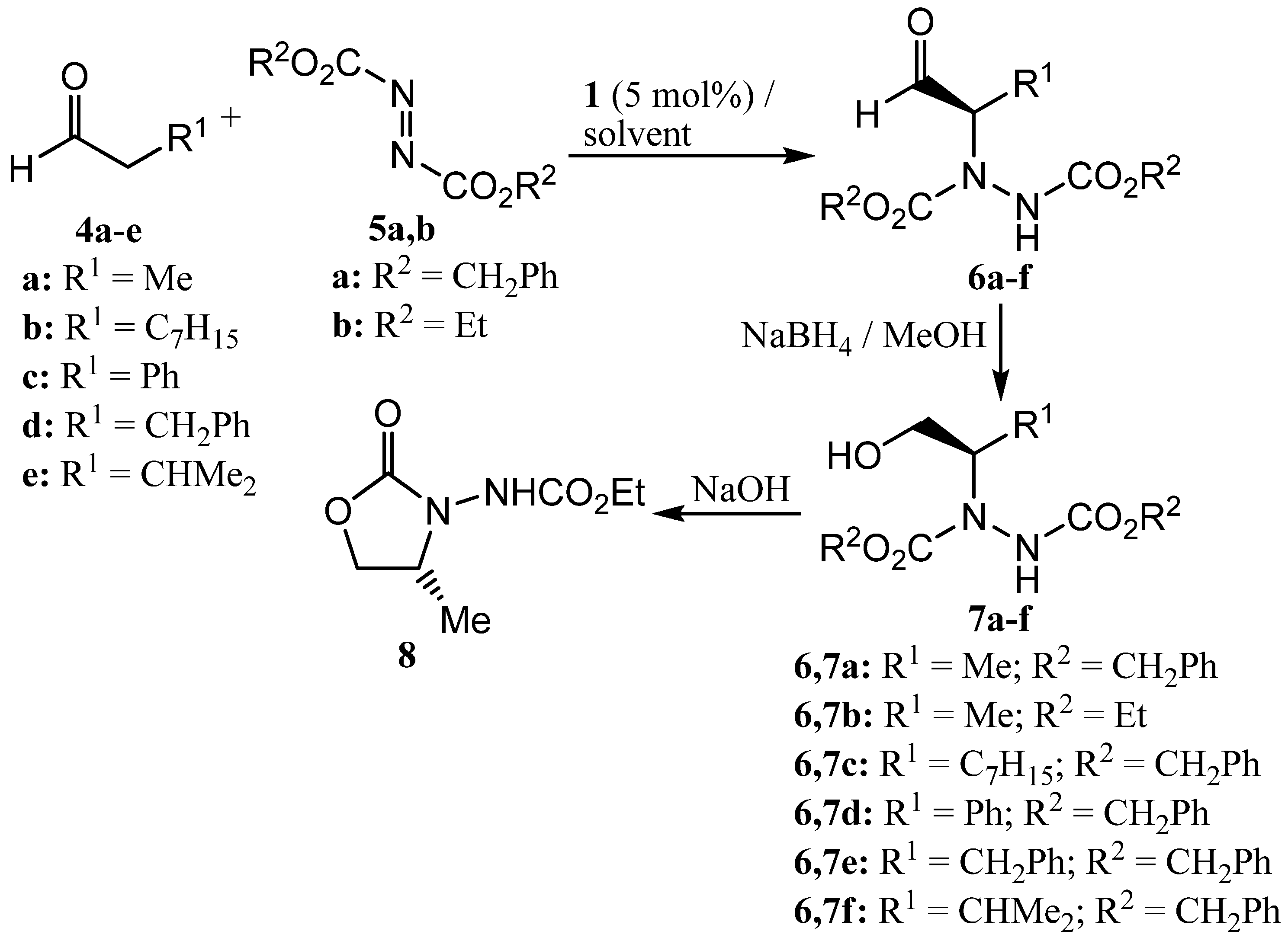
2. Results and Discussion
3. Experimental
3.1. Chemicals and instrumentation
3.2. Synthesis of compound 8 [46,63]
3.3. General procedure for the synthesis of alcohols 7
3.4. General procedure for the synthesis of ketones 10a-d
4. Conclusions
Acknowledgements
References and Notes
- Hayashi, T.; Asai, T.; Ogoshi, H. Conformational Analysis of β-Turn Structure in Tetrapeptides Containing Proline or Proline Analogs. Tetrahedron Lett. 1997, 38, 3039–3042. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. β-Turns in Proteins. J. Mol. Biol. 1997, 115, 135–175. [Google Scholar] [CrossRef]
- Jones, I.G.; Jones, W.; North, M. Conformational Analysis of Peptides and Pseudopeptides Incorporating an endo-(2S,3R)-Norborn-5-ene Residue as a Turn Inducer. J. Org. Chem. 1998, 63, 1505–1513. [Google Scholar] [CrossRef]
- Belokon, Y.N.; Bulychev, A.G.; Vitt, S.V.; Struchkov, Y.T.; Batsanov, A.S.; Timofeeva, T.V.; Tsyryapkin, V.A.; Ryzhov, M.G.; Lysova, L.A.; Bakhmutov, V.I.; Belikov, V.M. General method of diastereo- and enantio-selective synthesis of β-hydroxy-α-amino acids by condensation of aldehydes and ketones with glycine. J. Am. Chem. Soc. 1985, 107, 4252–4259. [Google Scholar] [CrossRef]
- North, M.; Zagotto, G. Asymmetric Desymmetrisation of Meso-Norbornene Anhydrides Utilising Methyl Prolinate as a Chiral Reagent. Synlett 1995, 639–640. [Google Scholar] [CrossRef]
- Albers, T.; Biagini, S.C.G.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.M.A.; North, M.; Uriarte, E.; Zagotto, G. Desymmetrisation of Meso-Anhydrides utilising (S)-Proline Derivatives. Synthesis 1996, 393–398. [Google Scholar] [CrossRef]
- List, B. Asymmetric Aminocatalysis. Synlett 2001, 1675–1686. [Google Scholar] [CrossRef]
- Gröger, H.; Wilken, J. The Application of L-Proline as an Enzyme Mimic and Further New Asymmetric Syntheses Using Small Organic Molecules as Chiral Catalysts. Angew. Chem. Int. Ed. 2001, 40, 529–532. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- List, B. Enamine Catalysis Is a Powerful Strategy for the Catalytic Generation and Use of Carbanion Equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Kazmaier, U. Amino Acids–Valuable Organocatalysts in carbohydrate Synthesis. Angew. Chem. Int. Ed. 2005, 44, 2186–2188. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Gaunt, M.J.; Johansson, C.C.C.; McNally, A.; Vo, N.T. Enantioselective organocatalysis. Drug Discov. Today 2007, 12, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis—Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent Developments in Asymmetric Organocatalysis; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Brogan, A.P.; Dickerson, T.J.; Janda, K.D. Enamine-Based Aldol Organocatalysis in Water: Are They Really “All Wet”? Angew. Chem. Int. Ed. 2006, 45, 8100–8102. [Google Scholar] [CrossRef] [PubMed]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Water in Stereoselective Organocatalytic Reactions. Adv. Synth. Catal. 2009, 351, 33–57. [Google Scholar] [CrossRef]
- Toma, Š.; Mečiarova, M.; Šebesta, R. Are Ionic Liquids Suitable Media for Organocatalytic Reactions? Eur. J. Org. Chem. 2009, 321–327. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Armstrong, A.; Coombe, V.; Wells, A. Water in Organocatalytic Processes: Debunking the Myths. Angew. Chem. Int. Ed. 2007, 46, 3798–3800. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, W.; Zhang, Y.; Wang, H. Do We Understand the Recyclability of Ionic Liquids? Chem. Eur. J. 2009, 15, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.; Franzke, A.; Armstrong, A.; Blackmond, D.G. Clarification of the Role of Water in Proline-Mediated Aldol Reactions. J. Am. Chem. Soc. 2007, 129, 15100–15101. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, B.; Rantanen, T.; Bolm, C. Solvent-Free Asymmetric Organocatalysis in a Ball Mill. Angew. Chem. Int. Ed. 2006, 46, 6924–6926. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, B.; Bruckmann, A.; Bolm, C. A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill. Chem. Eur. J. 2007, 13, 4710–4722. [Google Scholar] [CrossRef] [PubMed]
- Schäffner, B.; Schäffner, F.; Verevkin, S.P.; Börner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Omedes-Pujol, M. Catalytic, asymmetric cyanohydrin synthesis in propylene carbonate. Tetrahedron Lett. 2009, 50, 4452–4454. [Google Scholar] [CrossRef]
- North, M.; Omedes-Pujol, M. Kinetics and mechanism of vanadium catalysed asymmetric cyanohydrin synthesis in propylene carbonate. Belstein J. Org. Chem. 2010, 6, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Silva, L.B.; Freitas, L.C.G. Structural and thermodynamic properties of liquid ethylene carbonate and propylene carbonate by Monte Carlo Simulations. J. Mol. Structure Theochem. 2007, 806, 23–34. [Google Scholar] [CrossRef]
