Synthesis of Stable and Soluble One-Handed Helical Homopoly(substituted acetylene)s without the Coexistence of Any Other Chiral Moieties via Two-Step Polymer Reactions in Membrane State: Molecular Design of the Starting Monomer

 ,
,
Abstract
:1. Introduction



2. Results and Discussion
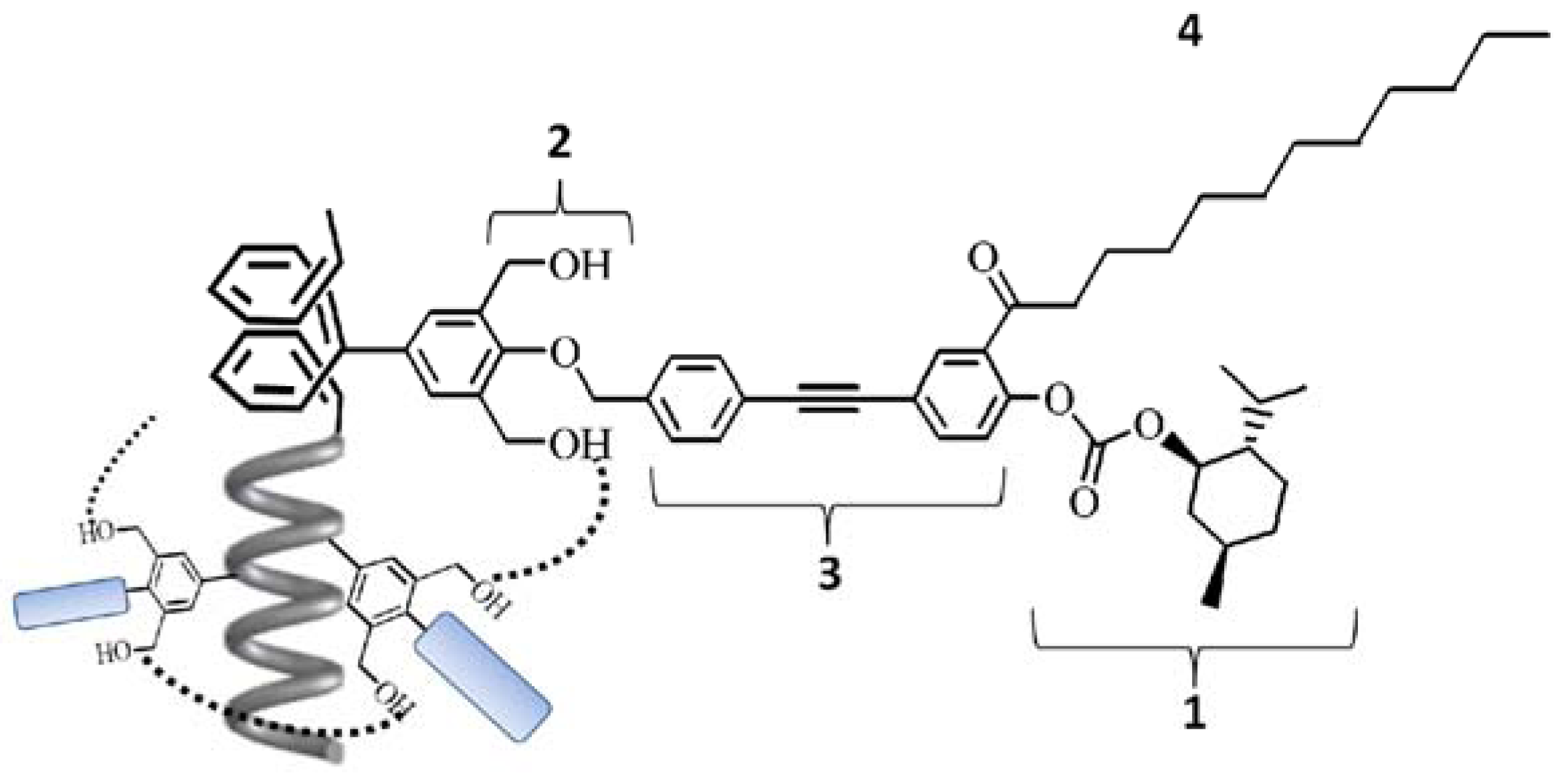
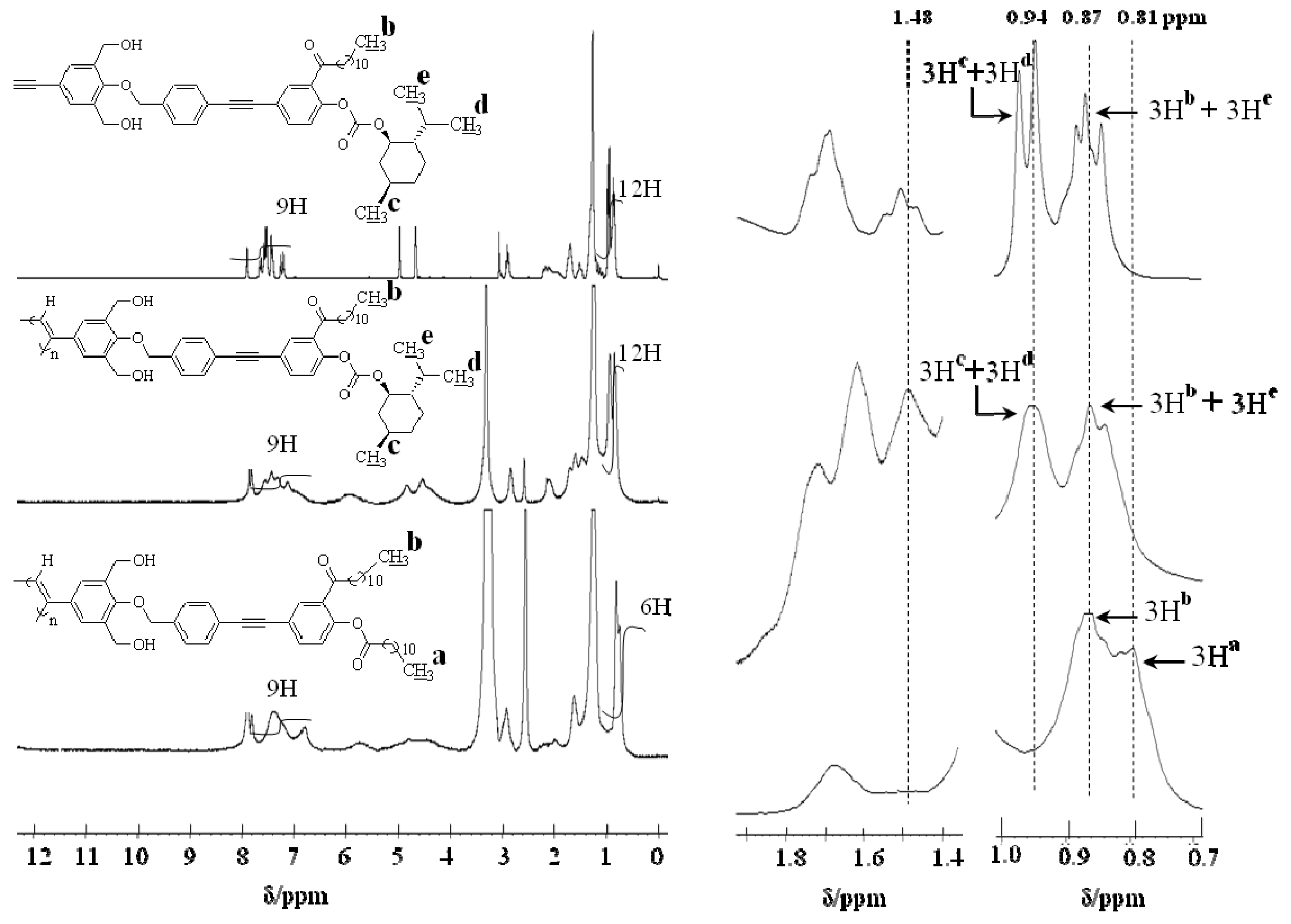
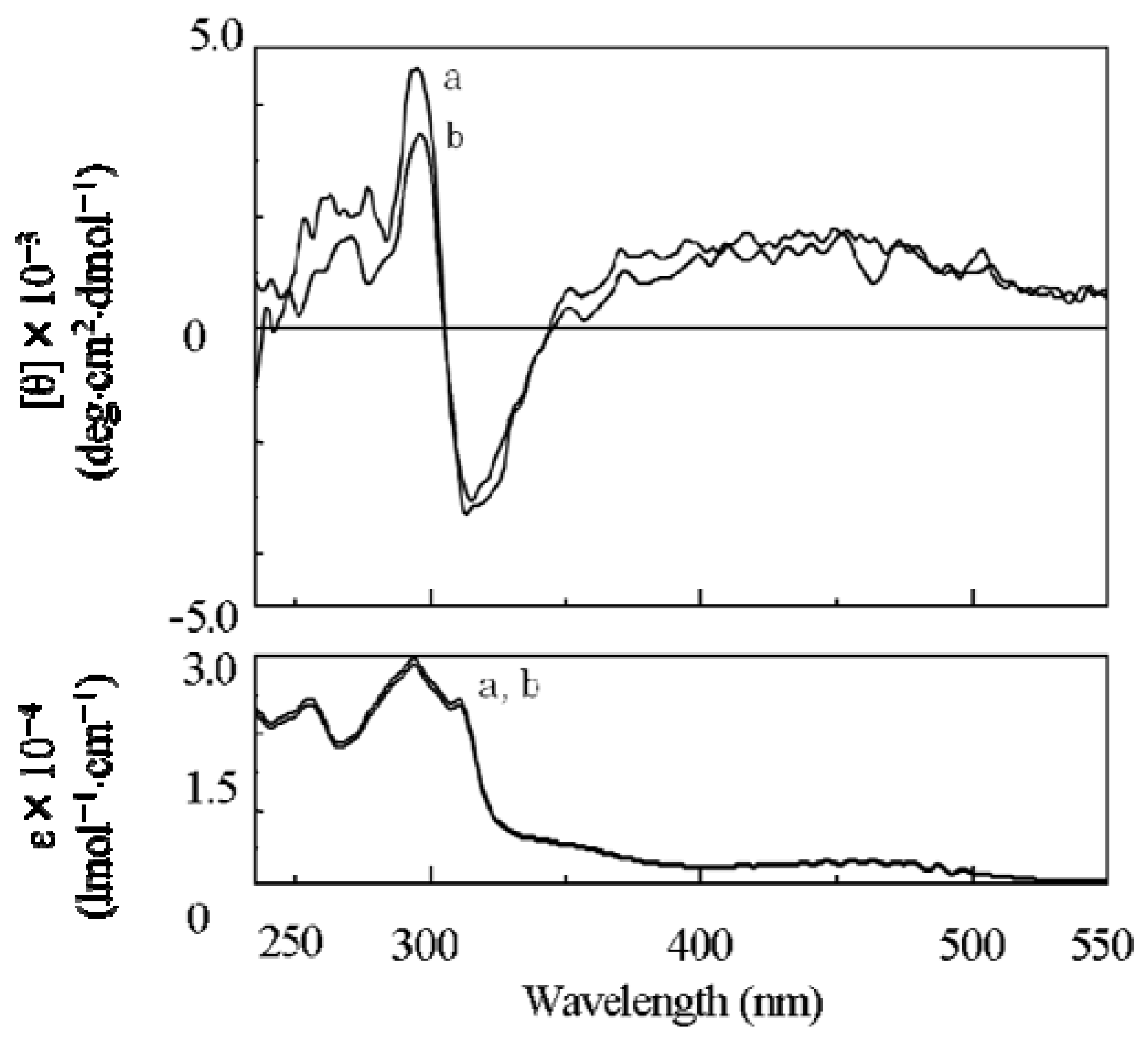
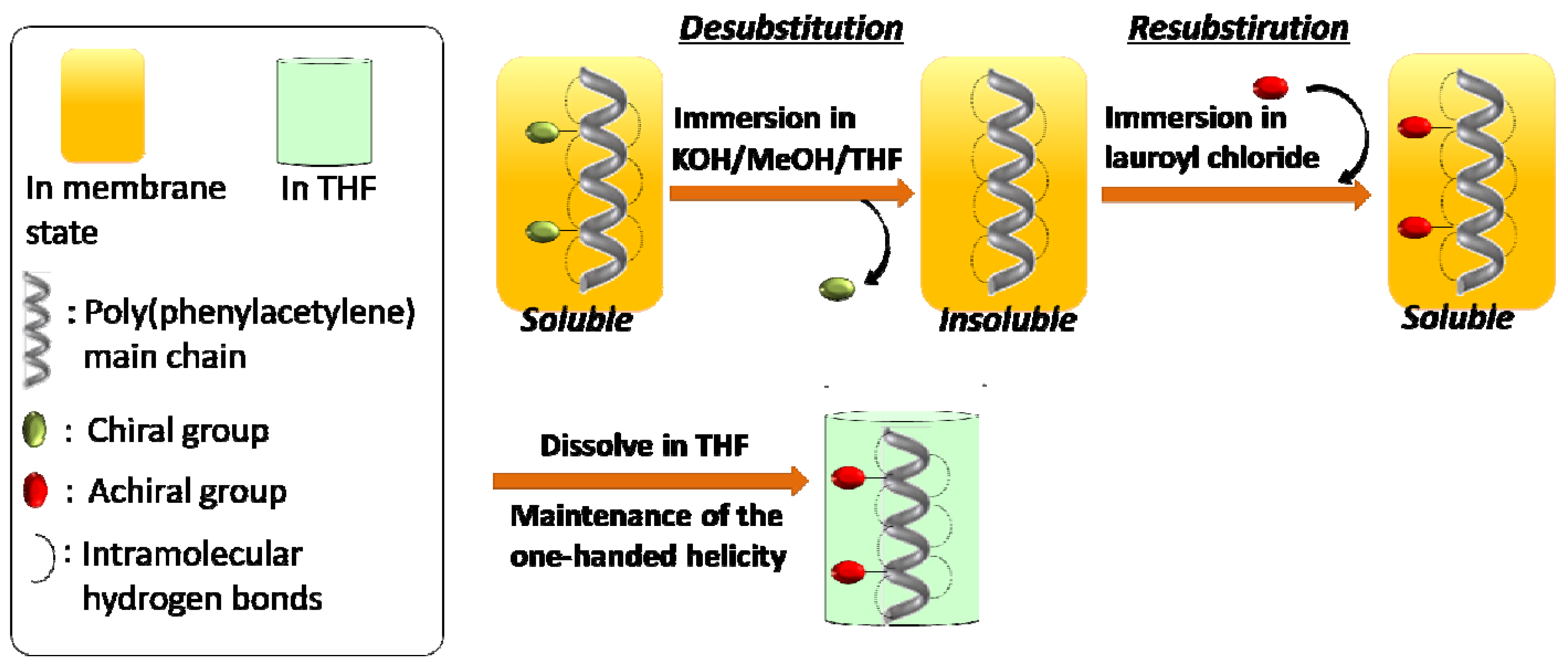
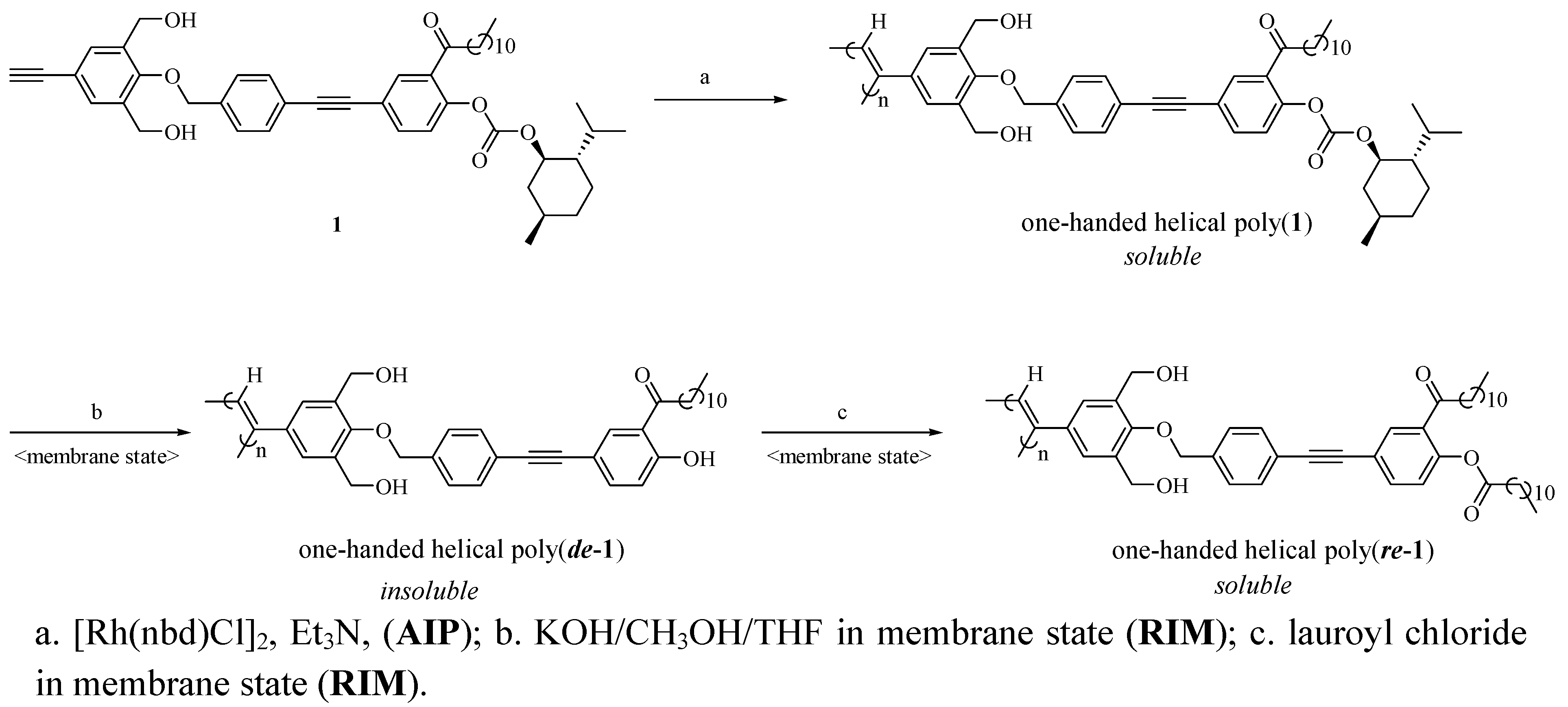
2.1. Synthesis of a Soluble One-Handed Helical Poly(1) Having Chiral Pendant Groups by Asymmetric-Induced Polymerization(AIP) of 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Yield b/% | Mw c/×105 | Mw/Mnc | [α]D20 d/° | g450e/×10−7 |
|---|---|---|---|---|---|
| 1 | - | - | - | −18.4 | 0 |
| poly(1) | 98.6 | 27 | 16 | −176 | 2.7 |
| poly(re-1) | 91.4 | 5.1 | 10 | 117 | 2.5 |



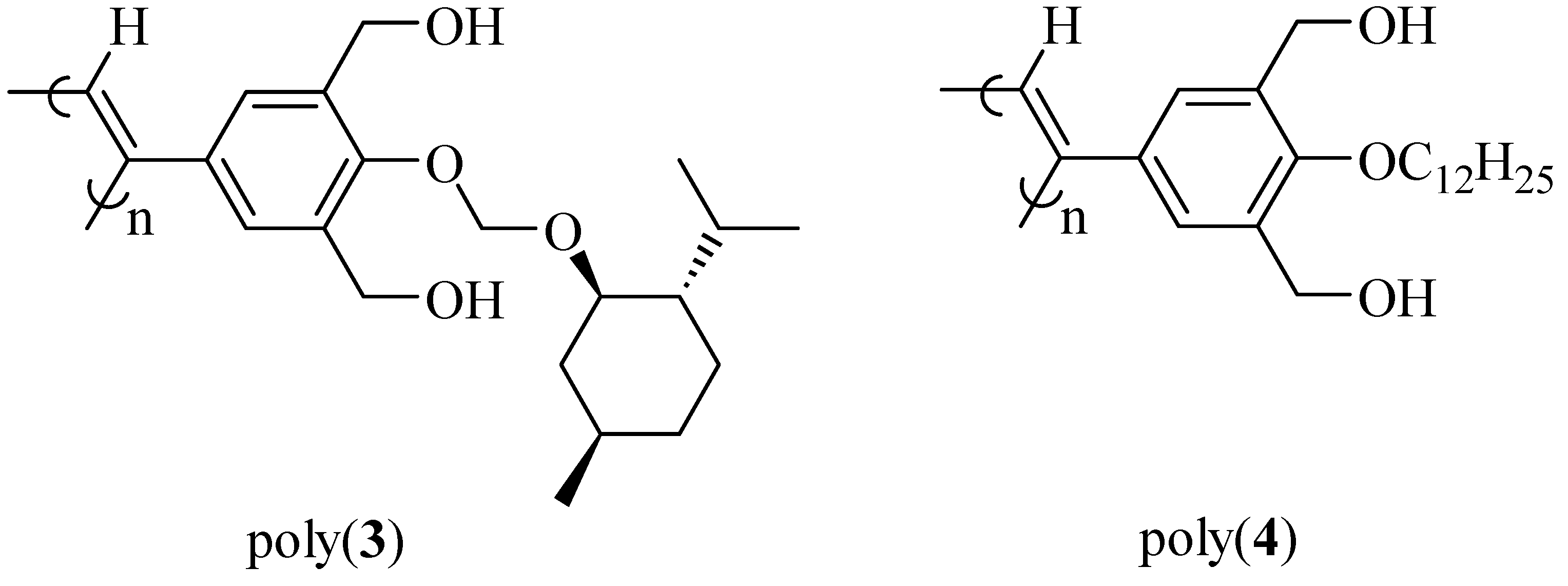
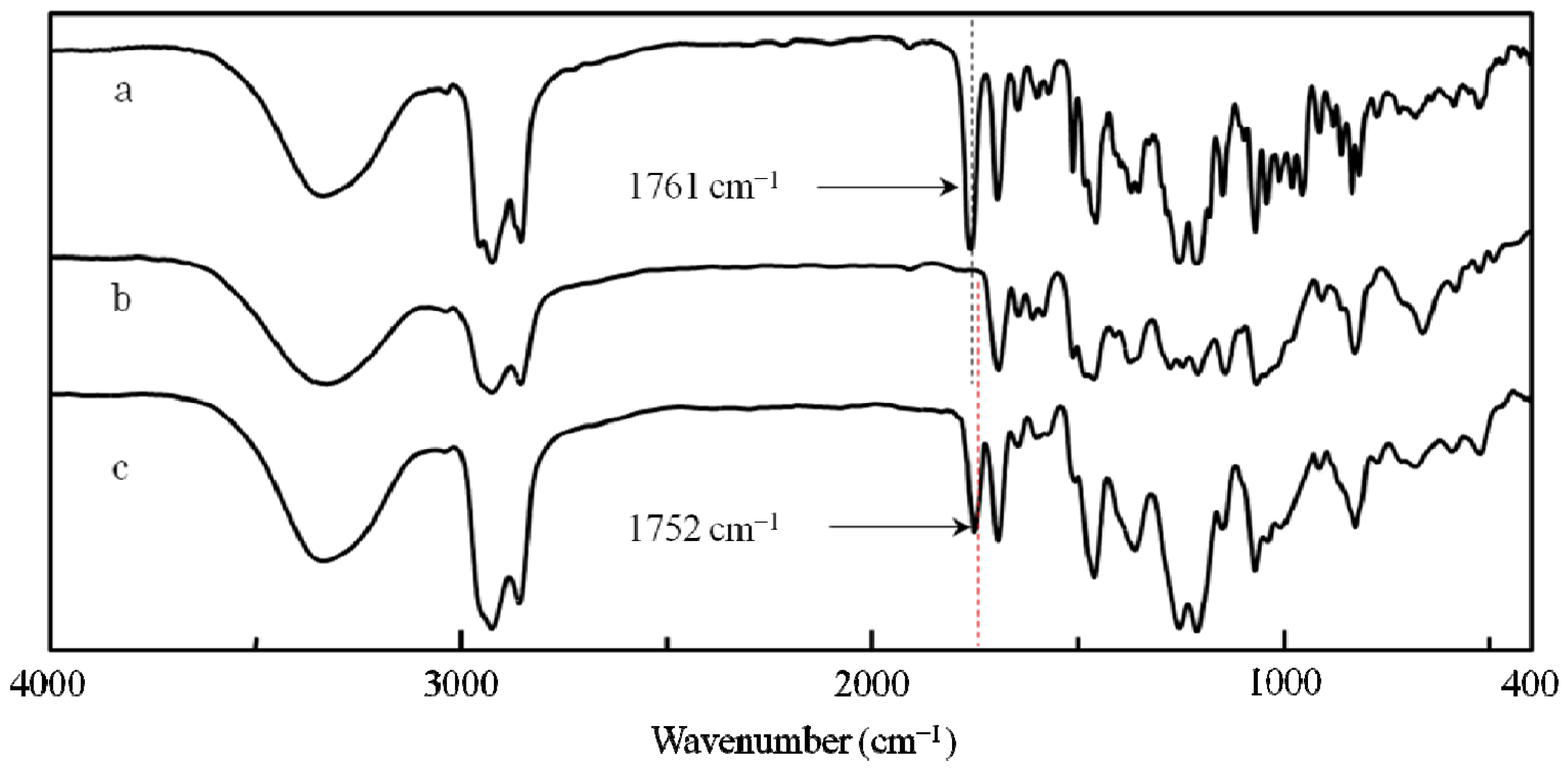
2.2. Synthesis of an Insoluble One-Handed Helical Poly(de-1) Having no Chiral Pendant Groups by Removing the Chiral Groups (Desubstitution) Quantitatively from Poly(1) in Membrane State (RIM)


2.3. Synthesis of a Soluble One-Handed Helical poly(re-1) Having no Chiral Pendant Groups by Quantitative Introduction of Achiral Groups (Resubstitution) to Poly(de-1) in Membrane State(RIM)
2.3.1. Quantitative Introduction of Achiral Groups (Resubstitution) by RIM


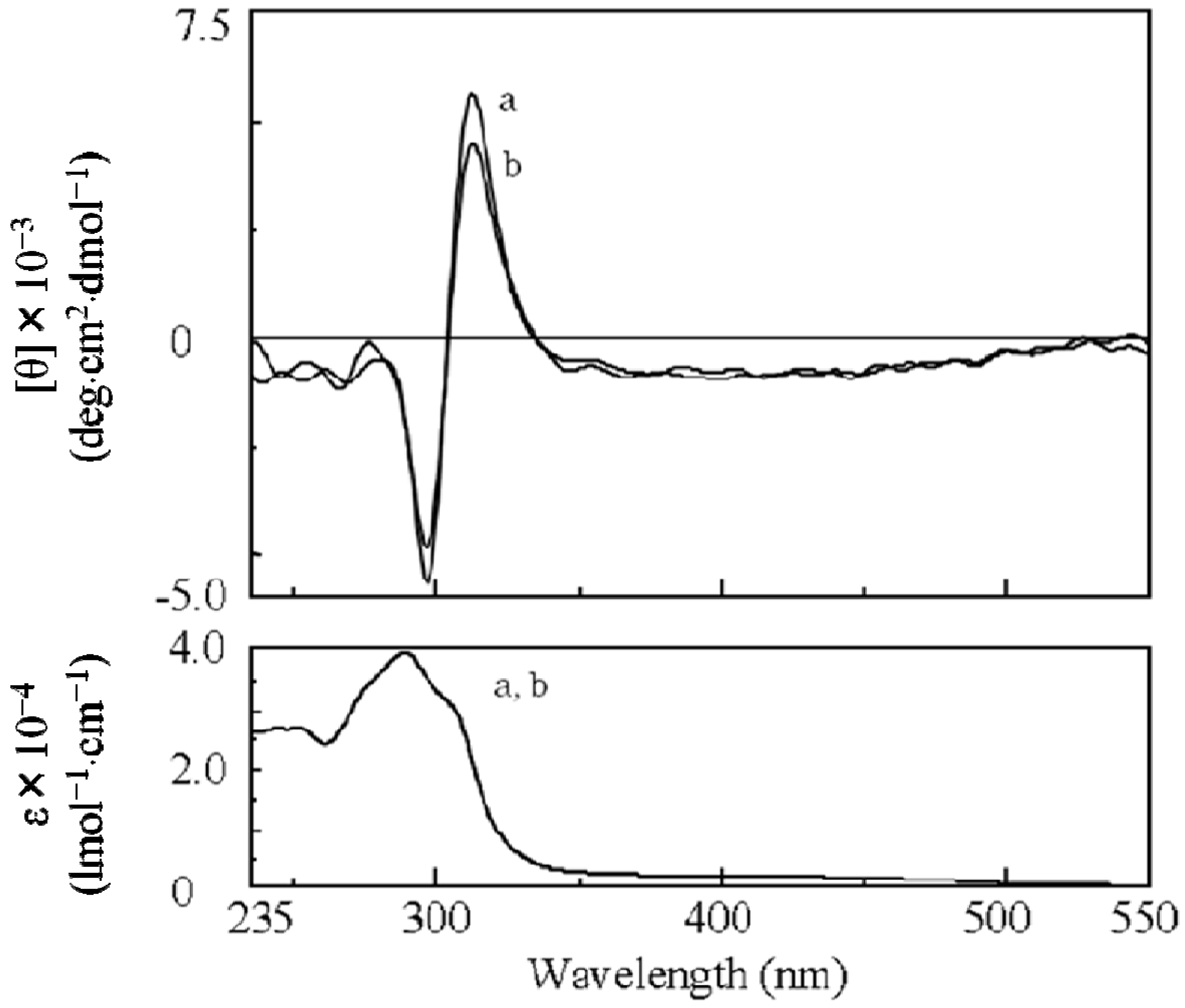
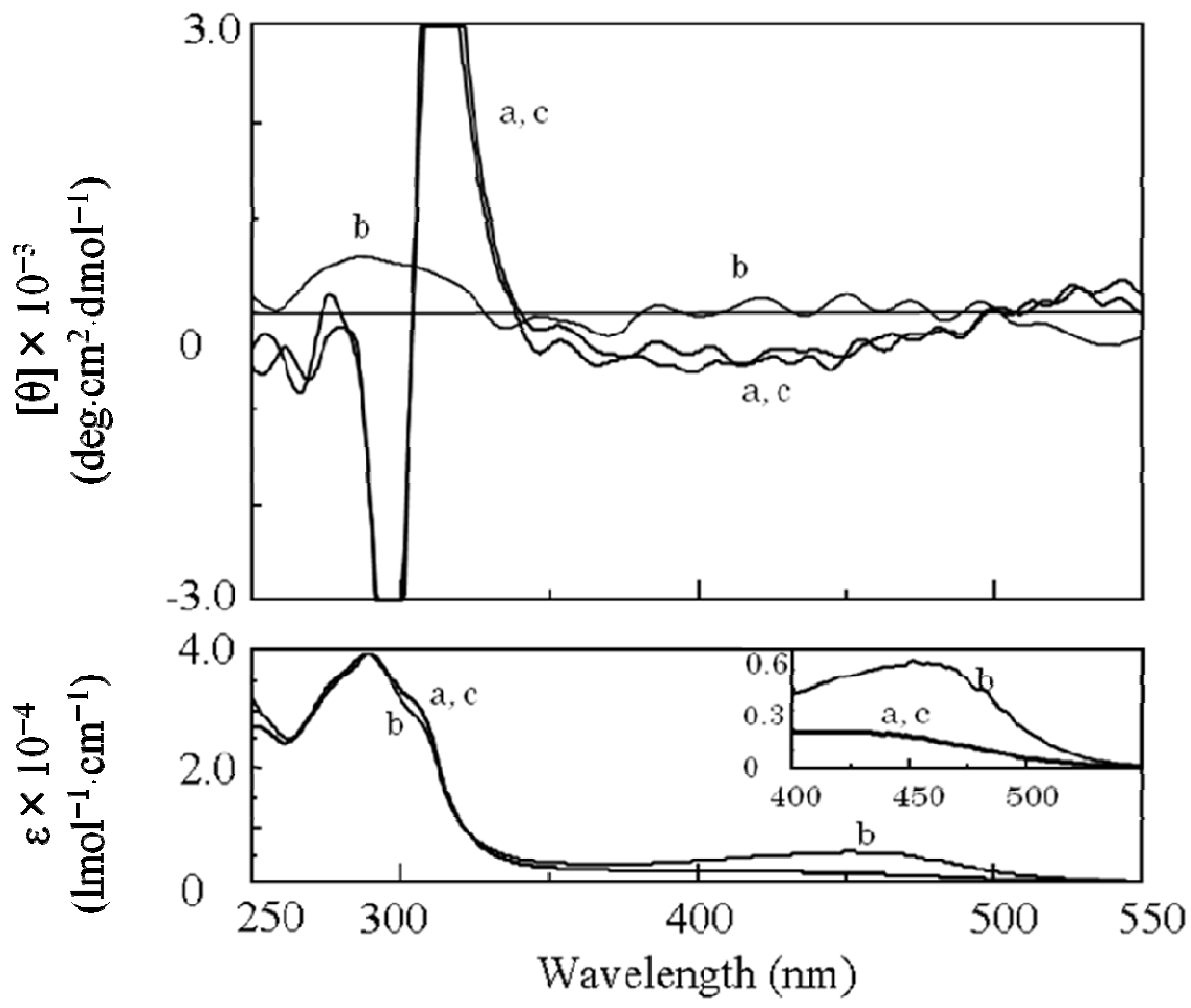
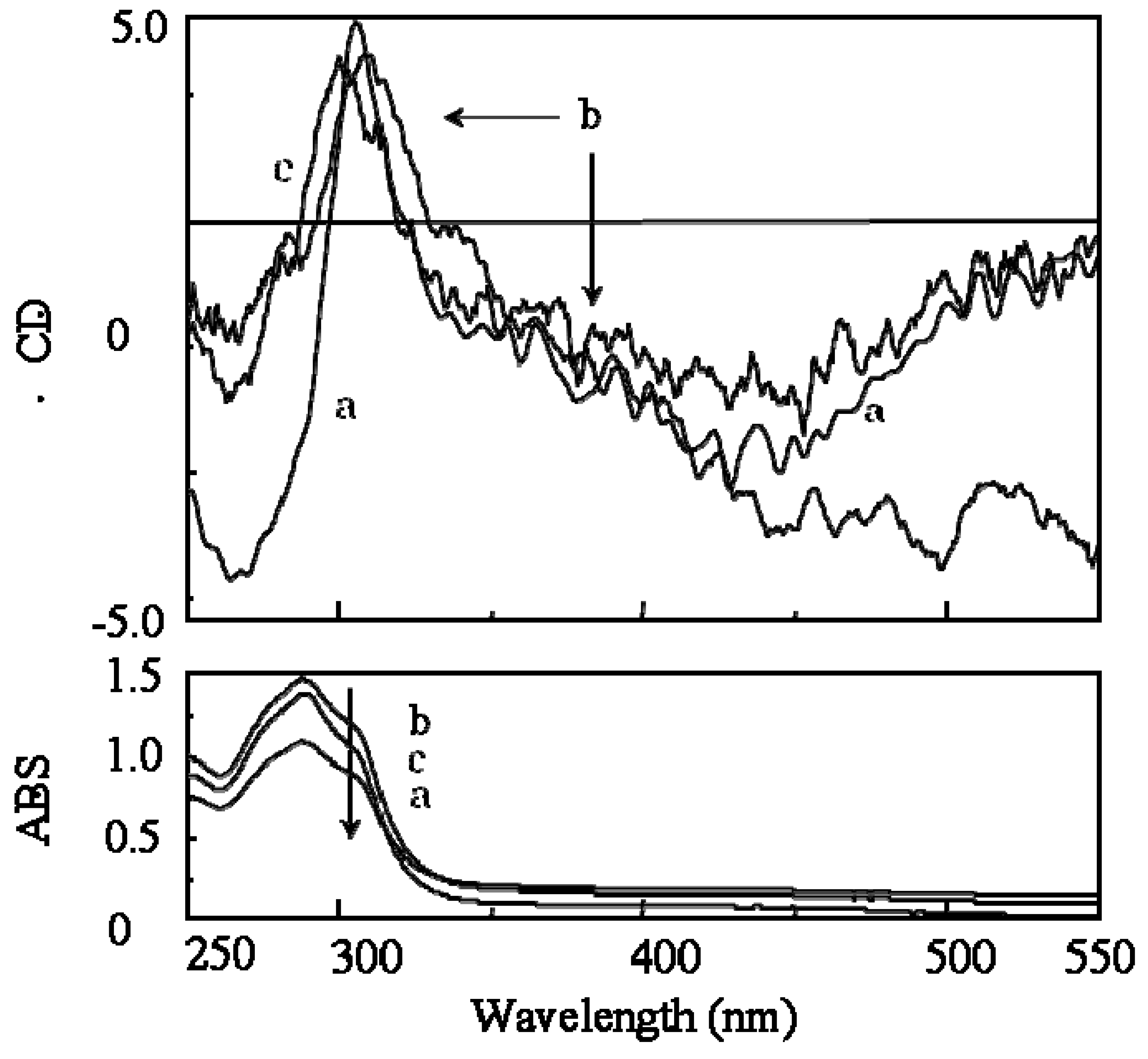
2.3.2. Evidence of the One-Handed Helicity of Poly(re-1) Prepared by Two-Step RIM


