Chiroptical Switches: Applications in Sensing and Catalysis


Abstract
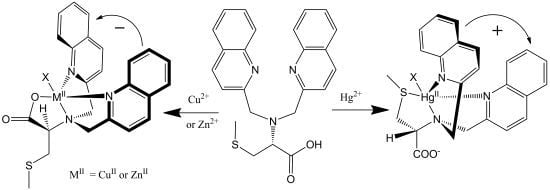
:
1. Introduction

| Type | Acronym | Definition |
|---|---|---|
| Optical rotation | α | The rotation of linearly polarized light as it travels through certain non-racemic materials. |
| Optical rotatory dispersion | ORD | The variation in the specific rotation of a substance with a change in the wavelength of light. May be used to determine the absolute configuration of metal complexes, for example. |
| Circular dichroism | CD | A form of spectroscopy based on the differential absorption (Δε) of left- and right-handed circularly polarized light (LCP and RCP) in non-racemic molecules. |
| Exction-coupled circular dichroism(Exciton Chirality) | ECCD | Circular dichroism coming from an exciton couplet: Two chromophores presenting in a molecule in close proximity to one another, who interact and give rise to differentiation of the energies of the transitions. |
| Fluorescence-detected CD | FDCD | Circular dichroism derived from the differential emission of light from a sample excited with LCP and RCP radiation when the analyte is chiral, fluorescent, and the quantum yield is the same for both circularly polarized components. |
| Diffrentialcircularly polarized fluorescence excitation | CPE(ΔF) | The differential emission of light from a sample excited with LCP and RCP radiation. The quantum yield does not have to be the same for both circularly polarized components |
| Circularly polarized luminescence | CPL | The anisotropic emission of circularly polarized light originated from non-polarized excitation. |
2. Chemically Triggered Chiroptical Switches
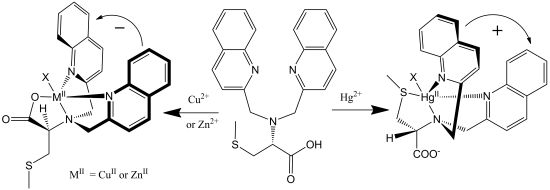
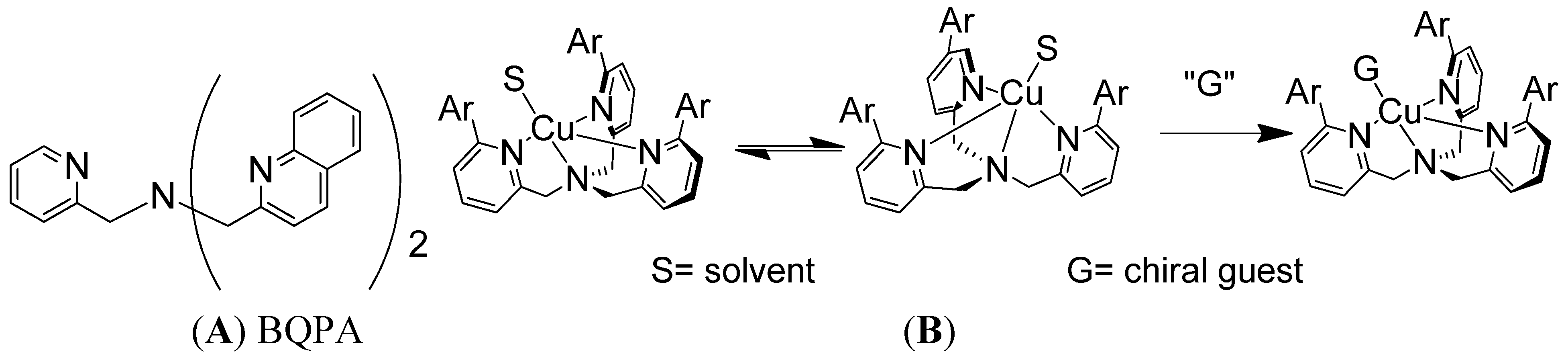
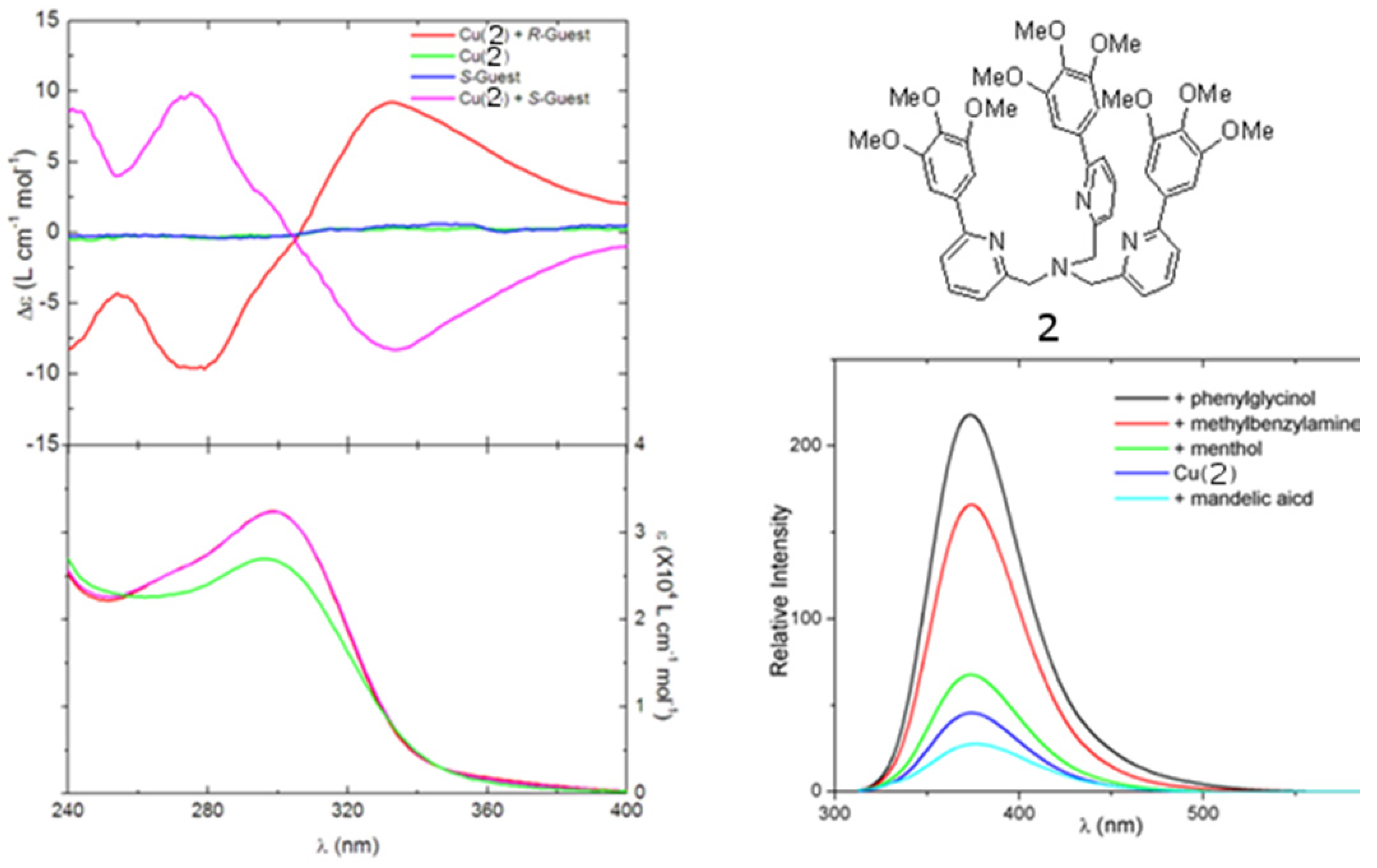
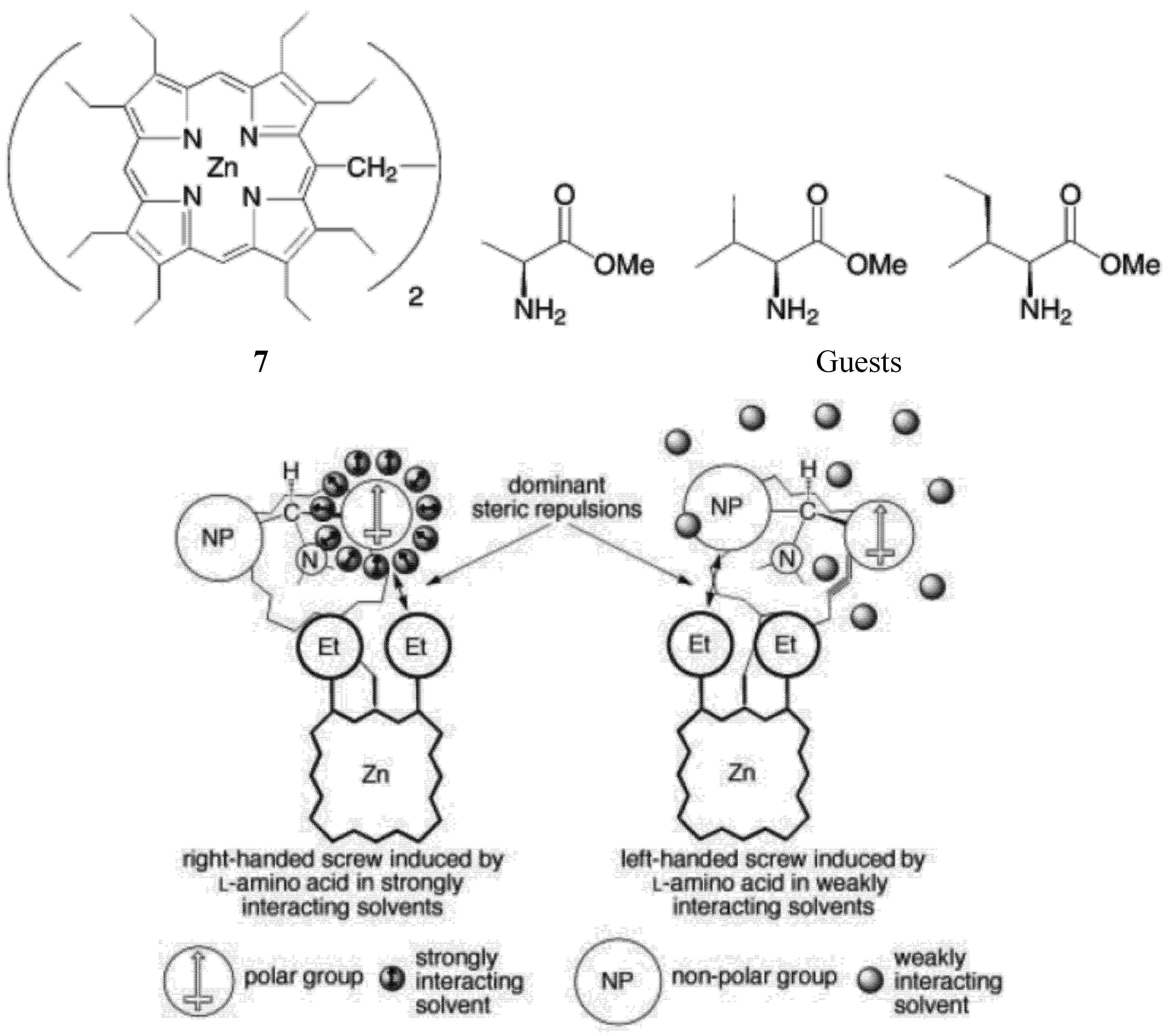
2.1. Chiroptical Switches Triggered by Chiral Guests




2.2. Chiroptical Switches Triggered by Achiral Guests
2.2.1. Chiroptical Switches Triggered by Achiral Organic Compounds

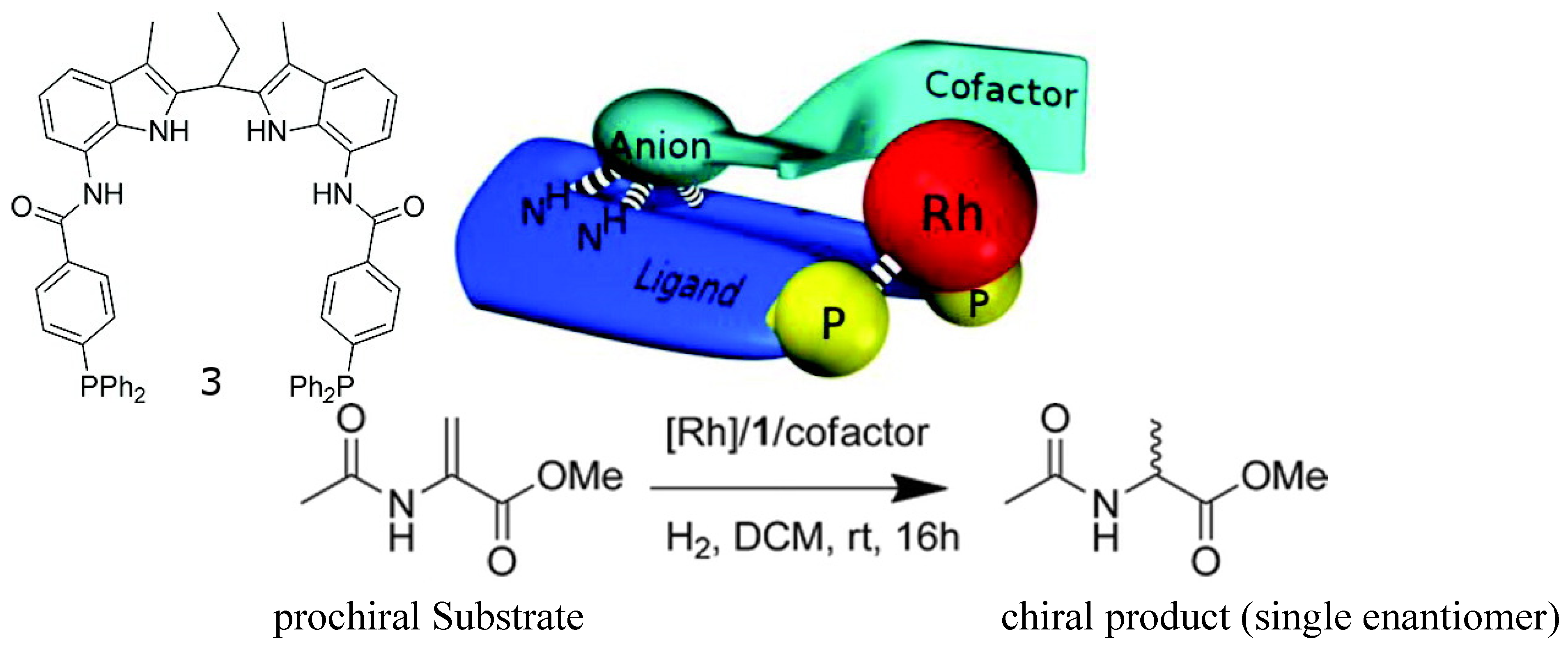
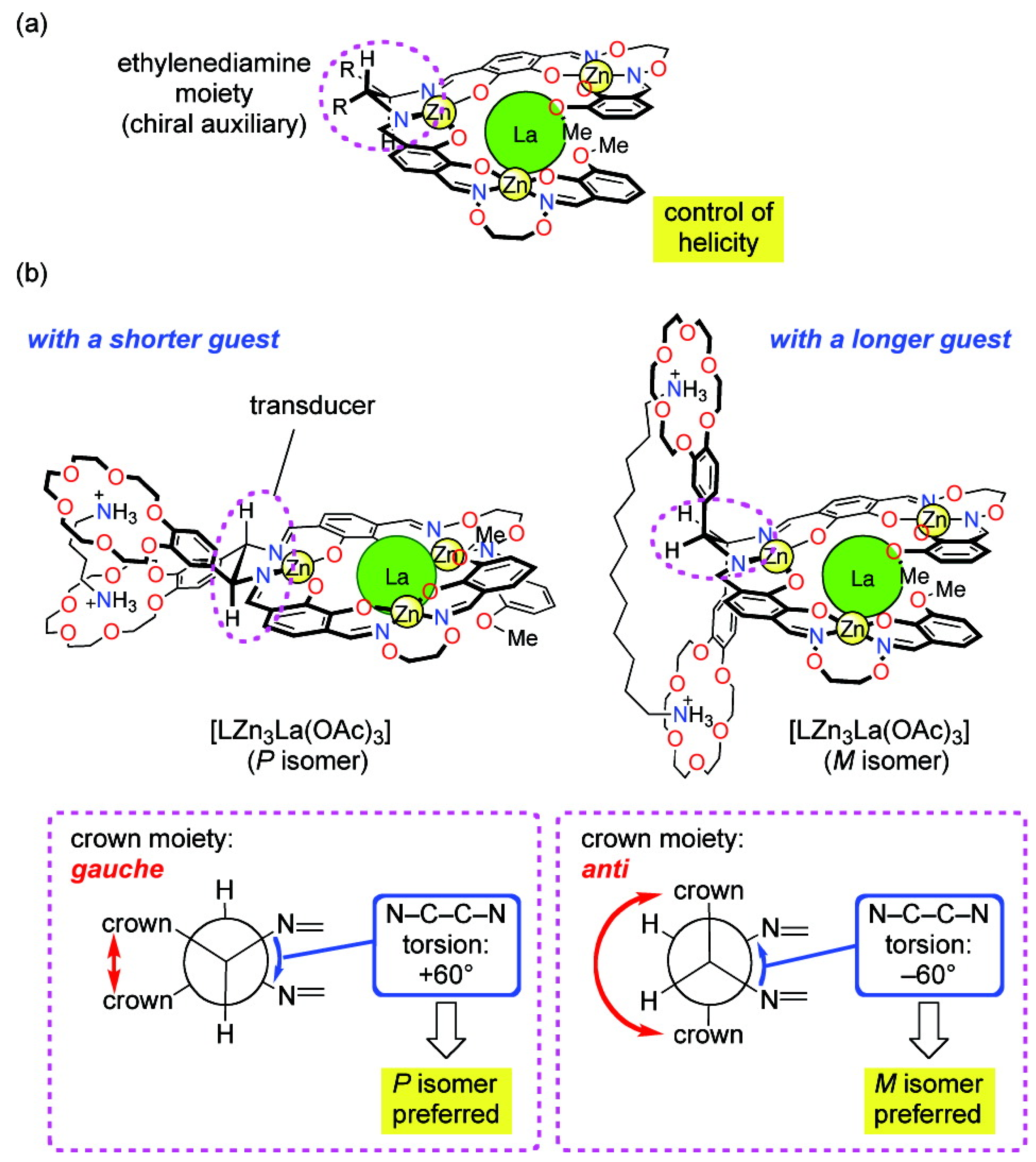
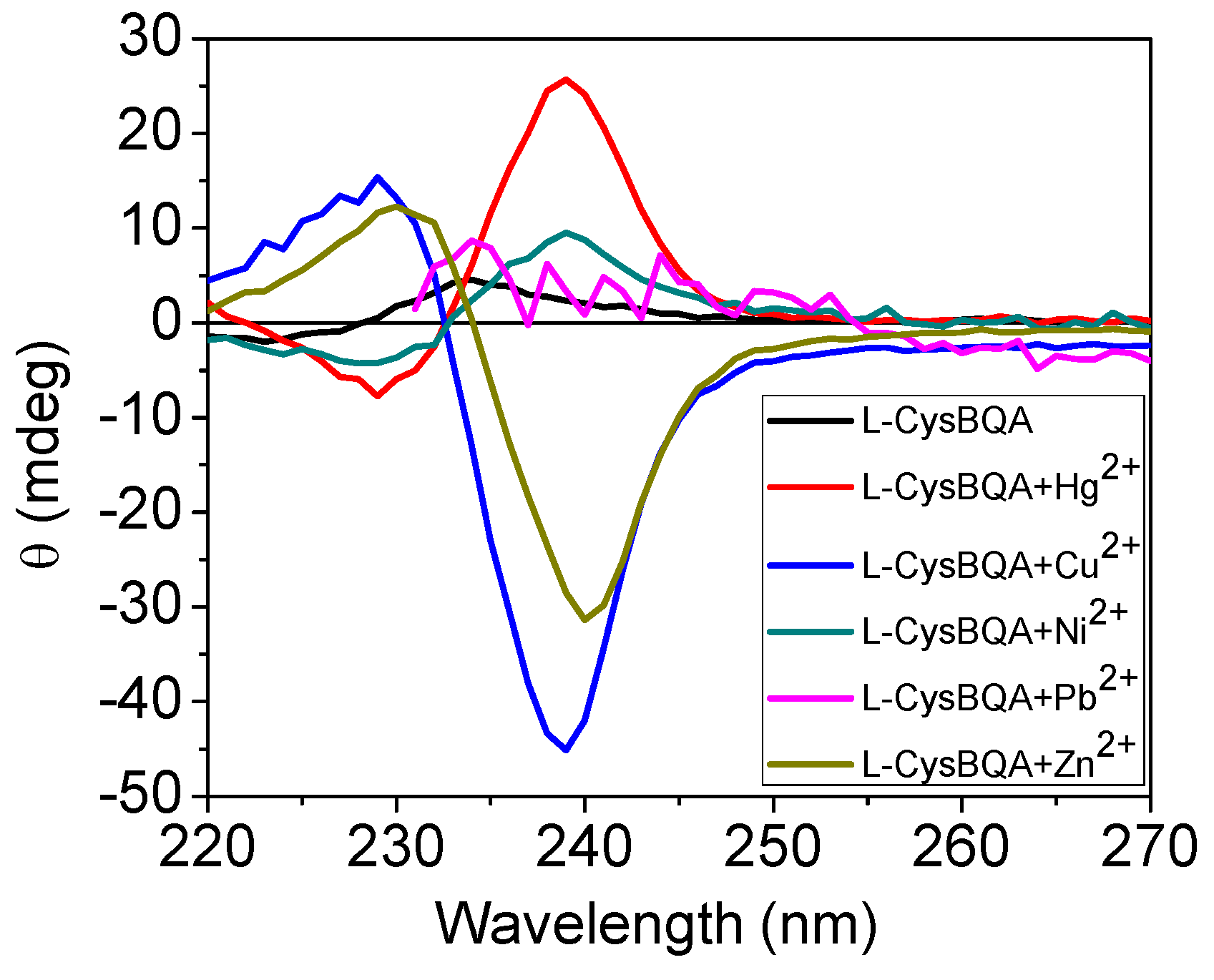
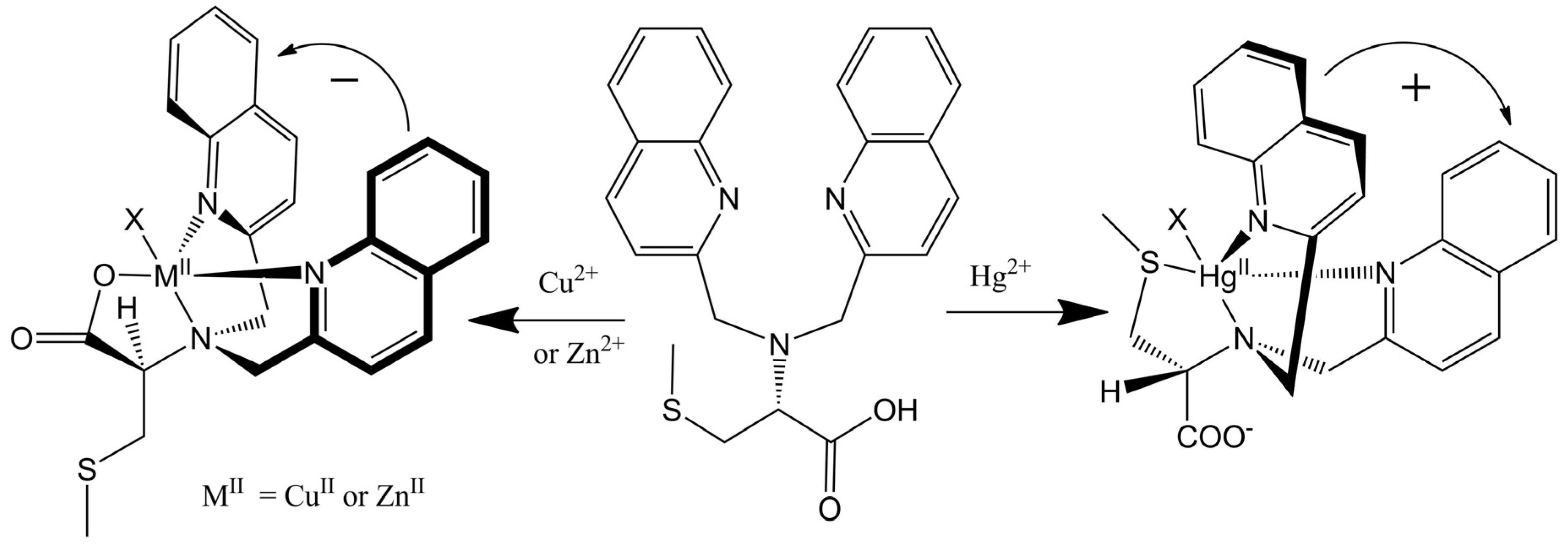
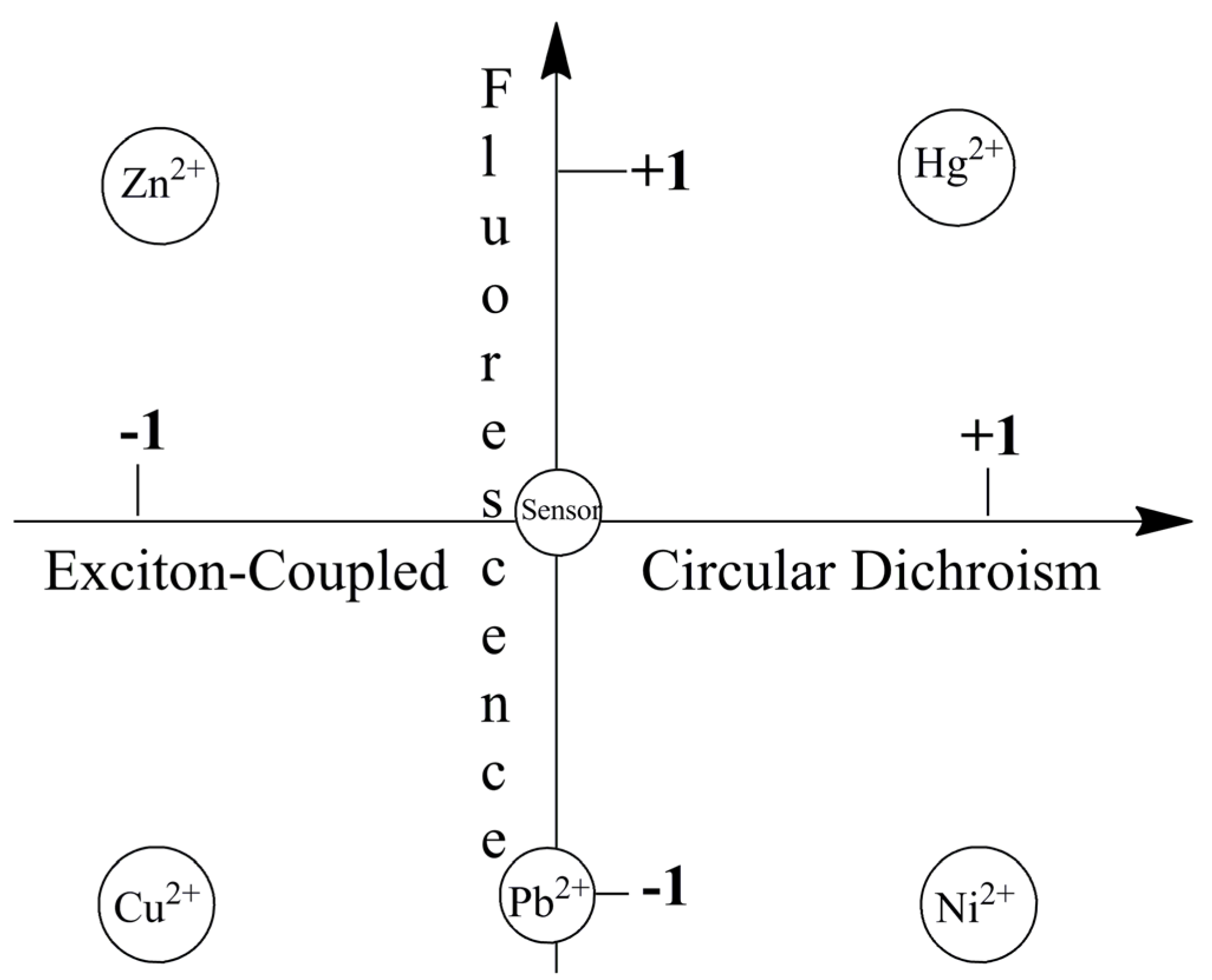
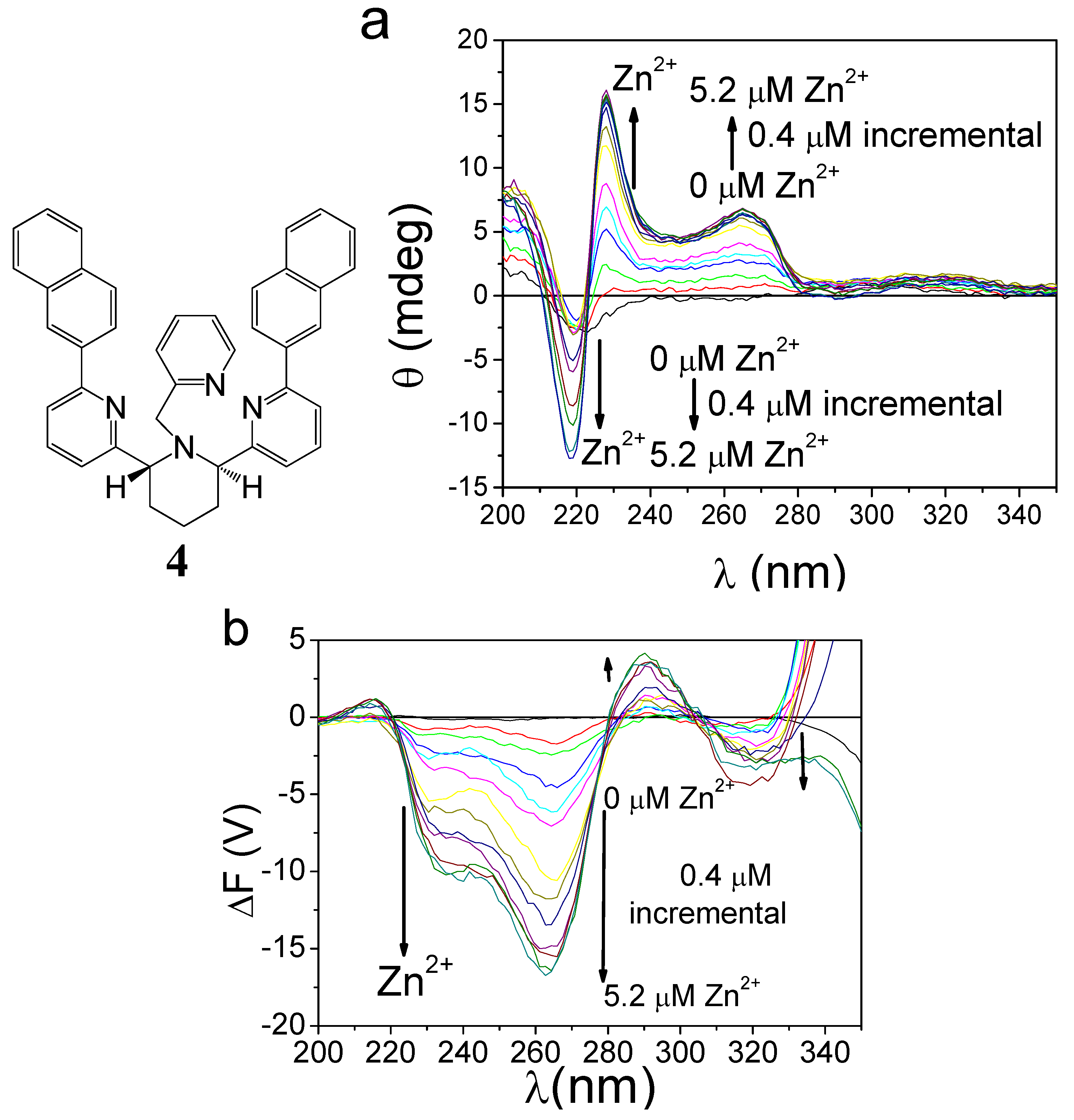
2.2.2. Chiroptical Switches Triggered by Metal Cations