- Yoshida, M.; Ihara, M. Novel Methodologies for the Synthesis of Cyclic Carbonates. Chem. Eur. J. 2004, 10, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-L.; Luo, S.-L.; Yin, S.-F.; Au, C.-T. The direct transformation of carbon dioxide to organic carbonates over heterogeneous catalysts. Appl. Cat. A 2009, 366, 2–12. [Google Scholar] [CrossRef]
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Pasquale, R.; Young, C. Synthesis of Cyclic Carbonates from Epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Ballivet-Tkatchenko, D.; Dibenedetto, A. Synthesis of Linear and Cyclic Carbonates. In Carbon Dioxide as Chemical Feedstock; Aresta, M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 169–212. [Google Scholar]
- Meléndez, J.; North, M.; Pasquale, R. Synthesis of cyclic carbonates from atmospheric pressure carbon dioxide using exceptionally active aluminium(salen) complexes as catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Meléndez, J.; North, M.; Pasquale, R. Mechanism of cyclic carbonate synthesis from epoxides and CO2. Angew. Chem., Int. Ed. 2009, 48, 2946–2948. [Google Scholar]
- Meléndez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic carbonate synthesis catalysed by bimetallic aluminium(salen) complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Villuendas, P.; Young, C. A gas phase flow reactor for ethylene carbonate synthesis from waste CO2. Chem. Eur. J. 2009, 11454–11457. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; North, M.; Villuendas, P.; Young, C. One-component bimetallic aluminium(salen)-based catalysts for cyclic carbonate synthesis and their immobilization. Dalton Trans. 2011, 40, 3885–3902. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, I.S.; North, M.; Pasquale, R.; Thursfield, A. An integrated approach to energy and chemicals production. Energy Environ. Sci. 2010, 3, 212–215. [Google Scholar] [CrossRef]
- North, M.; Pizzato, F.; Villuendas, P. Organocatalytic, asymmetric aldol reactions with a sustainable catalyst in a green solvent. ChemSusChem 2009, 2, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Harrington, R.W.; North, M.; Pizzato, F.; Villuendas, P. Cyclic carbonates as sustainable solvents for proline-catalysed aldol reactions. Tetrahedron: Asymmetry 2010, 21, 1262–1271. [Google Scholar] [CrossRef]