3. Experimental
3.1. General
3.2. Monomer Synthesis (see Scheme 4)

3.1.1. Synthesis of 4-Bromophenyl dodecanoate (1a)
3.1.2. Synthesis of 5-Bromo-2-hydroxydodecanophenone (1b)
3.1.3. Synthesis of 2-Hydroxy-5-trimethylsilylethynyldodecanophenone (1c)
3.1.4. Synthesis of 5-Ethynyl-2-hydroxydodecanophenone (1d)
3.1.5. Synthesis of 4-Bromo-2,6-bis(hydroxymethyl)phenol (1e)
3.1.6. Synthesis of 1,3-Bis(hydroxymethyl)-2-(4'-iodobenzyloxy)-5-bromobenzene (1f)
3.1.7. Synthesis of 1,3-Bis(tert-butyldimethylsilyloxymethyl)-2-(4'-iodobenzyloxy)-5-bromobenzene (1g)
3.1.8. Synthesis of 1,3-Bis(tert-butyldimethylsilyloxymethyl)-2-{4'-[4''-hydroxy-3''-(1-oxododecyl)phenyl-ethynyl]benzyloxy}-5-bromobenzene (1h)
3.1.9. Synthesis of 1,3-Bis(tert-butyldimethylsilyloxymethyl)-2-{4'-[4''-hydroxy-3''-(1-oxododecyl)phenyl-ethynyl]benzyloxy}-5-(trimethylsilylethynyl)benzene (1i)
3.1.10. Synthesis of 1,3-Bis(tert-butyldimethylsilyloxymethyl)-2-{4'-[4''-( L-menthoxycarbonyloxy)-3''-(1-oxododecyl)phenylethynyl]benzyloxy}-5-(trimethylsilylethynyl)benzene (1j)
3.1.11. Synthesis of 3,5-Bis(hydroxymethyl)-4-{4'-[4''-(L-menthoxycarbonyloxy)-3''-(1-oxododecyl)-phenylethynyl]benzyloxy}phenylacetylene (1)
3.2. Asymmetric-Induced Polymerization (AIP) and Polymer Reaction in Membrane State (RIM) (see Scheme 1 and Scheme 2)
3.2.1. Asymmetric-Induced Polymerization (AIP) of 1
3.2.2. Synthesis of Poly(de-1) by Desubstitution of the Chiral Groups from Poly(1) in Membrane State (RIM)
3.3.3. Synthesis of Poly(re-1) by Resubstitution of the Achiral Groups to poly(de-1) in Membrane State (RIM)
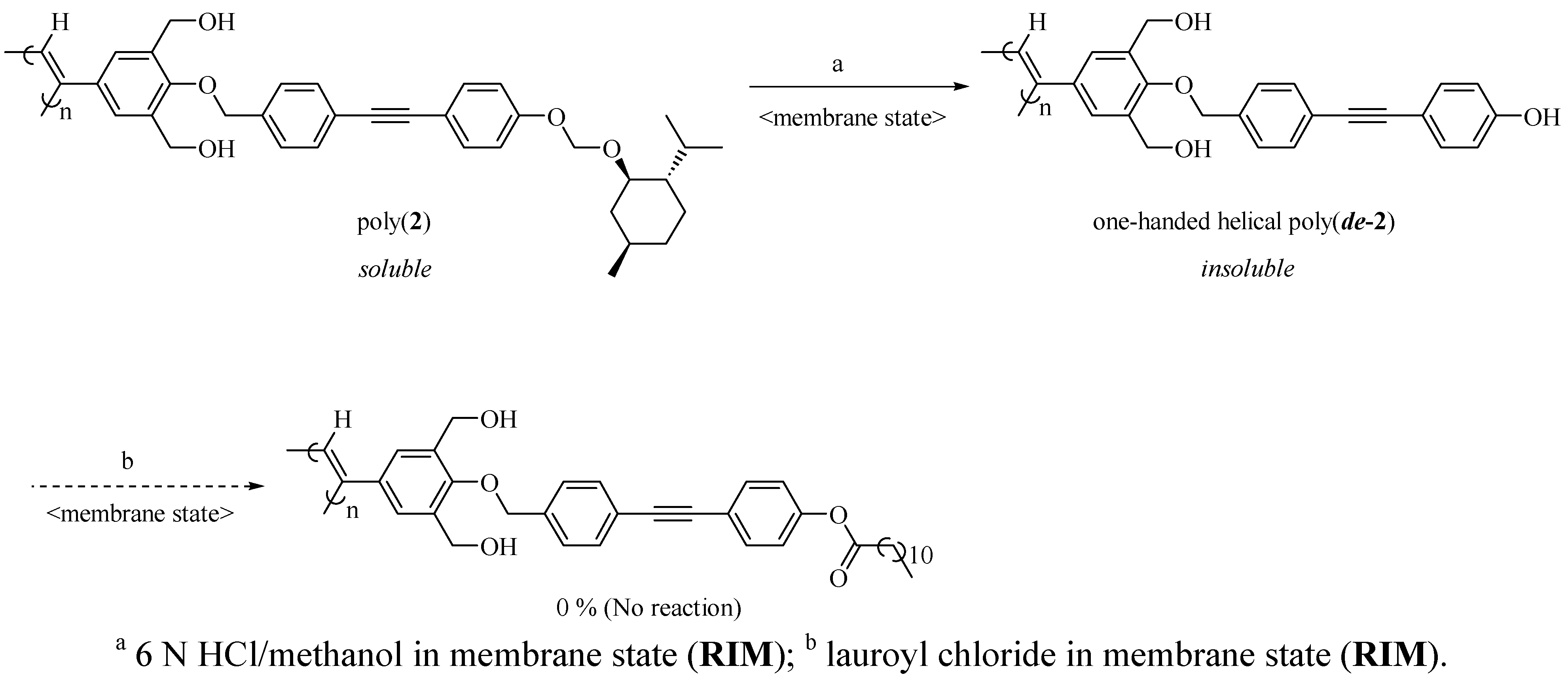