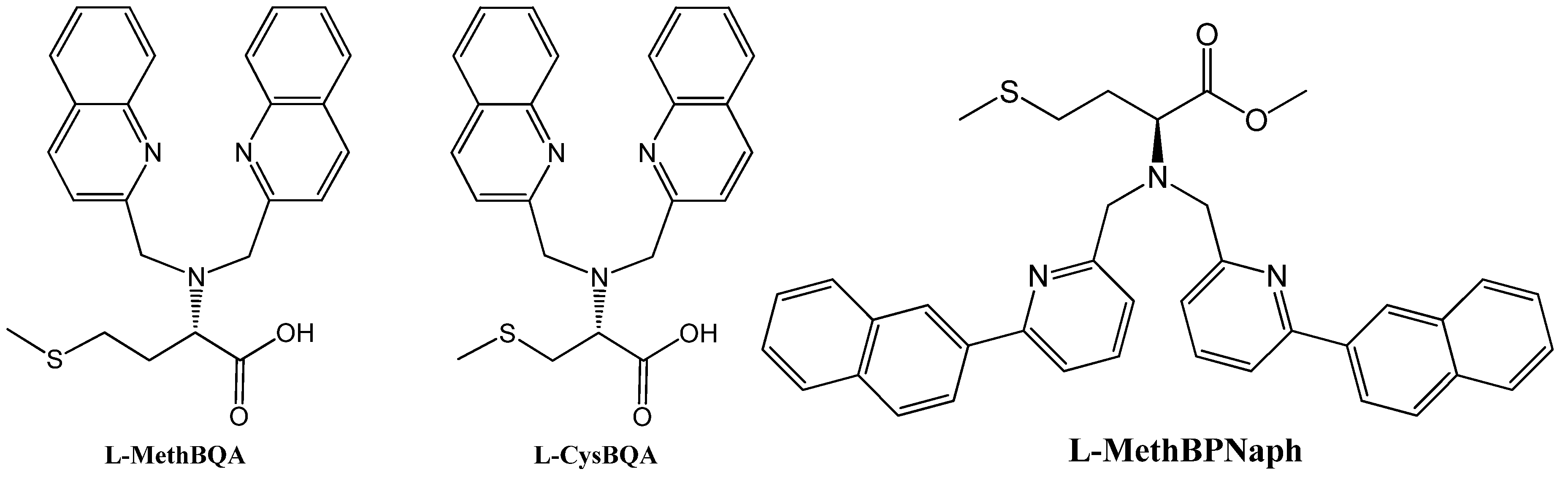
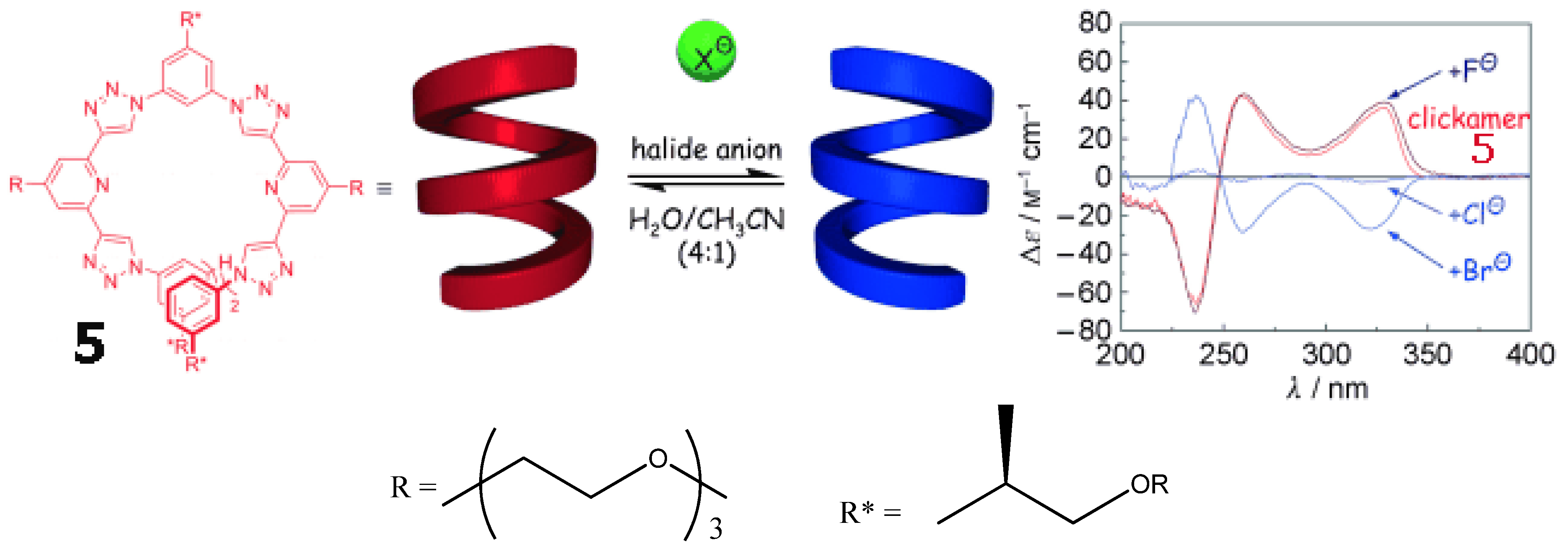
2.2.3. Chiroptical Switches Triggered by Achiral Anions


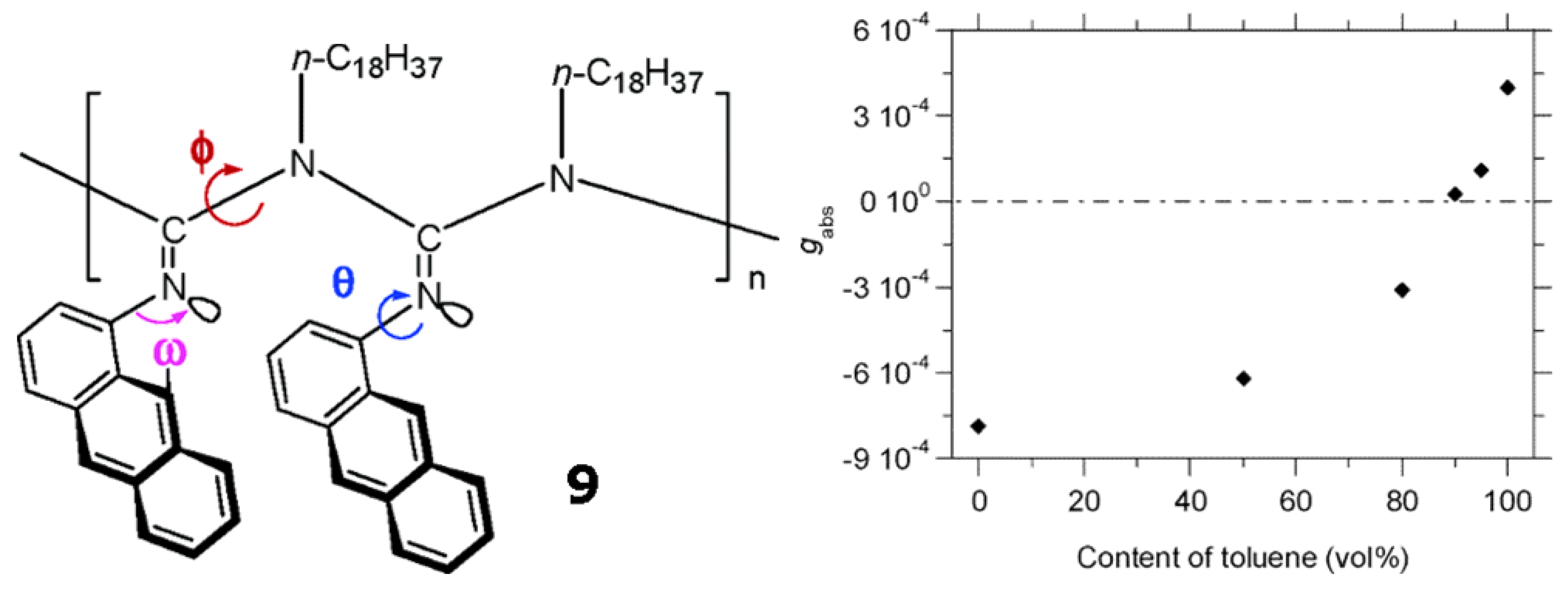
2.3. Solvent-Triggered Chiroptical Switches








2.4. Redox-Triggered Switches


2.5. Electrochemically Triggered Switches
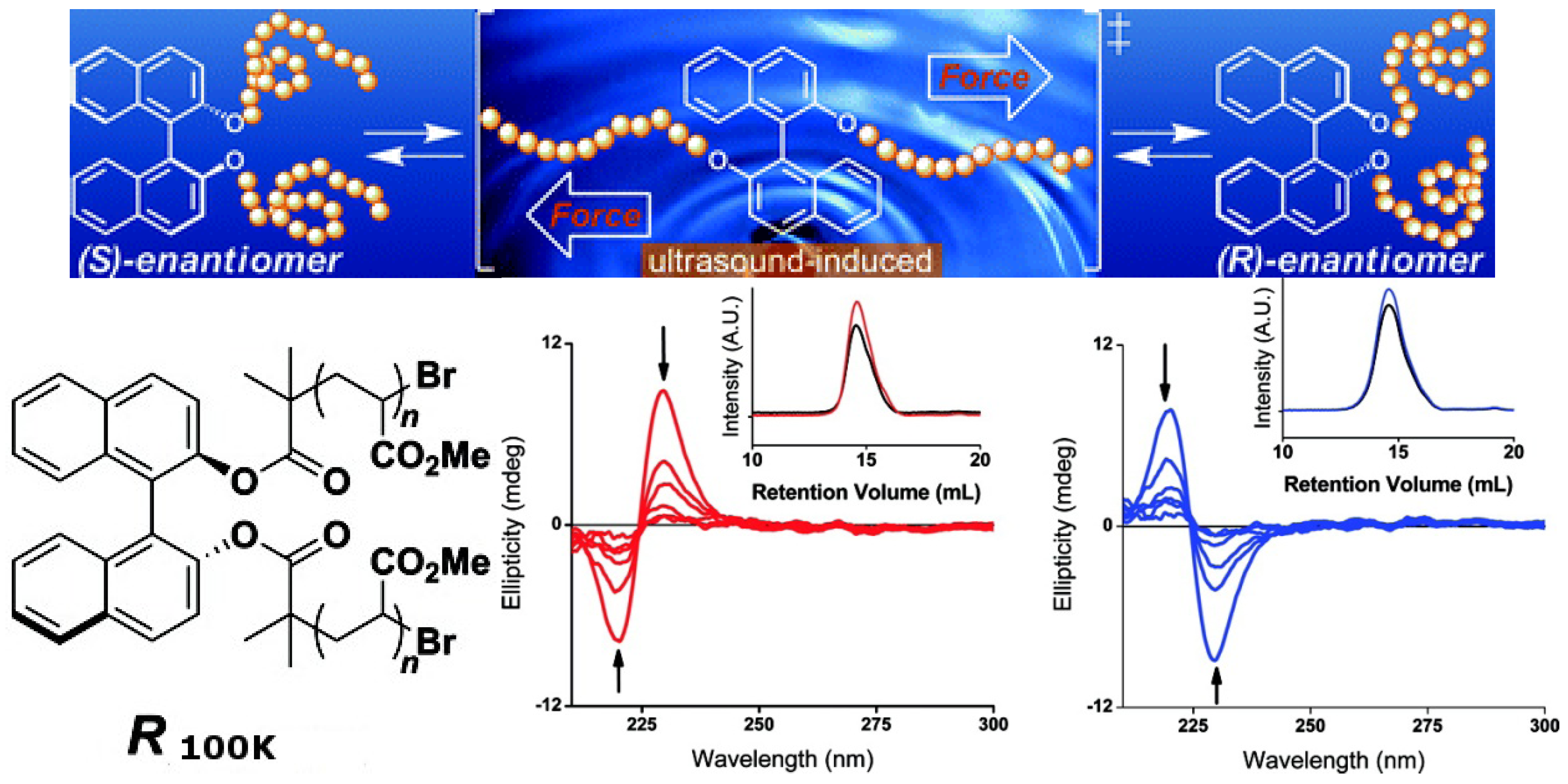
3. Mechanically Switchable Chiroptical Switches
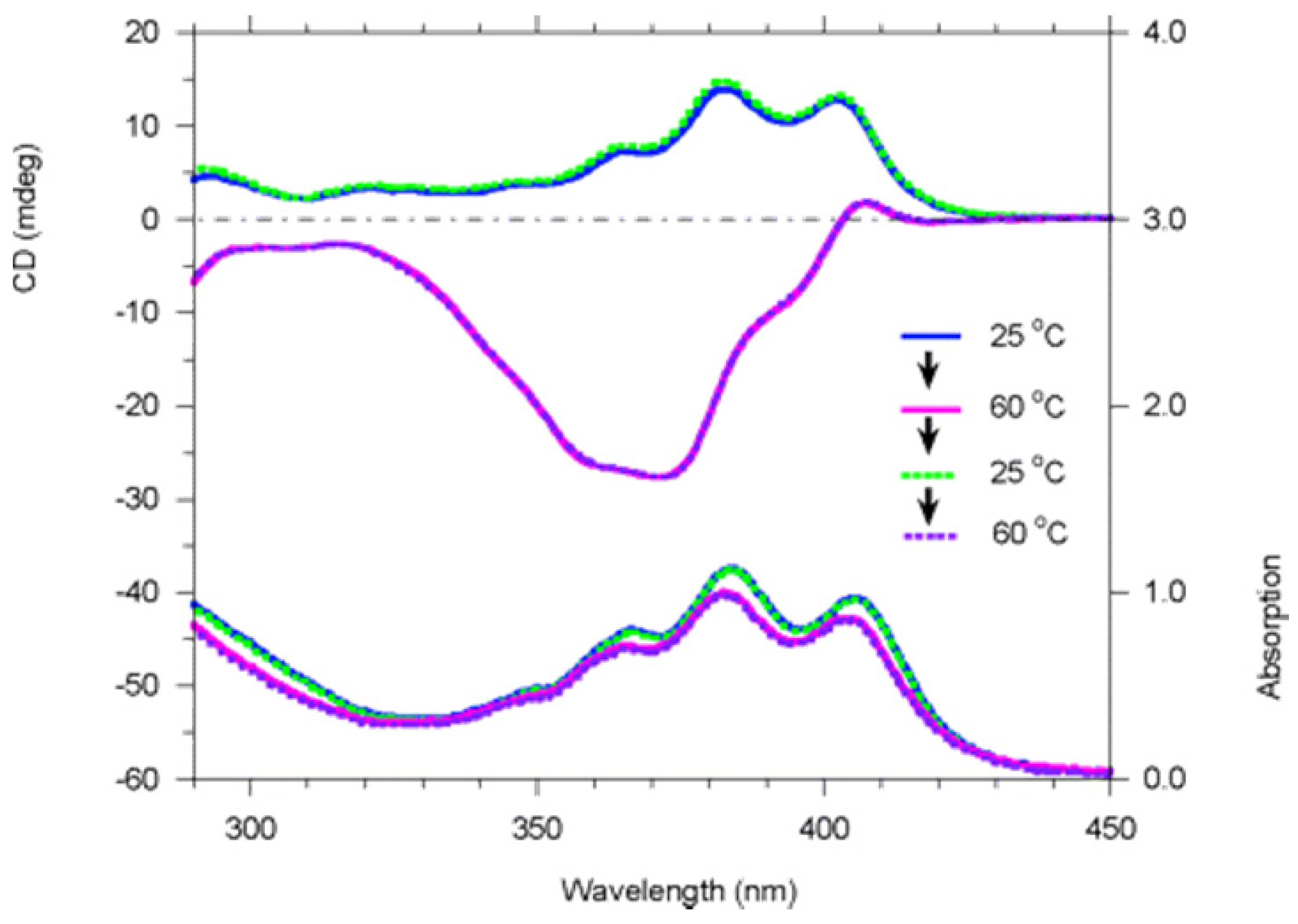
4. Temperature Triggered Switches



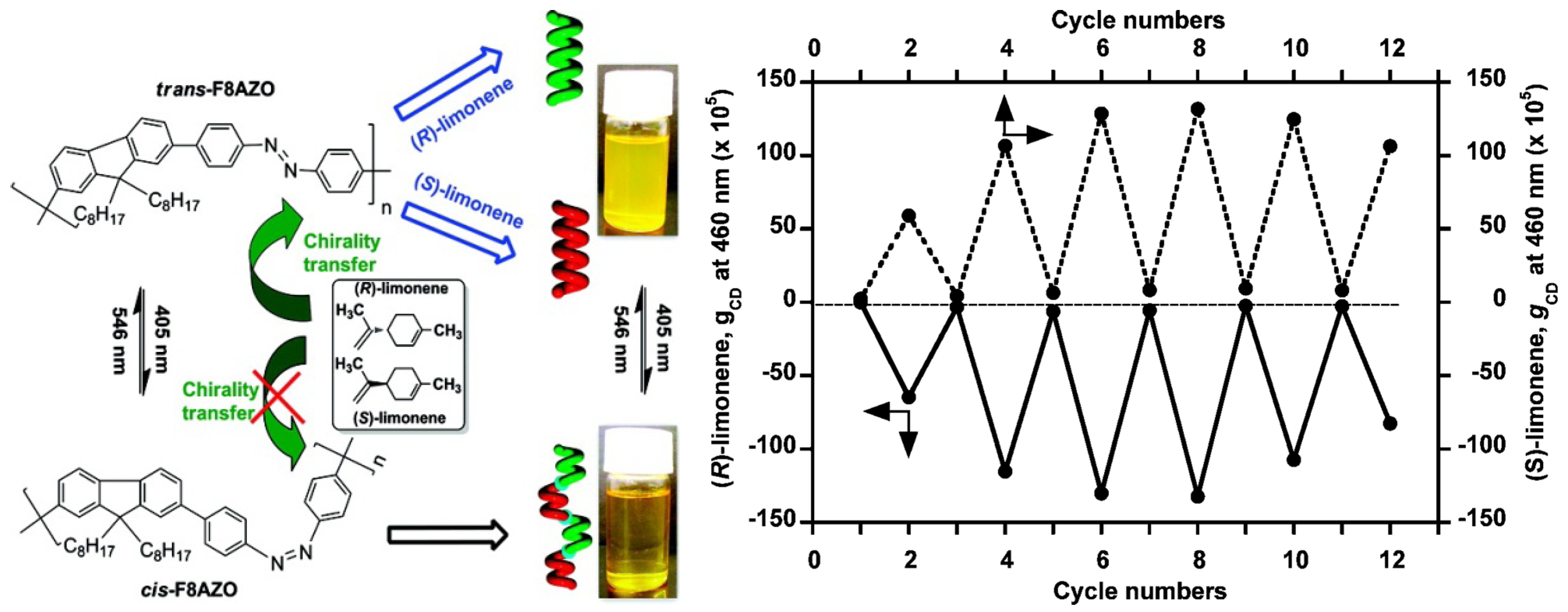
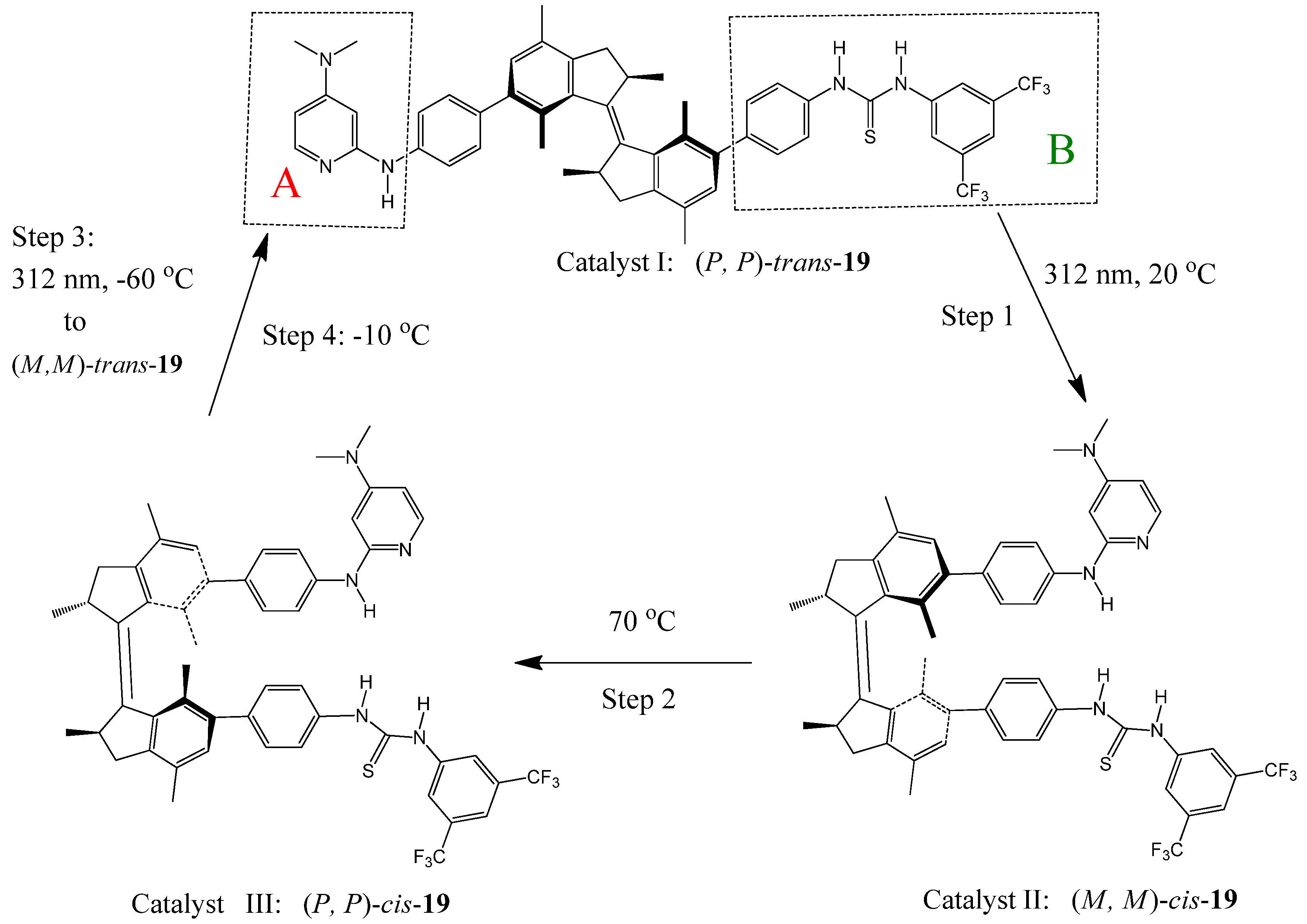
6. Photoswitchable Chiroptical Switches


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
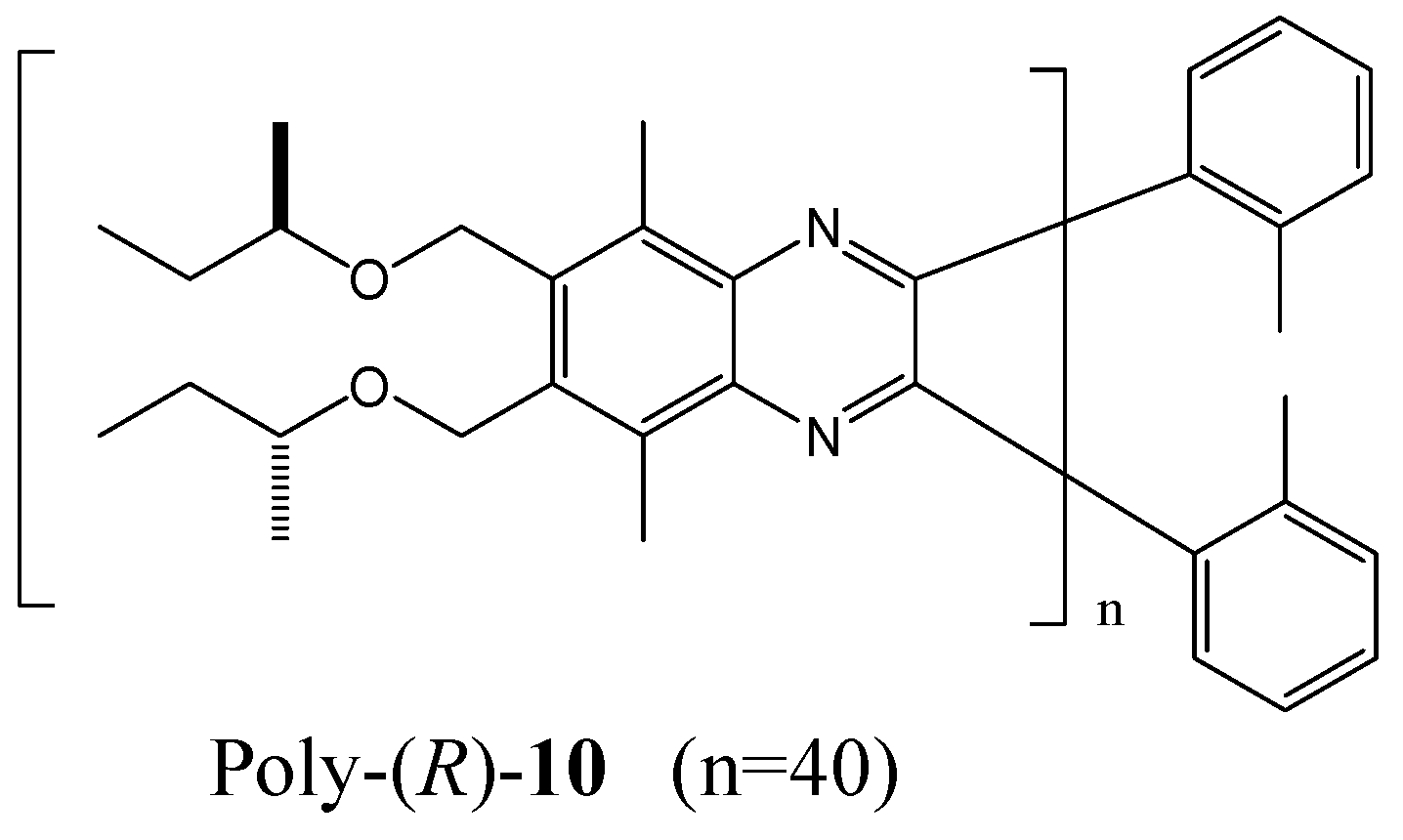{kind=link}
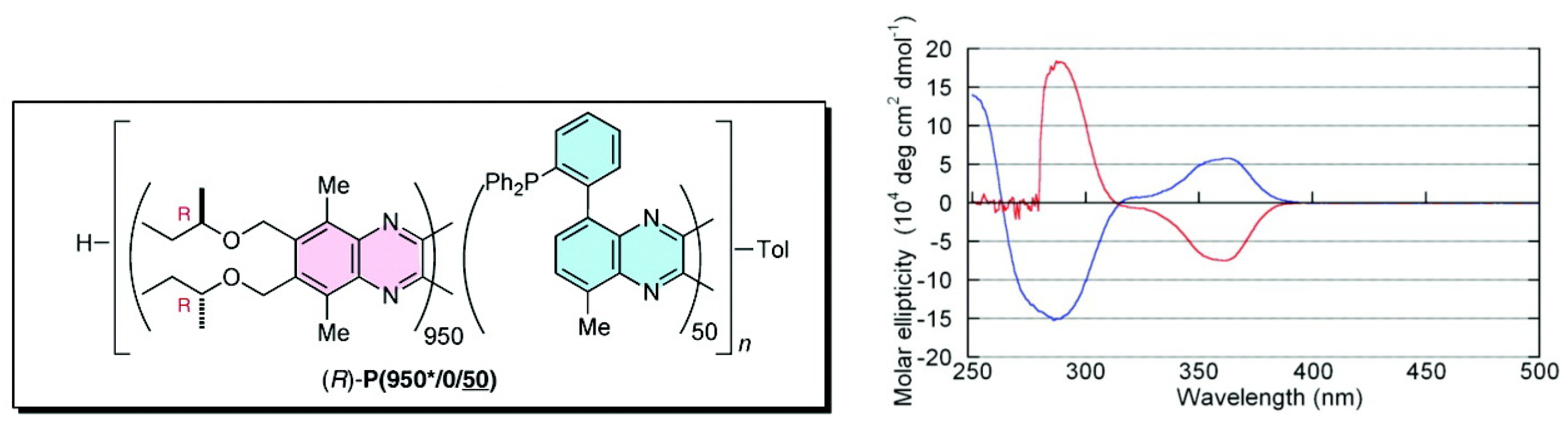{kind=link}
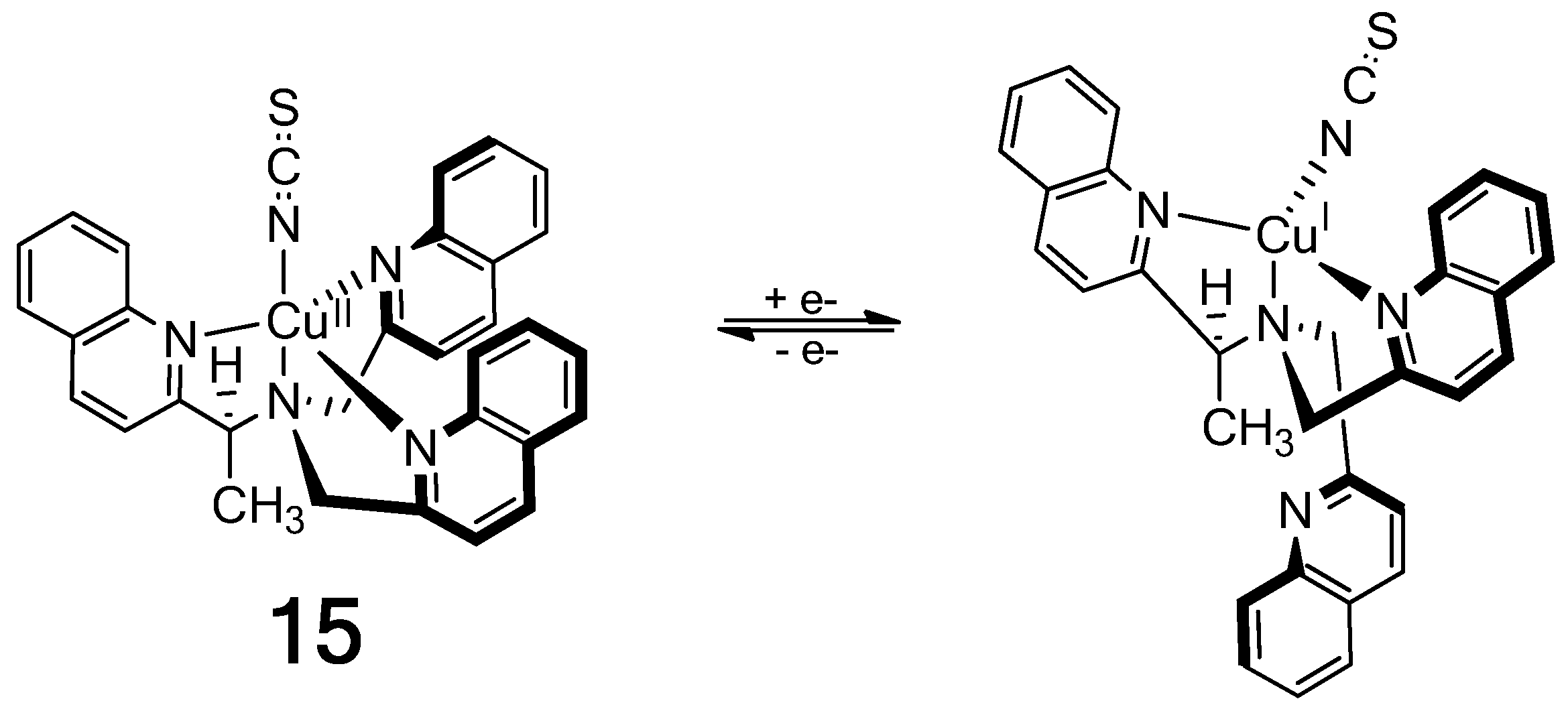{kind=link}
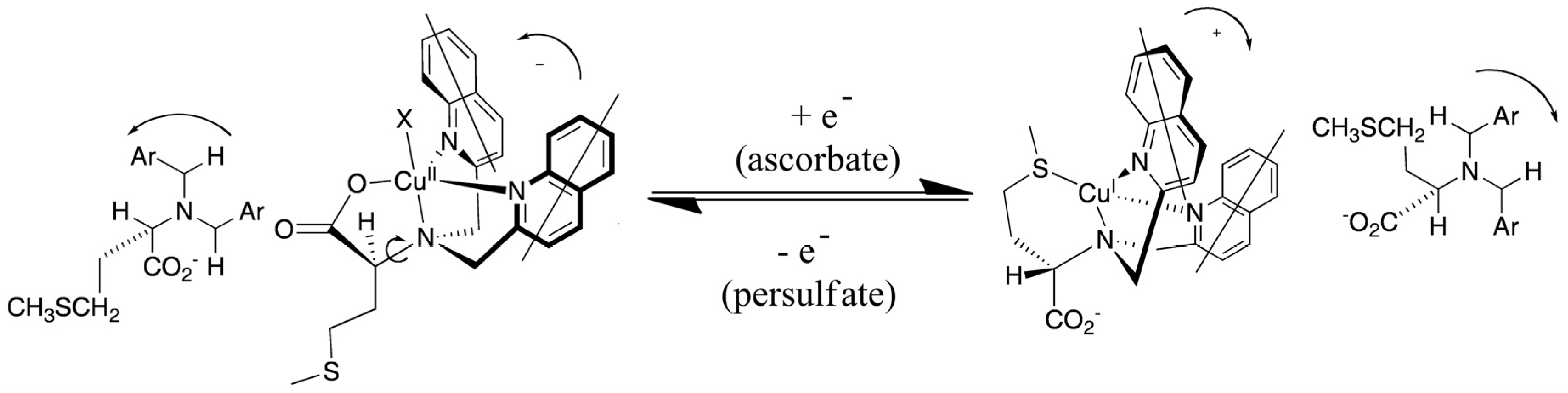{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
7. Conclusions
Acknowledgments
References
- Castagnetto, J.M.; Xu, X.; Berova, N.; Canary, J.W. Absolute configurational assignment of self-organizing asymmetric tripodal ligand-metal complexes. Chirality 1997, 9, 616–622. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; dos Santos, O.; Canary, J.W. Conformational control of propeller-like chirality in Zn(II) complexes: Tightly balanced steric bias. Tetrahedron 1999, 55, 12069–12078. [Google Scholar] [CrossRef]
- Stoll, R.S.; Hecht, S. Artificial light-gated catalyst systems. Angew. Chem. Int. Ed. 2010, 49, 5054–5075. [Google Scholar] [CrossRef]
- Bartók, M. Unexpected inversions in asymmetric reactions: Reactions with chiral metal complexes, chiral organocatalysts, and heterogeneous chiral catalysts. Chem. Rev. 2009, 110, 1663–1705. [Google Scholar] [CrossRef]
- Feringa, B.L. Molecular Switches; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Feringa, B.L. The art of building small: From molecular switches to molecular motors. J. Org. Chem. 2007, 72, 6635–6652. [Google Scholar] [CrossRef]
- Saha, S.; Stoddart, J.F. Photo-driven molecular devices. Chem. Soc. Rev. 2007, 36, 77–92. [Google Scholar] [CrossRef]
- Nakano, T.; Okamoto, Y. Synthetic helical polymers: Conformation and function. Chem. Rev. 2001, 101, 4013–4038. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Iida, H.; Furusho, Y.; Nagai, K. Helical polymers: Synthesis, structures, and functions. Chem. Rev. 2009, 109, 6102–6211. [Google Scholar] [CrossRef]