- North, M.; Villuendas, P. A Chiral Solvent Effect in Asymmetric Organocatalysis. Org. Lett. 2010, 12, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Bøgevig, A.; Juhl, K.; Kumaragurubaran, N.; Zhuang, W.; Jørgensen, K.A. Direct Organo-Catalytic Asymmetric α-Amination of Aldehydes–A Simple Approach to Optically Active α-Amino Aldehydes, α-Amino Alcohols, and α-Amino Acids. Angew. Chem. Int. Ed. 2002, 41, 1790–1793. [Google Scholar] [CrossRef]
- Iwamura, H.; Mathew, S.P.; Blackmond, D.G. In Situ Catalyst Improvement in the Proline-Mediated α-Amination of Aldehydes. J. Am. Chem. Soc. 2004, 126, 11770–11771. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, H.; Wells, D.H., Jr.; Mathew, S.P.; Klussmann, M.; Armstrong, A.; Blackmond, D.G. Probing the Active Catalyst in Product-Accelerated Proline-Mediated Reactions. J. Am. Chem. Soc. 2004, 126, 16312–16313. [Google Scholar] [CrossRef] [PubMed]
- Umbreen, S.; Brockhaus, M.; Ehrenberg, H.; Schmidt, B. Norstatines from Aldehydes by Sequential Organocatalytic α-Amination and Passerini Reaction. Eur. J. Org. Chem. 2006, 4585–4595. [Google Scholar] [CrossRef]
- Baumann, T.; Vogt, H.; Bräse, S. The Proline-Catalyzed Asymmetric Amination of Branched Aldehydes. Eur. J. Org. Chem. 2007, 266–282. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Kitajima, M.; Takayama, H. First Asymmetric Total Syntheses of Cernuane-Type Lycopodium Alkaloids, Cernuine, and Cermizine D. Org. Lett. 2008, 10, 1987–1990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, F.; Zhang, H.; Shao, Z. Organocatalytic Synthesis of Terminal Propargylamine Derivatives by Tandem Amination–Alkynylation. Synlett 2009, 3287–3290. [Google Scholar]
- Blackmond, D.G.; Moran, A.; Hughes, M.; Armstrong, A. Unusual Reversal of Enantioselectivity in the Proline-Mediated α-Amination of Aldehydes Induced by Tertiary Amine Additives. J. Am. Chem. Soc. 2010, 132, 7598–7599. [Google Scholar] [CrossRef] [PubMed]
- List, B. Direct Catalytic Asymmetric α-Amination of Aldehydes. J. Am. Chem. Soc. 2002, 124, 5656–5657. [Google Scholar] [CrossRef] [PubMed]
- Kumaragurubaran, N.; Juhl, K.; Zhuang, W.; Bøgevig, A.; Jørgensen, K.A. Direct L-Proline-Catalyzed Asymmetric α-Amination of Ketones. J. Am. Chem. Soc. 2002, 124, 6254–6255. [Google Scholar] [CrossRef] [PubMed]
- Suri, J.T.; Steiner, D.D.; Barbas, C.F., III. Organocatalytic Enantioselective Synthesis of Metabotropic Glutamate Receptor Ligands. Org. Lett. 2005, 7, 3885–3888. [Google Scholar] [CrossRef] [PubMed]