3.3.4. Synthesis of Poly(de-2) by Removing the Chiral Groups from Poly(2) in Membrane State (RIM) (Scheme 3)
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds 1a, 1e and 1f are available from the authors.
References and Notes
- Aoki, T.; Kaneko, T.; Teraguchi, M. Synthesis of functional π-conjugated polymers from aromatic acetylenes. Polymer 2006, 47, 4867–4892. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Iida, H.; Furusho, Y.; Nagai, K. Helical polymers: Synthesis, structures, and functions. Chem. Rev. 2009, 109, 6102–6211. [Google Scholar] [CrossRef]
- Ito, S.; Nozaki, K. Asymmetric polymerization. In Catalytic Asymmetric Synthesis, 3rd; Ojima, I., Ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 931–985. [Google Scholar]
- Aoki, T.; Kaneko, T.; Maruyama, N.; Sumi, A.; Takahashi, M.; Sato, T.; Teraguchi, M. Helix-sense-selective polymerization of achiral phenylacetylene having two hydroxy groups using a chiral catalytic system. J. Am. Chem. Soc. 2003, 125, 6346–6347. [Google Scholar] [CrossRef]
- Sato, T.; Aoki, T.; Teraguchi, M.; Kaneko, T.; Kim, S.Y. Role of chiral amines in helix-sense-selective polymerization of a phenylacetylene using chiral amines as cocatalysts. Polymer 2004, 45, 8109–8114. [Google Scholar] [CrossRef]
- Hadano, S.; Teraguchi, M.; Kaneko, T.; Aoki, T. Enantioselective permeability through membranes from a poly(substituted phenylacetylene) having a chiral helical backbone and achiral bidentate ligands as pendant groups. Chem. Lett. 2007, 36, 220–221. [Google Scholar] [CrossRef]