- Canary, J.W. Redox-triggered chiroptical molecular switches. Chem. Soc. Rev. 2009, 38, 747–756. [Google Scholar] [CrossRef]
- Canary, J.W.; Mortezaei, S.; Liang, J. Transition metal-based chiroptical switches for nanoscale electronics and sensors. Coord. Chem. Rev. 2010, 254, 2249–2266. [Google Scholar] [CrossRef]
- Canary, J.W.; Mortezaei, S.; Liang, J. Redox-reconfigurable tripodal coordination complexes: Stereodynamic molecular switches. Chem. Commun. 2010, 46, 5850–5860. [Google Scholar] [CrossRef]
- Canary, J.W.; Dai, Z. Dynamic stereochemistry and chiroptical spectroscopy of metallo-organic compounds. In Comprehensive Chiroptical Spectroscopy; Berova, N., Woody, R.W., Polavarapu, P., Nakanishi, K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 2. [Google Scholar]
- Canary, J.W.; Dai, Z.; Mortezaei, S. Spectroscopic analysis: Chiroptical sensors. In Comprehensive Chirality; Tranter, G., Ed.; Elsevier: Oxford, UK, 2012. [Google Scholar]
- Miyake, H.; Tsukube, H. Helix architecture and helicity switching via dynamic metal coordination chemistry. Supramol. Chem. 2005, 17, 53–59. [Google Scholar] [CrossRef]
- Nakanishi, K.; Berova, N.; Woody, R. Circular Dichroism: Principles and Applications; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Yoneda, H.; Nakashima, Y.; Sakaguchi, U. Laser photochemistry. I. Partial photoresolution of tris(acetylacetonato)chromium(III) and related complexes. Chem. Lett. 1973, 1343–1346. [Google Scholar]
- Urry, D.W.; Eyrin, H. Optical rotatory dispersion studies of L-histidine chelation. J. Am. Chem. Soc. 1964, 86, 4574–4580. [Google Scholar] [CrossRef]
- Taniguchi, T.; Monde, K.; Nishimura, S.-I.; Yoshida, J.; Sato, H.; Yamagishi, A. Rewinding of helical systems by use of the Cr(III) complex as a photoresponsive chiral dopant. Mol. Cryst. Liq. Cryst. 2006, 460, 107–116. [Google Scholar] [CrossRef]
- Montgomery, C.P.; New, E.J.; Parker, D.; Peacock, R.D. Enantioselective regulation of a metal complex in reversible binding to serum albumin: Dynamic helicity inversion signalled by circularly polarised luminescence. Chem. Commun. 2008, 4261–4263. [Google Scholar]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar] [CrossRef]
- Canary, J.W.; Holmes, A.E.; Liu, J. Prospects for circular dichroism detection of nonracemic extraterrestrial organic molecules. Enantiomer 2001, 6, 181–188. [Google Scholar]
