Phosphane-Based Cyclodextrins as Mass Transfer Agents and Ligands for Aqueous Organometallic Catalysis


Abstract
:1. Introduction

2. Results and Discussion
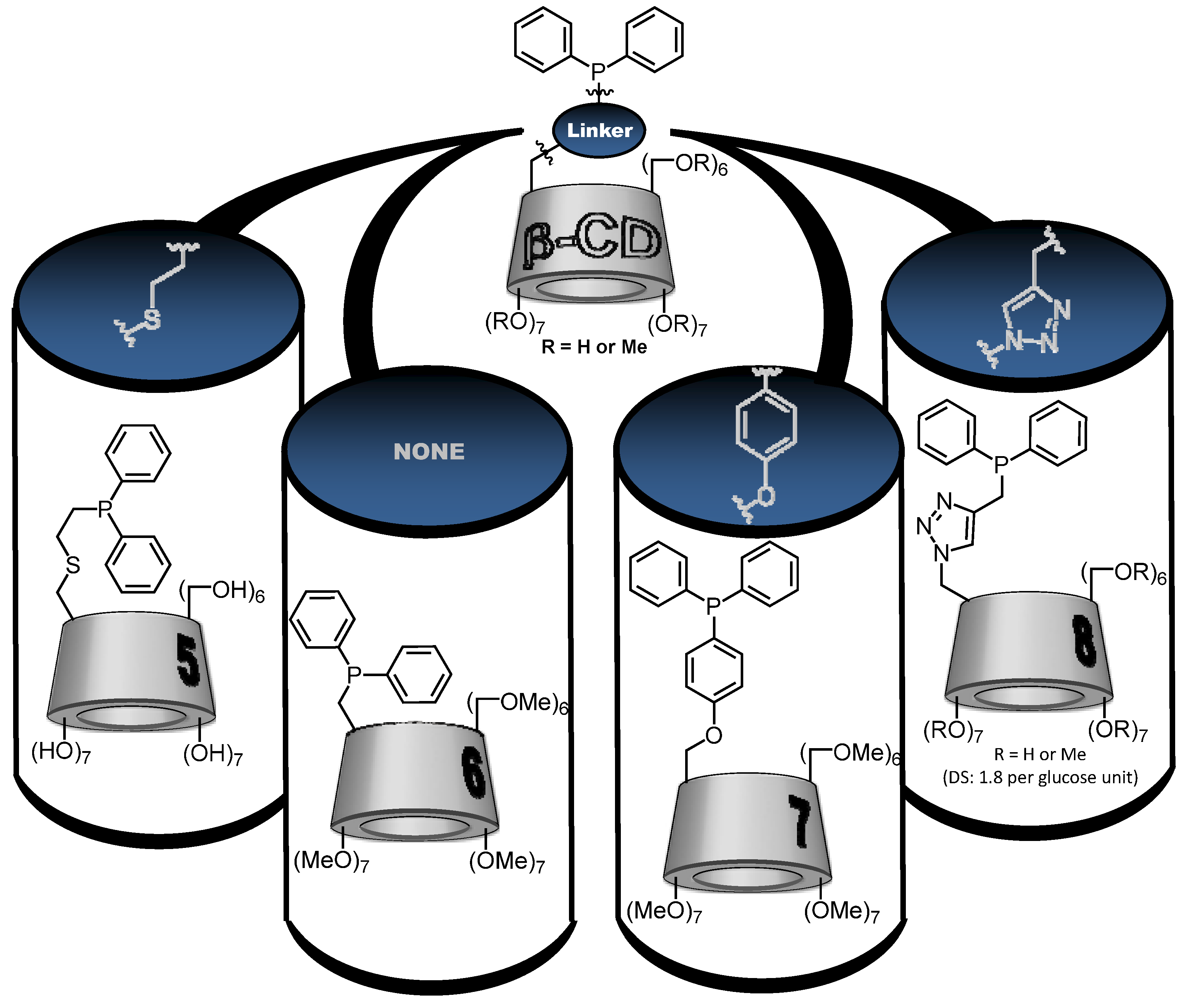
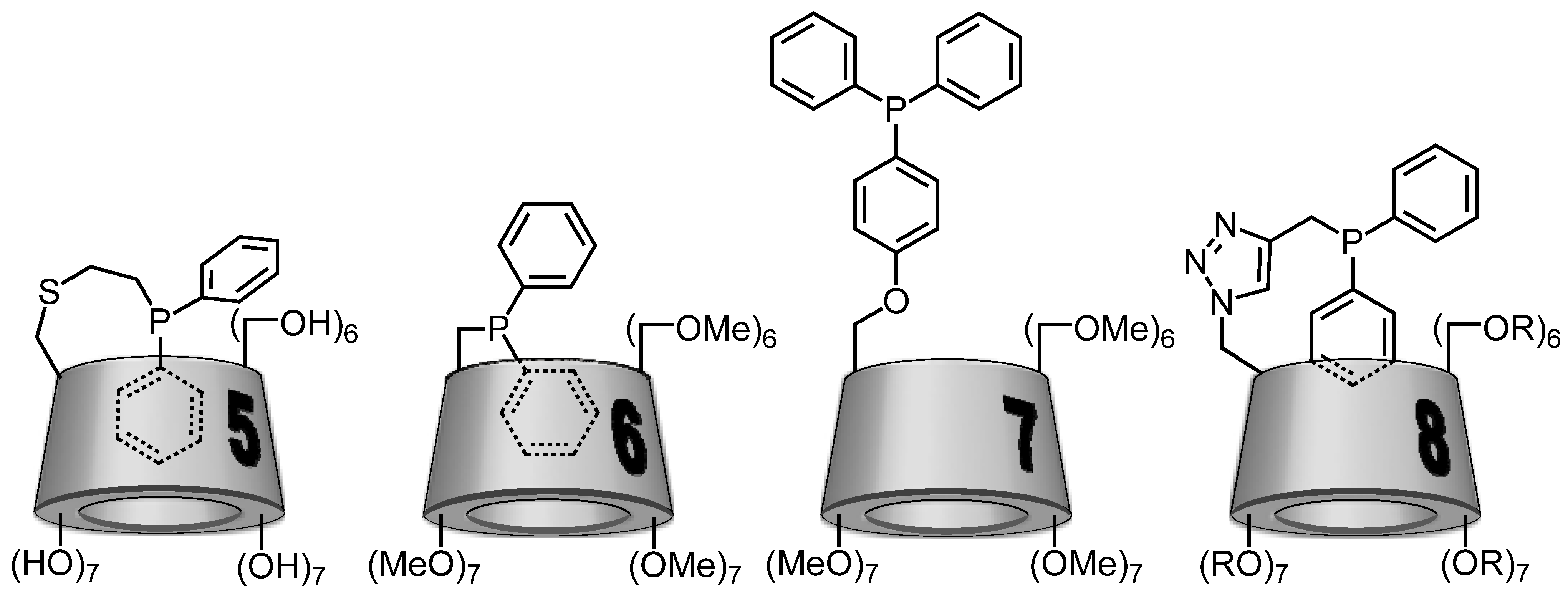
2.1. Cyclodextrin-Diphosphanes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
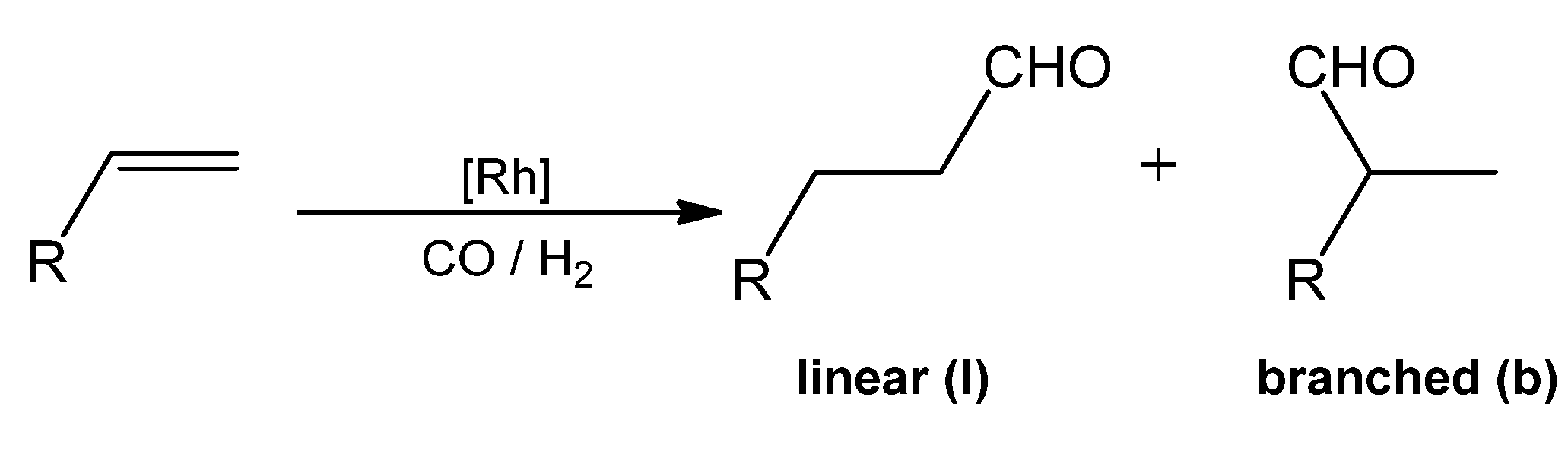
 | |||
| Entry | Ligand | Solvent | A/B |
| 1 | PhN(CH2PPh2)2 | DMF | 50/50 |
| 2 | 1 | DMF | 68/32 |
| 3 | 2a | DMF | 74/26 |
| 4 | 2b | DMF | 71/29 |
| 5 | 2c | DMF | 66/34 |
| 6 | 1 | DMF/H2O (30/70) | 82/18 |
| 7 | 2a | DMF/H2O (30/70) | 81/19 |
| 8 | 3 | DMF/H2O (30/70) | 87/13 |
| 9 * | 3 | DMF/H2O (30/70) | 57/43 |


2.2. Cyclodextrin-Monophosphanes



3. Conclusions
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Cornils, B.; Herrmann, W.A. Aqueous-Phase Organometallic Catalysis; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2004; p. 1. [Google Scholar]
- Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. Cyclodextrins and their applications in aqueous-phase metal-catalyzed reactions. C.R. Chim. 2011, 14, 149–166. [Google Scholar] [CrossRef]
- Shaughnessy, K.H. Hydrophilic ligands and their application in aqueous-phase metal-catalyzed reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar]
- Gramage-Doria, R.; Rodriguez-Lucena, D.; Armspach, D.; Egloff, C.; Jouffroy, M.; Matt, D.; Toupet, L. A cavity-shaped diphosphane displaying oschelating behaviour. Angew. Chem. Int. Ed. 2011, 50, 1554–1559. [Google Scholar]