- Katagiri, H.; Kaneko, T.; Teraguchi, M.; Aoki, T. Copper(I) iodide accelerates catalytic activation in rhodium complex-catalyzed helix-sense-selective polymerization of achiral phenylacetylene monomers. Chem. Lett. 2008, 37, 390–391. [Google Scholar] [CrossRef]
- Umeda, Y.; Kaneko, T.; Teraguchi, M.; Aoki, T. Helix-sense-selective polymerization of a phenylacetylene bearing an achiral and bulky galvinoxyl moiety. Chem. Lett. 2005, 34, 854–855. [Google Scholar] [CrossRef]
- Kaneko, T.; Umeda, Y.; Yamamoto, T.; Teraguchi, M.; Aoki, T. Assignment of helical sense for poly(phenylacetylene) bearing achiral galvinoxyl chromophore synthesized by helix-sense-selective polymerization. Macromolecules 2005, 38, 9420–9426. [Google Scholar]
- Kaneko, T.; Umeda, Y.; Jia, H.; Hadano, S.; Teraguchi, M.; Aoki, T. Helix-sense tunability induced by achiral diene ligands in the chiral catalytic system for the helix-sense-selective polymerization of achiral and bulky phenylacetylene monomers. Macromolecules 2007, 40, 7098–7102. [Google Scholar] [CrossRef]
- Hadano, S.; Teraguchi, M.; Kaneko, T.; Aoki, T. Enantioselective permeability through membranes from a poly(substituted phenylacetylene) having a chiral helical backbone and achiral bidentate ligands as pendant groups. Chem. Lett. 2007, 36, 220–221. [Google Scholar] [CrossRef]
- Hadano, S.; Kishimoto, T.; Hattori, T.; Tanioka, D.; Teraguchi, M.; Aoki, T.; Kaneko, T.; Namikoshi, T.; Marwanta, E. Helix-sense-selective polymerization of achiral bis(hydroxyl-methyl)phenylacetylenes having an alkyl group of different lengths. Macromol. Chem. Phys. 2009, 210, 717–727. [Google Scholar] [CrossRef]