- Joyce, L.A.; Maynor, M.S.; Dragna, J.M.; da Cruz, G.M.; Lynch, V.M.; Canary, J.W.; Anslyn, E.V. A simple method for the determination of enantiomeric excess and identity of chiral carboxylic acids. J. Am. Chem. Soc. 2011, 133, 13746–13752. [Google Scholar]
- Nieto, S.; Lynch, V.M.; Anslyn, E.V.; Kim, H.; Chin, J. Rapid enantiomeric excess and concentration determination using simple racemic metal complexes. Org. Lett. 2008, 10, 5167–5170. [Google Scholar] [CrossRef]
- Nieto, S.; Lynch, V.M.; Anslyn, E.V.; Kim, H.; Chin, J. High-throughput screening of identity, enantiomeric excess, and concentration using MLCT transitions in CD spectroscopy. J. Am. Chem. Soc. 2008, 130, 9232–9233. [Google Scholar] [CrossRef]
- Zhang, J. Redox Triggered Molecular Mechanics. Ph.D. Dissertation, New York University, New York, NY, USA, 2005. [Google Scholar]
- Dai, Z.; Xu, X.; Canary, J.W. Rigidified tripodal chiral ligands in the asymmetric recognition of amino compounds. Chirality 2005, 17, S227–S233. [Google Scholar] [CrossRef]
- Berova, N.; Pescitelli, G.; Petrovic, A.G.; Proni, G. Probing molecular chirality by CD-sensitive dimeric metalloporphyrin hosts. Chem. Commun. 2009, 5958–5980. [Google Scholar]
- Li, X.; Borhan, B. Prompt determination of absolute configuration for epoxy alcohols via exciton chirality protocol. J. Am. Chem. Soc. 2008, 130, 16126–16127. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Lintuluoto, J.M.; Inoue, Y. Supramolecular chirogenesis in bis(zincporphyrin): An absolute configuration probe highly sensitive to guest structure. Org. Lett. 2000, 2, 1565–1568. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Origin, control, and application of supramolecular chirogenesis in bisporphyrin-based systems. Acc. Chem. Res. 2004, 37, 449–459. [Google Scholar] [CrossRef]
- Anderson, M.; Wilcox, K.; Guericke, M.; Chu, H.; Wilson, M.; Wilson, E.; Lucas, K.; Holmes, A.E. Enantio-discrimination of methamphetamine by circular dichroism using porphyrin tweezers. Chirality 2010, 22, 398–402. [Google Scholar]
- Ishii, H.; Chen, Y.; Miller, R.A.; Karady, S.; Nakanishi, K.; Berova, N. Chiral recognition of cyclic alpha-hydroxyketones by CD-sensitive zinc tetraphenylporphyrin tweezer. Chirality 2005, 17, 305–315. [Google Scholar] [CrossRef]