- Zaborova, E.; Deschamp, J.; Guieu, S.; Blériot, Y.; Poli, G.; Ménand, M.; Madec, D.; Prestat, G.; Sollogoub, M. Cavitand supported tetraphosphine: Cyclodextrin offers a useful platform for Suzuki-Miyaura cross-coupling. Chem. Commun. 2011, 47, 9206–9208. [Google Scholar]
- Gramage-Doria, R.; Rodriguez-Lucena, D.; Armspach, D.; Egloff, C.; Jouffroy, M.; Matt, D.; Toupet, L. Regioselective double capping of cyclodextrin scaffolds. Chem. Eur. J. 2011, 17, 3911–3921. [Google Scholar]
- Guieu, S.; Zaborova, E.; Blériot, Y.; Poli, G.; Jutand, A.; Madec, D.; Prestat, G.; Sollogoub, M. Can hetero-polysubstituted cyclodextrins be considered as inherently chiral concave molecules? Angew. Chem. Int. Ed. 2010, 49, 2314–2318. [Google Scholar]
- Armspach, D.; Matt, D.; Toupet, L. Self-mediated, stereoselective oxidation of thia-capped cyclodextrins. Angew. Chem. Int. Ed. 2009, 48, 4555–4558. [Google Scholar]
- Poorters, L.; Armspach, D.; Matt, D.; Toupet, L.; Jones, P.G. A metallocavitand functioning as a container for anions: formation of noncovalent linear assemblies mediated by a cyclodextrin-entrapped NO3−ion. Angew. Chem. Int. Ed. 2007, 46, 2663–2665. [Google Scholar] [CrossRef]
- Poorters, L.; Armspach, D.; Matt, D.; Toupet, L.; Choua, S.; Turek, P. Synthesis and properties of TRANSDIP, A rigid chelator built upon a cyclodextrin cavity: Is TRANSDIP an authentic trans-spanning ligand? Chem. Eur. J. 2007, 13, 9448–9461. [Google Scholar] [CrossRef]