- Jia, H.; Hadano, S.; Teraguchi, M.; Aoki, T.; Abe, Y.; Kaneko, T.; Namikoshi, T.; Marwanta, E. Two modes of asymmetric polymerization of phenylacetylene having a L-valinol residue and two hydroxy groups. Macromolecules 2009, 42, 17–19. [Google Scholar]
- Liu, L.; Zang, Y.; Hadano, S.; Aoki, T.; Teraguchi, M.; Kaneko, T.; Namikoshi, T. New achiral phenylacetylene monomers having an oligosiloxanyl group most suitable for helix-sense-selective polymerization and for obtaining good optical resolution membrane materials. Macromolecules 2010, 43, 9268–9276. [Google Scholar] [CrossRef]
- Liu, L.; Oniyama, Y.; Zang, Y.; Hadano, S.; Aoki, T.; Teraguchi, M.; Kaneko, T.; Namikoshi, T.; Marwanta, E. Synthesis and helix-sense-selective polymerization of a novel phenylacetylene having a trisiloxanyl group and two hydroxyl groups and enantioselective permeability of the resulting chiral polymeric membrane: Effect of the trisiloxanyl group on the polymerization and enantioselective permeability. Polymer (Comm.) 2010, 51, 2460–2464. [Google Scholar]
- Nolte, R.J.M.; Van Beijnen, A.J.M.; Drenth, W. Chirality in polyisocyanides. J. Am. Chem. Soc. 1974, 96, 5932–5933. [Google Scholar] [CrossRef]
- Okamoto, Y.; Suzuki, K.; Ohta, K.; Hatada, K.; Yuki, H. Optically active poly(triphenylmethyl methacrylate) with one-handed helical conformation. J.Am.Chem.Soc. 1979, 101, 4763–4765. [Google Scholar] [CrossRef]
- Nakano, T.; Okamoto, Y.; Hatada, K. Asymmetric polymerization of triphenylmethyl methacrylate leading to a one-handed helical polymer: Mechanism of polymerization. J. Am. Chem. Soc. 1992, 114, 1318–1329. [Google Scholar] [CrossRef]