- Guo, Y.M.; Oike, H.; Aida, T. Chiroptical transcription of helical information through supramolecular harmonization with dynamic helices. J. Am. Chem. Soc. 2004, 126, 716–717. [Google Scholar]
- Li, W.S.; Jiang, D.L.; Suna, Y.; Aida, T. Cooperativity in chiroptical sensing with dendritic zinc porphyrins. J. Am. Chem. Soc. 2005, 127, 7700–7702. [Google Scholar] [CrossRef]
- Aimi, J.; Oya, K.; Tsuda, A.; Aida, T. Chiroptical sensing of asymmetric hydrocarbons using a homochiral supramolecular box from a bismetalloporphyrin rotamer. Angew. Chem. Int. Ed. 2007, 46, 2031–2035. [Google Scholar]
- Choi, J.K.; Sargsyan, G.; Shabbir-Hussain, M.; Holmes, A.E.; Balaz, M. Chiroptical detection of condensed nickel(II)-Z-DNA in the presence of the B-DNA via porphyrin exciton coupled circular dichroism. J. Phys. Chem. B 2011, 115, 10182–10188. [Google Scholar] [CrossRef]
- Iwaniuk, D.P.; Wolf, C. A stereodynamic probe providing a chiroptical response to substrate-controlled induction of an axially chiral arylacetylene framework. J. Am. Chem. Soc. 2011, 133, 2414–2417. [Google Scholar] [CrossRef]
- Katoono, R.; Kawai, H.; Fujiwara, K.; Suzuki, T. Dynamic Molecular propeller: Supramolecular chirality sensing by enhanced chiroptical response through the transmission of point chirality to mobile helicity. J. Am. Chem. Soc. 2009, 131, 16896–16904. [Google Scholar]
- Fenniri, H.; Deng, B.-L.; Ribbe, A.E. Helical rosette nanotubes with tunable chiroptical properties. J. Am. Chem. Soc. 2002, 124, 11064–11072. [Google Scholar] [CrossRef]
- Muller, G. Luminescent chiral lanthanide(iii) complexes as potential molecular probes. Dalton Trans. 2009, 9692–9707. [Google Scholar] [CrossRef]
- Dydio, P.; Rubay, C.; Gadzikwa, T.; Lutz, M.; Reek, J.N.H. Cofactor”-controlled enantioselective catalysis. J. Am. Chem. Soc. 2011, 133, 17176–17179. [Google Scholar] [CrossRef]
- Akine, S.; Hotate, S.; Nabeshima, T. A Molecular leverage for helicity control and helix inversion. J. Am. Chem. Soc. 2011, 133, 13868–13871. [Google Scholar] [CrossRef]
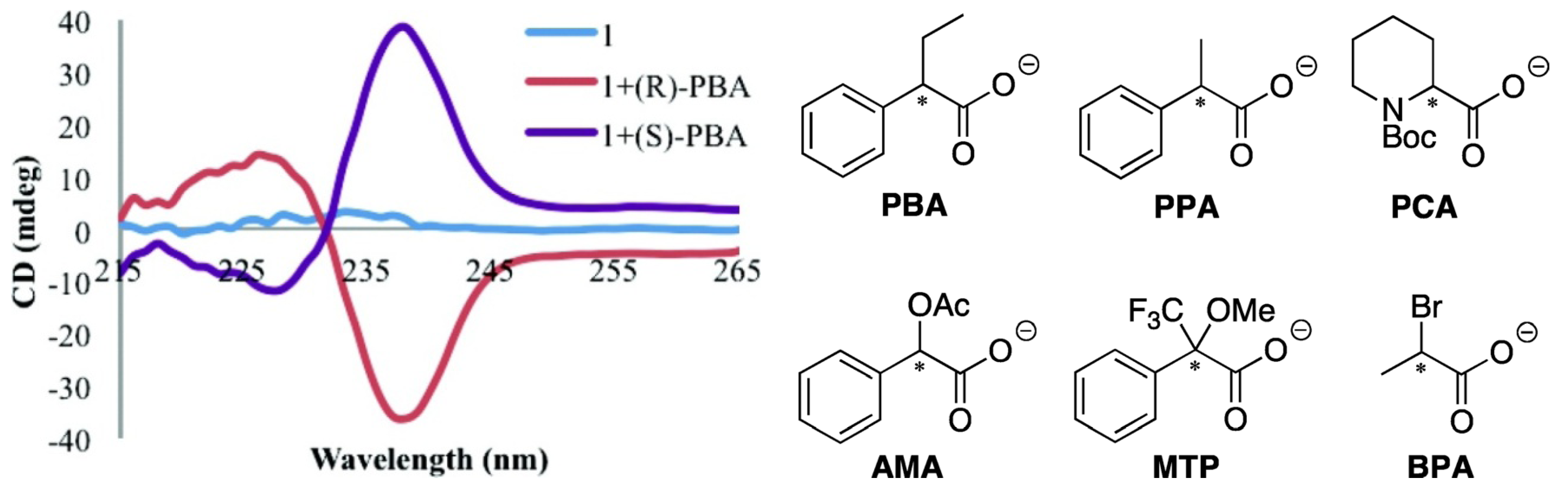
- Carney, P.; Lopez, S.; Mickley, A.; Grinberg, K.; Zhang, W.; Dai, Z. Multimode selective detection of mercury by chiroptical fluorescent sensors based on methionine/cysteine. Chirality 2011, 23, 916–920. [Google Scholar] [CrossRef]