- Kotkar, S.P.; Chavan, V.B.; Sudalai, A. Organocatalytic Sequential α-Amination-Horner-Wadsworth-Emmons Olefination of Aldehydes: Enantioselective Synthesis of γ-Amino-α,β-Unsaturated Esters. Org. Lett. 2007, 9, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Bächle, M.; Hartmann, C.; Bräse, S. Thermal Effects in the Organocatalytic Asymmetric α-Amination of Disubstituted Aldehydes with Azodicarboxylates: A High-Temperature Organocatalysis. Eur. J. Org. Chem. 2008, 2207–2212. [Google Scholar] [CrossRef]
- Hartmann, C.E.; Gross, P.J.; Nieger, M.; Bräse, S. Towards an asymmetric synthesis of the bacterial peptide deformylase (PDF) inhibitor fumimycin. Org. Biomol. Chem. 2009, 7, 5059–5062. [Google Scholar] [CrossRef] [PubMed]
- Ait-Youcef, R.; Sbargoud, K.; Moreau, X.; Greck, C. Asymmetric α-Amination of Chiral Protected β-Hydroxyaldehydes Catalyzed by Proline. Synlett 2009, 3007–3010. [Google Scholar]
- Kalch, D.; De Rycke, N.; Moreau, X.; Greck, C. Efficient syntheses of enantioenriched (R)-pipecolic acid and (R)-proline via electrophilic organocatalytic amination. Tetrahedron Lett. 2009, 50, 492–494. [Google Scholar] [CrossRef]
- Hartmann, C.E.; Baumann, T.; Bächle, M.; Bräse, S. Asymmetric synthesis of deuterated and fluorinated aromatic α,α-disubstituted amino acid derivatives. Tetrahedron: Asymmetry 2010, 21, 1341–1349. [Google Scholar] [CrossRef]
- Kotrusz, P.; Alemayehu, S.; Toma, Š.; Schmalz, H.-G.; Adler, A. Enantioselective Organocatalysis in Ionic Liquids: Addition of Aliphatic Aldehydes and Ketones to Diethyl Azodicarboxylate. Eur. J. Org. Chem. 2005, 4904–4911. [Google Scholar] [CrossRef]
- Liu, P.-M.; Chang, C.; Reddy, R.J.; Ting, Y.-F.; Kuan, H.-H.; Chen, K. Remarkable Reaction Rate and Excellent Enantioselective Direct α-Amination of Aldehydes with Azodicarboxylates Catalyzed by Pyrrolidinylcamphor-Derived Organocatalysts. Eur. J. Org. Chem. 2010, 42–46. [Google Scholar] [CrossRef]
- Gosiewska, S.; Soni, R.; Clarkson, G.J.; Wills, M. Synthesis and use of a stable aminal derived from TsDPEN in asymmetric organocatalysis. Tetrahedron Lett. 2010, 51, 4214–4217. [Google Scholar] [CrossRef]
- Lacoste, E.; Vaique, E.; Berlande, M.; Pianet, I.; Vincent, J.-M.; Landais, Y. Benzimidazole-pyrrolidine/H+ (BIP/H+), a Highly Reactive Organocatalyst for Asymmetric Processes. Eur. J. Org. Chem. 2007, 167–177. [Google Scholar] [CrossRef]