- Nakano, T.; Nakagawa, O.; Tsuji, M.; Tanikawa, M.; Yade, T.; Okamoto, Y. Poly(2,7-di-n-pentyldibenzofulvene) showing chiroptical properties in the solid state based purely on a chiral conformation. Chem.Commun. 2004, 144–145. [Google Scholar]
- Su, S.J.; Kuramoto, N. In situ synthesis of optically active poly(o-ethoxyzniline) in organic media and its chiroptical properties. Chem. Mater. 2001, 13, 4787–4793. [Google Scholar] [CrossRef]
- Oishi, T.; Kagawa, K.; Nagata, H. Synthesis and polymerization of maleimide containing an L-menthoxy group. Polymer 1997, 38, 1461–1469. [Google Scholar] [CrossRef]
- Nakano, T.; Tamada, D.; Miyazaki, J.; Kakiuchi, K.; Okamoto, Y. Asymmetric radical polymerization of maleimides using a chiral cobalt(II) complex. Macromolecules 2000, 33, 1489–1491. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Kwak, G.; Masuda, T. Synthesis of poly(1-β-naphthyl-2-phenylacetylene) membranes through desilylation and their properties. Polymer 2002, 43, 3937–3942. [Google Scholar] [CrossRef]
- Wulff, G. Main-chain chirality and optical activity in polymers consisting of C-C chains. Angew. Chem. Int. Ed. Engl. 1989, 28, 21–37. [Google Scholar] [CrossRef]
- Wulff, G.; Dhal, P.K. Template monomer control of the chirality induction in the polymer backbone during asymmetric vinyl polymerization. Macromolecules 1990, 23, 4525–4527. [Google Scholar] [CrossRef]
- Wulff, G.; Schmidt, H.; Witt, H.; Zentel, R. Cooperativity and transfer of chirality in liquid-crystalline polymers. Angew. Chem. Int. Ed. Engl. 1994, 33, 188–191. [Google Scholar] [CrossRef]