- Sanji, T.; Sato, Y.; Kato, N.; Tanaka, M. Solvophobically and metal-coordination-driven chirality induction in poly(m-phenylenedisilanylene). Macromolecules 2007, 40, 4747–4749. [Google Scholar] [CrossRef]
- Otsuka, I.; Sakai, R.; Satoh, T.; Kakuchi, R.; Kaga, H.; Kakuchi, T. Metal-cation-induced chiroptical switching for poly(phenylacetylene) bearing a macromolecular ionophore as a graft chain. J. Polymer Sci. A: Polymer Chem. 2005, 43, 5855–5863. [Google Scholar] [CrossRef]
- Pagel, K.; Vagt, T.; Kohajda, T.; Koksch, B. From alpha-helix to beta-sheet-A reversible metal ion induced peptide secondary structure switch. Org. Biomol. Chem. 2005, 3, 2500–2502. [Google Scholar] [CrossRef]
- Shirotani, D.; Suzuki, T.; Kaizaki, S. Novel configurational structures of sodium tetrakis(3-heptafluorobutylryl-(+)-camphorato) Ln(III) complexes with a trapped Na+ by Na+·FC interactions in the solid state and in solution. Inorg. Chem. 2006, 45, 6111–6113. [Google Scholar] [CrossRef]
- Gregolinski, J.; Starynowicz, P.; Hua, K.T.; Lunkley, J.L.; Muller, G.; Lisowski, J. Helical lanthanide(III) complexes with chiral nonaaza macrocycle. J. Am. Chem. Soc. 2008, 130, 17761–17773. [Google Scholar]
- Barcena, H.S.; Liu, B.; Mirkin, M.V.; Canary, J.W. An electrochiroptical molecular switch: Mechanistic and kinetic studies. Inorg. Chem. 2005, 44, 7652–7660. [Google Scholar] [CrossRef]
- Das, D.; Dai, Z.; Holmes, A.E.; Canary, J.W. Exploring the scope of redox-triggered chiroptical switches: Syntheses, X-ray structures and circular dichroism of cobalt and nickel complexes of N,N-bis(arylmethyl)methionine derivatives. Chirality 2008, 20, 585–591. [Google Scholar] [CrossRef]
- Zahn, S.; Das, D.; Canary, J.W. Redox-induced ligand reorganization and helicity inversion in copper complexes of N,N-dialkylmethionine derivatives. Inorg. Chem. 2006, 45, 6056–6063. [Google Scholar] [CrossRef]
- Holmes, A.E.; Zahn, S.; Canary, J.W. Synthesis and circular dichroism studies of N,N-bis(2-quinolylmethyl)amino acid Cu(II) complexes: Determination of absolute configuration and enantiomeric excess by the exciton coupling method. Chirality 2002, 14, 471–477. [Google Scholar] [CrossRef]
- Catagnetto, J.M.; Canary, J.W. A chiroptically enhanced fluorescent chemosensor. Chem. Commun. 1998, 203–204. [Google Scholar]
- Lee, J.; Dai, Z. Unpublished work, Pace University: New York, NY, USA, 2012.