- Thomassigny, C.; Primm, D.; Geck, C. Amino acid-catalyzed asymmetric α-amination of carbonyls. Tetrahedron Lett. 2006, 47, 1117–1119. [Google Scholar] [CrossRef]
- George, S.; Suryavanshi, G.S.; Sudalai, A. A short enantioselective synthesis of (–)-bestatin via L-proline-catalyzed α-amination of an aldehyde. Tetrahedron Lett. 2008, 49, 6791–6793. [Google Scholar] [CrossRef]
- Ait-Youcef, R.; Kalch, D.; Moreau, X.; Thomassigny, C.; Greck, C. Asymmetric α-Amination of Aldehydes and Ketones Catalyzed by tert-Butoxy-L-Proline. Lett. Org. Chem. 2009, 6, 377–380. [Google Scholar] [CrossRef]
Sample Availability: Not Available. |






{kind=link}
{kind=link}
{kind=link}
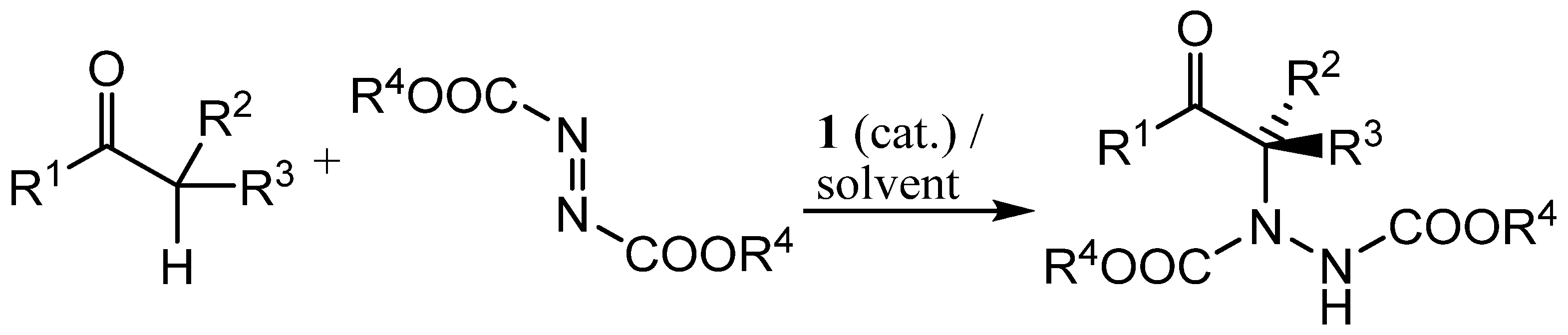{kind=link}
{kind=link}
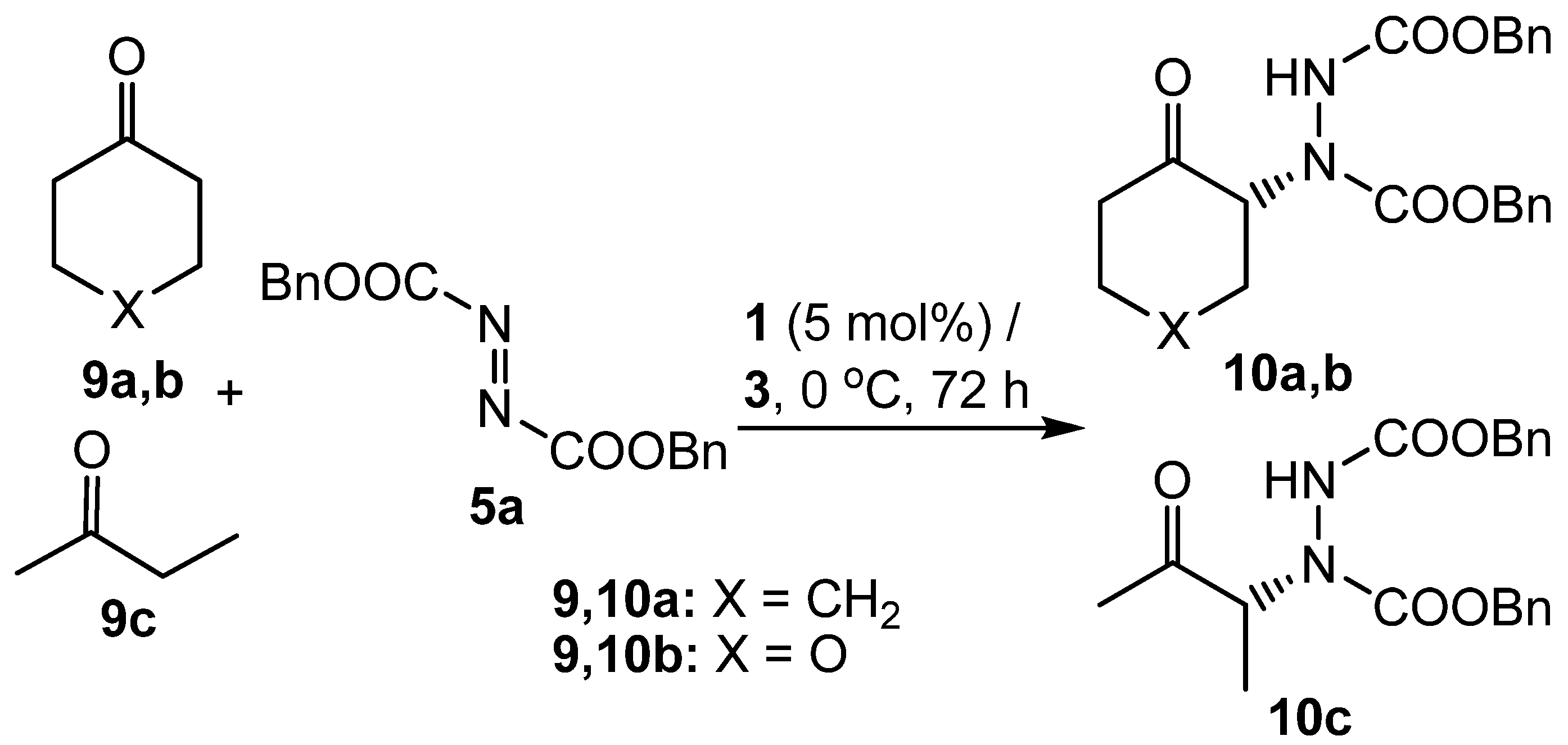{kind=link}
| Entry | Solvent | Aldehyde | Product | Time (h) | T (°C) | Yield (%) | ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | CH2Cl2 | 4a | 7a | 2 | RT | 86 | 98 (R)a |
| 2 | 2 | 4a | 7a | 2 | RT | 74 | 69 (R)a |
| 3 | 3 | 4a | 7a | 2 | RT | 81 | 80 (R)a |
| 4 | 3 | 4a | 7a | 2 | 0 | 18 | 99 (R)a |
| 5 | 3 | 4a | 7a | 24 | 0 | 69 | 97 (R)a |
| 6 | 3 | 4a | 7b | 24 | 0 | 39c | 66 (R)b |
| 7 | 3 | 4b | 7c | 24 | 0 | 41 | 90 (R)d |
| 8 | 3 | 4c | 7d | 24 | 0 | 70 | 36 (R)d |
| 9 | 3 | 4d | 7e | 24 | 0 | 76 | 99 (R)a |
| 10 | 3 | 4e | 7f | 24 | 0 | 87 | 92 (R)a |
| Entry | Solvent | Ketone | Product | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | Racemic-3 | 9a | 10a | 71 | 77 (R)a |
| 2 | Racemic-3 | 9b | 10b | 51 | 72 (R)b |
| 3 | Racemic-3 | 9c | 10c | 31 | 52 (R)a |
| 4 | (R)-3 | 9a | 10a | 81 | 74 (R)a |
| 5c | (R)-3 | 9a | 10a | 78 | 75 (S)a |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beattie, C.; North, M.; Villuendas, P. Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules 2011, 16, 3420-3432. https://doi.org/10.3390/molecules16043420
Beattie C, North M, Villuendas P. Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules. 2011; 16(4):3420-3432. https://doi.org/10.3390/molecules16043420
Chicago/Turabian StyleBeattie, Christopher, Michael North, and Pedro Villuendas. 2011. "Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents" Molecules 16, no. 4: 3420-3432. https://doi.org/10.3390/molecules16043420
APA StyleBeattie, C., North, M., & Villuendas, P. (2011). Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules, 16(4), 3420-3432. https://doi.org/10.3390/molecules16043420