- Kakuchi, T.; Narumi, A.; Kaga, H.; Ishibashi, T.; Obata, M.; Yokota, K. Chirality induction in cyclocopolymerization. 13. Structural effect of 1,3-diol as chiral templates in the cyclocopolymerization of bis(4-vinylbenzoate)s with styrene. Macromolecules 2000, 33, 3964–3969. [Google Scholar] [CrossRef]
- Kakuchi, T.; Narumi, A.; Kaga, H.; Yamauchi, Y.; Obata, M.; Uesaka, T.; Yokota, K. Chirality induction in cyclocopolymerization. 14. Template effect of 1,2-cycloalkanediol in the cyclocopolymerization of bis(4-vinylbenzoate)s with styrene. Macromolecules 2001, 34, 38–43. [Google Scholar] [CrossRef]
- Yu, Z.; Wan, X.; Zhang, H.; Chen, X.; Zhou, Q. A free radical initiated optically active vinyl polymer with memory of chirality after removal of the inducing stereogenic center. Chem. Commun. 2003, 974–975. [Google Scholar]
- Abe, Y.; Aoki, T.; Jia, H.; Hadano, S.; Namikoshi, T.; Kakihana, Y.; Liu, L.; Zang, Y.; Teraguchi, M.; Kaneko, T. Polymer 2012. submitted.
- Amabilino, D.B.; Ramos, E.; Serrano, J.L.; Sierra, T.; Veciana, J. Long-range chiral induction in chemical systems with helical organization. Promesogenic monomers in the formation of poly(isocyanide)s and in the organization of liquid crystals. J. Am. Chem. Soc. 1998, 120, 9126–9134. [Google Scholar]
- In the case of poly(3) without the spacer, it was insoluble.
- Kaneko, T. Chem. Lett. 2012, in press.
- The sign of the CD signals for poly(re-1) was opposite to those for poly(1) and poly(de-1). The reason is not clear at present but since they are not mirror images each other, it did not show inversion of the helical sense.