- Nehira, T.; Parish, C.A.; Jockusch, S.; Turro, N.J.; Nakanishi, K.; Berova, N. Fluoresence-detected exciton-coupled circular dichroism: Scope and limitation in structural studies of organic molecules. J. Am. Chem. Soc. 1999, 121, 8681–8691. [Google Scholar]
- Dai, Z.; Proni, G.; Mancheno, D.; Karimi, S.; Berova, N.; Canary, J.W. Detection of zinc ion by differential circularly polarized fluorescence excitation. J. Am. Chem. Soc. 2004, 126, 11760–11761. [Google Scholar]
- Hassey, R.; Swain, E.J.; Hammer, N.I.; Venkataraman, D.; Barnes, M.D. Probing the chiroptical response of a single molecule. Science 2006, 314, 1437–1439. [Google Scholar] [CrossRef]
- Yamada, T.; Shinoda, S.; Sugimoto, H.; Uenishi, J.; Tsukube, H. Luminescent lanthanide complexes with stereo-controlled tris(2-pyridylmethyl)amine ligands: Chirality effects on lanthalide complexation and luminescent properties. Inorg. Chem. 2003, 42, 7932–7937. [Google Scholar] [CrossRef]
- Masaki, M.E.; Paul, D.; Nakamura, R.; Kataoka, Y.; Shinoda, S.; Tsukube, H. Chiral tripode approach toward multiple anion sensing with lanthanide complexes. Tetrahedron 2009, 65, 2525–2530. [Google Scholar] [CrossRef]
- Miyake, H.; Yoshida, K.; Sugimoto, H.; Tsukube, H. Dynamic helicity inversion by achiral anion stimulus in synthetic labile cobalt(II) complex. J. Am. Chem. Soc. 2004, 126, 6524–6525. [Google Scholar] [CrossRef]
- Meudtner, R.M.; Hecht, S. Helicity Inversion in Responsive foldamers induced by achiral halide ion guests. Angew. Chem. Int. Ed. 2008, 47, 4926–4930. [Google Scholar] [CrossRef]
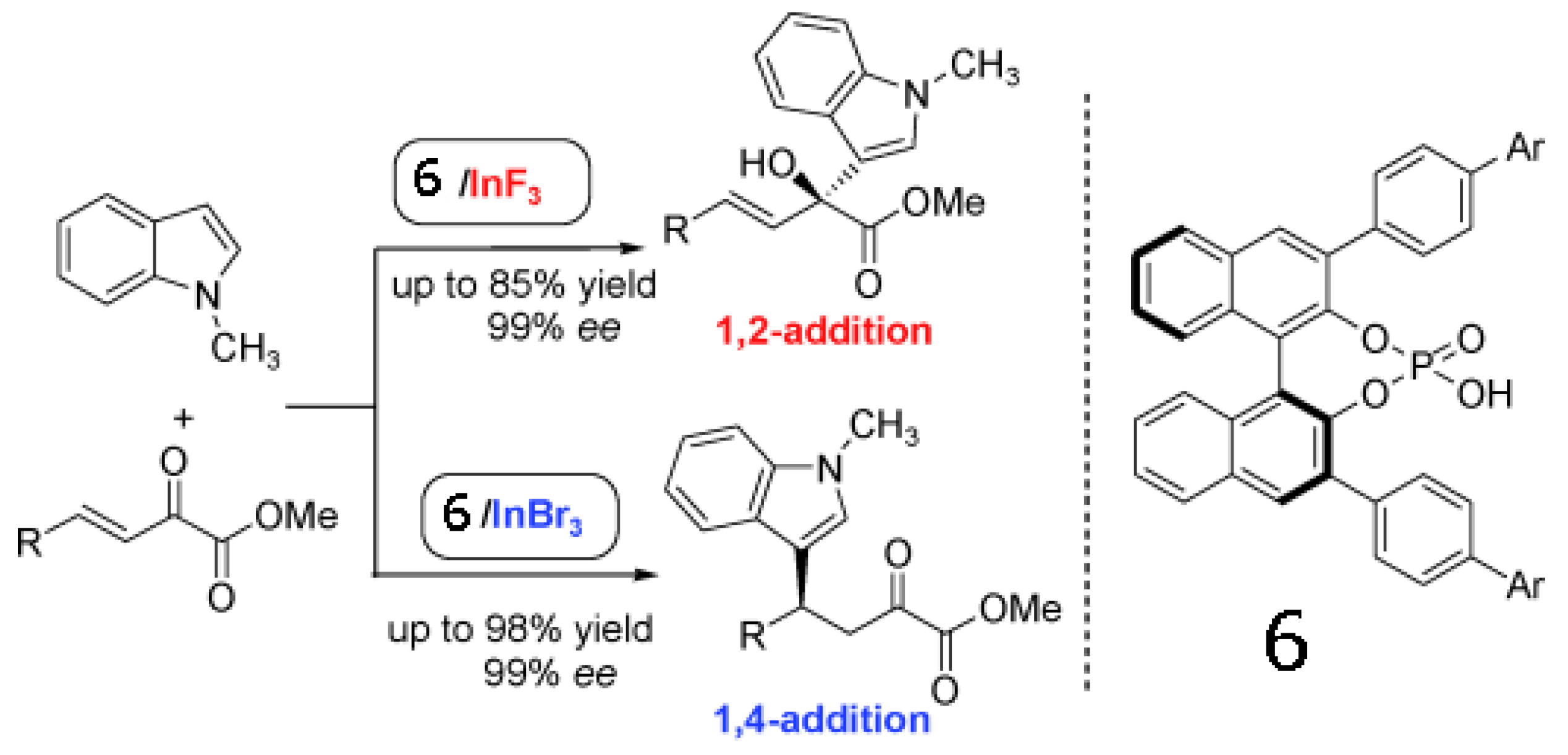
- Lv, J.; Zhang, L.; Zhou, Y.; Nie, Z.; Luo, S.; Cheng, J.-P. Asymmetric binary acid catalysis: A regioselectivity switch between enantioselective 1,2- and 1,4-addition through different counteranions of InIII. Angew. Chem. Int. Ed. 2011, 50, 6610–6614. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. The origin of solvent-controlled supramolecular chirality switching in a bis(zinc porphyrin) system. Angew. Chem. Int. Ed. 2003, 42, 5310–5314. [Google Scholar] [CrossRef]
- Hutin, M.; Nitschke, J. Solvent-tunable inversion of chirality transfer from carbon to copper. Chem. Commun. 2006, 16, 1724–1726. [Google Scholar] [CrossRef]
- Gregolinski, J.; Slepokura, K.; Lisowski, J. Lanthanide complexes of the chiral hexaaza macrocycle and its meso-type isomer: Solvent-controlled helicity inversion. Inorg. Chem. 2007, 46, 7923–7934. [Google Scholar] [CrossRef]
- Lama, M.; Mamula, O.; Kottas, G.S.; De Cola, L.; Stoeckli-Evans, H.; Shova, S. Enantiopure, supramolecular helices containing 3D tetranuclear lanthanide(III) arrays: Synthesis, structure, properties and solvent-driven trinuclear/tetranuclear interconversion. Inorg. Chem. 2008, 47, 8000–8015. [Google Scholar] [CrossRef]
- Nakako, H.; Nomura, R.; Masuda, T. Helix inversion of poly(propiolic esters). Macromolecules 2001, 34, 1496–1502. [Google Scholar] [CrossRef]
- Kawagoe, Y.; Fujiki, M.; Nakano, Y. Limonene magic: Noncovalent molecular chirality transfer leading to ambidextrous circularly polarised luminescent π-conjugated polymers. New J. Chem. 2010, 34, 637–647. [Google Scholar] [CrossRef]
- Tang, H.-Z.; Boyle, P.D.; Novak, B.M. Chiroptical switching polyguanidine synthesized by helix-sense-selective polymerization using [(R)-3,3′-Dibromo-2,2′-binaphthoxy](di-tert-butoxy)titanium(IV) catalyst. J. Am. Chem. Soc. 2005, 127, 2136–2142. [Google Scholar]
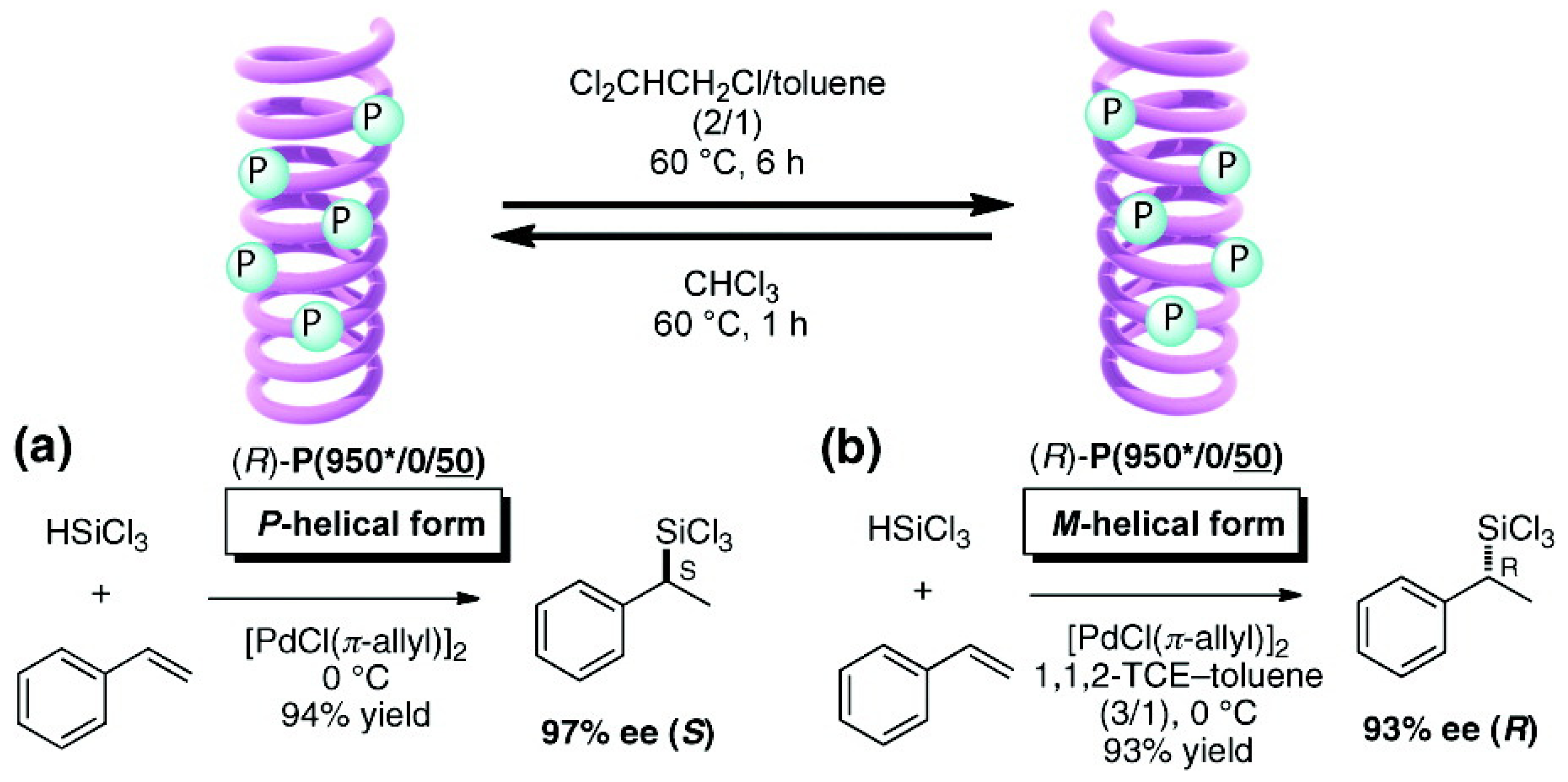
- Yamada, T.; Nagata, Y.; Suginome, M. Non-hydrogen-bonding-based, solvent-dependent helix inversion between pure P-helix and pure M-helix in poly(quinoxaline-2,3-diyl)s bearing chiral side chains. Chem. Commun. 2010, 46, 4914–4916. [Google Scholar] [CrossRef]
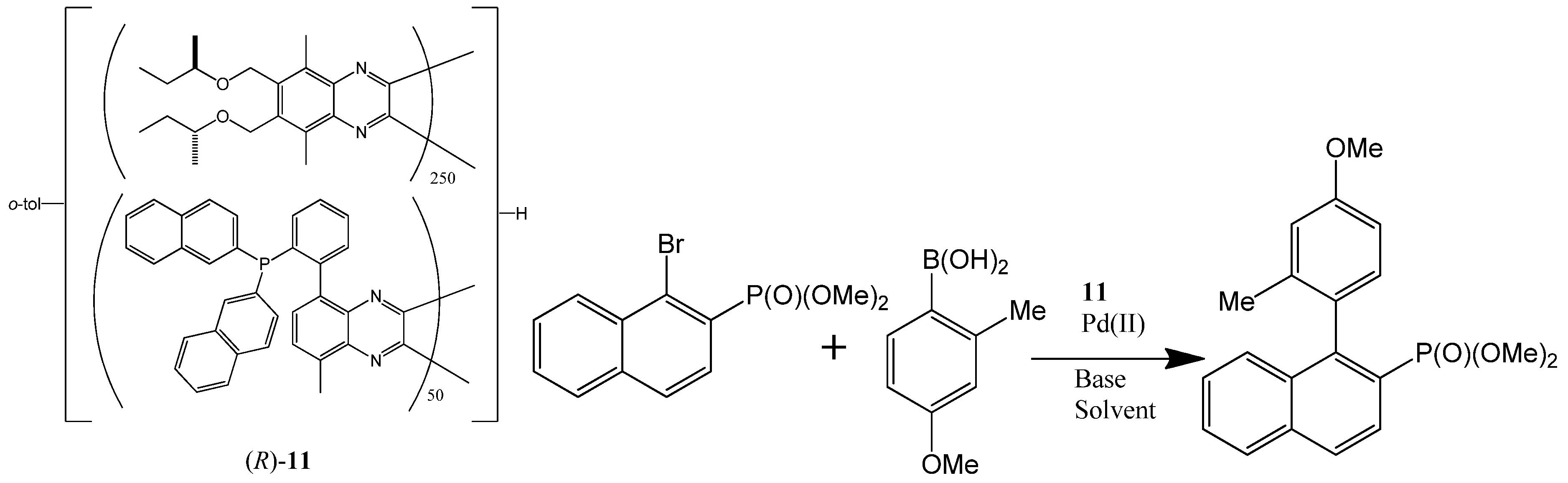
- Yamamoto, T.; Yamada, T.; Nagata, Y.; Suginome, M. High-molecular-weight polyquinoxaline-based helically chiral phosphine (PQXphos) as chirality-switchable, reusable, and highly enantioselective monodentate ligand in catalytic asymmetric hydrosilylation of styrenes. J. Am. Chem. Soc. 2010, 132, 7899–7901. [Google Scholar] [CrossRef]
- Yamamoto, T.; Akai, Y.; Nagata, Y.; Suginome, M. Highly enantioselective synthesis of axially chiral biarylphosphonates: Asymmetric Suzuki-Miyaura coupling using high-molecular-weight, helically chiral polyquinoxaline-based phosphines. Angew. Chem. Int. Ed. 2011, 50, 8844–8847. [Google Scholar] [CrossRef]
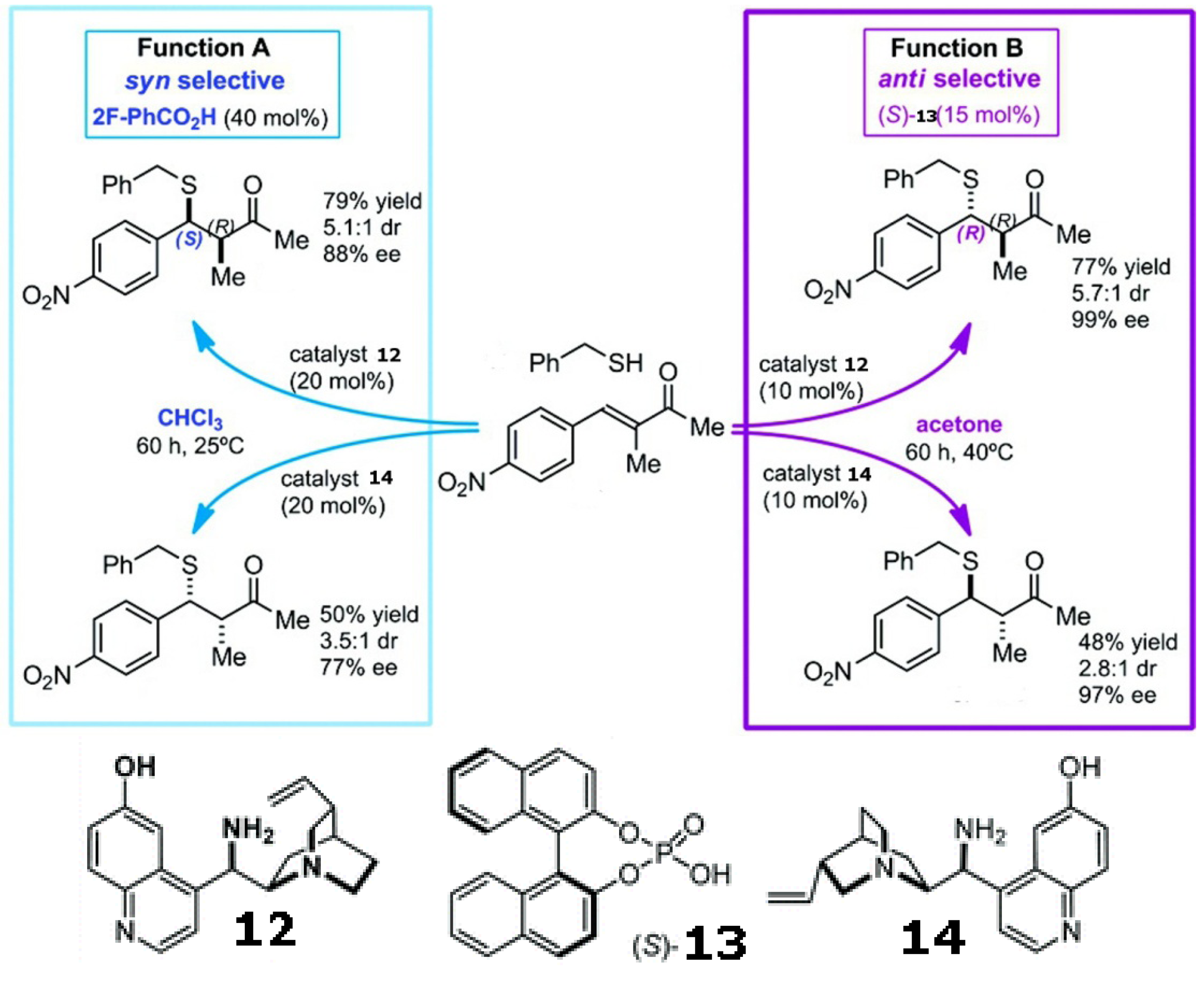
- Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. Diastereodivergent asymmetric sulfa-Michael additions of α-branched enones using a single chiral organic catalyst. J. Am. Chem. Soc. 2011, 133, 17934–17941. [Google Scholar]
- Zelikovich, L.; Libman, J.; Shanzer, A. Molecular redox switches based on chemical triggering of iron translocation in triple-stranded helical complexes. Nature 1995, 374, 790–792. [Google Scholar] [CrossRef]
- Company, A.; Guell, M.; Popa, D.; Benet-Buchholz, J.; Parella, T.; Fontrodona, X.; Llobet, A.; Miquel Sola, M.; Xavi Ribas, X.; Luis, J.M.; Costas, M. Redox-controlled molecular flipper based on a chiral Cu complex. Inorg. Chem. 2006, 45, 9643–9645. [Google Scholar]
- Li, D.; Wang, Z.Y.; Ma, D. Electrically-controlled near-infrared chiroptical switching of enantiomeric dinuclear ruthenium complexes. Chem. Commun. 2009, 1529–1531. [Google Scholar]