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abe, Y.; Aoki, T.; Jia, H.; Hadano, S.; Namikoshi, T.; Kakihana, Y.; Liu, L.; Zang, Y.; Teraguchi, M.; Kaneko, T. Synthesis of Stable and Soluble One-Handed Helical Homopoly(substituted acetylene)s without the Coexistence of Any Other Chiral Moieties via Two-Step Polymer Reactions in Membrane State: Molecular Design of the Starting Monomer. Molecules 2012, 17, 433-451. https://doi.org/10.3390/molecules17010433
Abe Y, Aoki T, Jia H, Hadano S, Namikoshi T, Kakihana Y, Liu L, Zang Y, Teraguchi M, Kaneko T. Synthesis of Stable and Soluble One-Handed Helical Homopoly(substituted acetylene)s without the Coexistence of Any Other Chiral Moieties via Two-Step Polymer Reactions in Membrane State: Molecular Design of the Starting Monomer. Molecules. 2012; 17(1):433-451. https://doi.org/10.3390/molecules17010433
Chicago/Turabian StyleAbe, Yunosuke, Toshiki Aoki, Hongge Jia, Shingo Hadano, Takeshi Namikoshi, Yuriko Kakihana, Lijia Liu, Yu Zang, Masahiro Teraguchi, and Takashi Kaneko. 2012. "Synthesis of Stable and Soluble One-Handed Helical Homopoly(substituted acetylene)s without the Coexistence of Any Other Chiral Moieties via Two-Step Polymer Reactions in Membrane State: Molecular Design of the Starting Monomer" Molecules 17, no. 1: 433-451. https://doi.org/10.3390/molecules17010433
APA StyleAbe, Y., Aoki, T., Jia, H., Hadano, S., Namikoshi, T., Kakihana, Y., Liu, L., Zang, Y., Teraguchi, M., & Kaneko, T. (2012). Synthesis of Stable and Soluble One-Handed Helical Homopoly(substituted acetylene)s without the Coexistence of Any Other Chiral Moieties via Two-Step Polymer Reactions in Membrane State: Molecular Design of the Starting Monomer. Molecules, 17(1), 433-451. https://doi.org/10.3390/molecules17010433