- Yamaguchi, S.; Katoh, T.; Shinokubo, H.; Osuka, A. Pt(II)- and Pt(IV)-bridged cofacial diporphyrins via carbon-transition metal σ-bonds. J. Am. Chem. Soc. 2008, 130, 14440–14441. [Google Scholar] [CrossRef]
- Zahn, S.; Canary, J.W. Redox-switched exciton-coupled circular dichroism: A novel strategy for binary molecular switching. Angew. Chem. Int. Ed. 1998, 37, 305–307. [Google Scholar] [CrossRef]
- Zhang, J.; Canary, J.W. Redox-triggered interconversion between piperidine chair conformations in a Cu(I/II) complex. Org. Lett. 2006, 8, 3907–3910. [Google Scholar] [CrossRef]
- Zahn, S.; Proni, G.; Spada, G.P.; Canary, J.W. Supramolecular detection of metal ion binding: Ligand conformational control of cholesteric induction in nematic liquid crystalline phases. Chem.-Eur. J. 2001, 7, 88–93. [Google Scholar] [CrossRef]
- Zahn, S.; Canary, J.W. Absolute configurations of N,N-dialkyl alpha-amino acids and beta-amino alcohols from exciton-coupled circular dichroism spectra of Cu(II) complexes. Org. Lett. 1999, 1, 861–864. [Google Scholar] [CrossRef]
- Zahn, S.; Canary, J.W. Electron-induced inversion of helical chirality in copper complexes of N,N-dialkylmethionines. Science 2000, 288, 1404–1407. [Google Scholar] [CrossRef]
- Holmes, A.E.; Das, D.; Canary, J.W. Chelation-enhanced circular dichroism of tripodal bisporphyrin ligands. J. Am. Chem. Soc. 2007, 129, 1506–1507. [Google Scholar]
- Barcena, H.S.; Holmes, A.E.; Canary, J.W. Inversion of helicity in propeller shaped molecules derived from S-methylcysteine and methioninol. Org. Lett. 2003, 709–711. [Google Scholar]
- Goto, H.; Yashima, E. Electron-induced switching of the supramolecular chirality of optically active polythiophene aggregates. J. Am. Chem. Soc. 2002, 124, 7943–7949. [Google Scholar] [CrossRef]
- Ohta, E.; Sato, H.; Ando, S.; Kosaka, A.; Fukushima, T.; Hashizume, D.; Yamasaki, M.; Hasegawa, K.; Muraoka, A.; Ushiyama, H.; et al. Redox-responsive molecular helices with highly condensed π-clouds. Nat. Chem. 2011, 3, 68–73. [Google Scholar] [CrossRef]
- Gregson, C.K.A.; Gibson, V.C.; Long, N.J.; Marshall, E.L.; Oxford, P.J.; White, A.J.P. Redox control within single-site polymerization catalysts. J. Am. Chem. Soc. 2006, 128, 7410–7411. [Google Scholar]
- Iida, H.; Mizoguchi, T.; Oh, S.-D.; Yashima, E. Redox-triggered switching of helical chirality of poly(phenylacetylene)s bearing riboflavin pendants. Polymer Chem. 2010, 1, 841–848. [Google Scholar] [CrossRef]
- Lorkovic, I.M.; Duff, R.R.; Wrighton, M.S. Use of the redox-active ligand 1,1'-bis(diphenylphosphino)cobaltocene to reversibly alter the rate of the rhodium(I)-catalyzed reduction and isomerization of ketones and alkenes. J. Am. Chem. Soc. 1995, 117, 3617–3618. [Google Scholar] [CrossRef]
- Wiggins, K.M.; Hudnall, T.W.; Shen, Q.; Kryger, M.J.; Moore, J.S.; Bielawski, C.W. Mechanical reconfiguration of stereoisomers. J. Am. Chem. Soc. 2010, 132, 3256–3257. [Google Scholar]
- Tashiro, R.; Sugiyama, H. Biomolecule-based switching devices that respond inversely to thermal stimuli. J. Am. Chem. Soc. 2005, 127, 2094–2097. [Google Scholar]
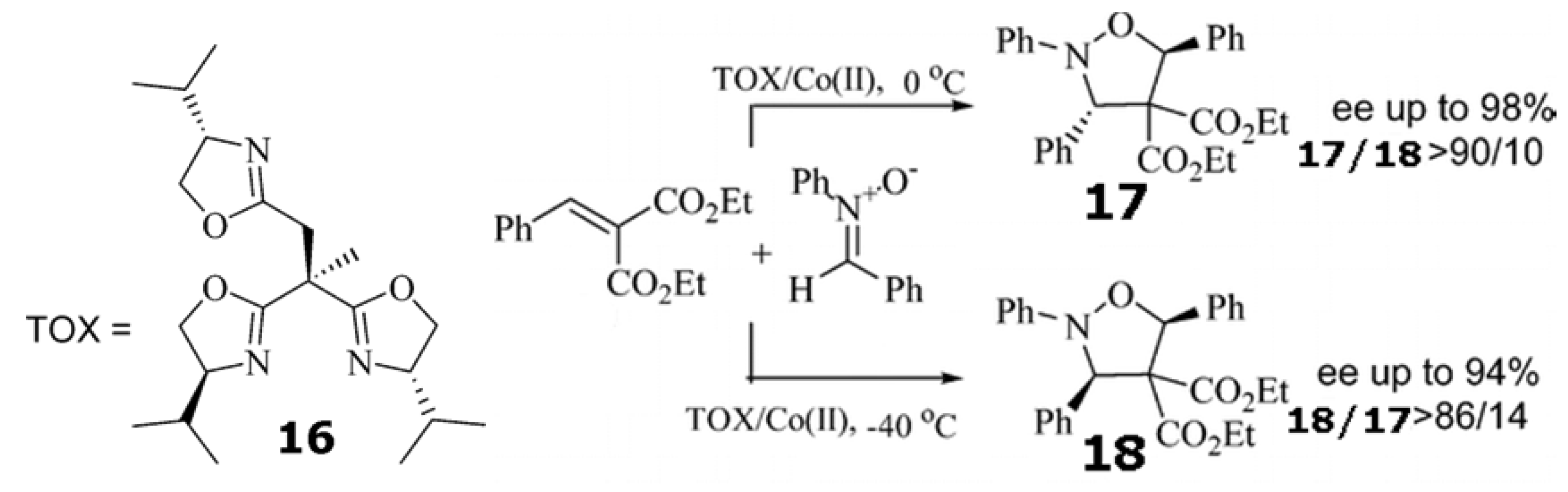
- Huang, Z.-Z.; Kang, Y.-B.; Zhou, J.; Ye, M.-C.; Tang, Y. Diastereoselectivity-switchable and highly enantioselective 1,3-dipolar cycloaddition of nitrones to alkylidene malonates. Org. Lett. 2004, 6, 1677–1679. [Google Scholar] [CrossRef]
- Ben-Asuly, A.; Tzur, E.; Diesendruck, C.E.; Sigalov, M.; Goldberg, I.; Lemcoff, N.G. A thermally switchable latent ruthenium olefin metathesis catalyst. Organometallics 2008, 27, 811–813. [Google Scholar] [CrossRef]
- Mathews, M.; Tamaoki, N. Planar chiral azobenzenophanes as chiroptic switches for photon mode reversible reflection color control in induced chiral nematic liquid crystals. J. Am. Chem. Soc. 2008, 130, 11409–11416. [Google Scholar] [CrossRef]
- Zhang, W.; Yoshida, K.; Fujiki, M.; Zhu, X. Unpolarized-light-driven amplified chiroptical modulation between chiral aggregation and achiral disaggregation of an azobenzene-alt-fluorene copolymer in limonene. Macromolecules 2011, 44, 5105–5111. [Google Scholar] [CrossRef]
- Stoll, R.S.; Peters, M.V.; Kuhn, A.; Heiles, S.; Goddard, R.; Bühl, M.; Thiele, C.M.; Hecht, S. Photoswitchable catalysts: Correlating structure and conformational dynamics with reactivity by a combined experimental and computational approach. J. Am. Chem. Soc. 2008, 131, 357–367. [Google Scholar]
- Wang, J.; Feringa, B.L. Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor. Science 2011, 331, 1429–1432. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dai, Z.; Lee, J.; Zhang, W. Chiroptical Switches: Applications in Sensing and Catalysis. Molecules 2012, 17, 1247-1277. https://doi.org/10.3390/molecules17021247
Dai Z, Lee J, Zhang W. Chiroptical Switches: Applications in Sensing and Catalysis. Molecules. 2012; 17(2):1247-1277. https://doi.org/10.3390/molecules17021247
Chicago/Turabian StyleDai, Zhaohua, Jennifer Lee, and Wenyao Zhang. 2012. "Chiroptical Switches: Applications in Sensing and Catalysis" Molecules 17, no. 2: 1247-1277. https://doi.org/10.3390/molecules17021247
APA StyleDai, Z., Lee, J., & Zhang, W. (2012). Chiroptical Switches: Applications in Sensing and Catalysis. Molecules, 17(2), 1247-1277. https://doi.org/10.3390/molecules17021247