- Poorters, L.; Armspach, D.; Matt, D.; Toupet, L. α-TEPHOS: A cyclodextrin-derived tetraphosphine for multiple metal binding. Dalton Trans. 2007, 7, 3195–3202. [Google Scholar]
- Hapiot, F.; Tilloy, S.; Monflier, E. Cyclodextrins as Supramolecular Hosts for Organometallic Complexes. Chem. Rev. 2006, 106, 767–781. [Google Scholar]
- Engeldinger, E.; Poorters, L.; Armspach, D.; Matt, D.; Toupet, L. Diastereospecific synthesis of phosphinidene-capped cyclodextrins leading to “introverted” ligands. Chem. Commun. 2004, 21, 634–635. [Google Scholar]
- Engeldinger, E.; Armspach, D.; Matt, D.; Jones, P.G. Cyclodextrin phosphanes as first and second coordination Sphere Cavitands. Chem. Eur. J. 2003, 9, 3091–3105. [Google Scholar] [CrossRef]
- Poorters, L.; Armspach, D.; Matt, D. Selective tetrafunctionalisation of alpha-cyclodextrin using the supertrityl protecting group. Synthesis of the first C2-symmetric tetraphosphine based on a cavitand (alpha-TEPHOS). Eur. J. Org. Chem. 2003, 2003, 1377–1381. [Google Scholar]
- Engeldinger, E.; Armspach, D.; Matt, D. Capped cyclodextrins. Chem. Rev. 2003, 103, 4147–4173. [Google Scholar] [CrossRef]
- Engeldinger, E.; Armspach, D.; Matt, D.; Toupet, L.; Wesolek, M. . Synthesis of large chelate rings with diphosphites built on a cyclodextrin scaffold. Unexpected formation of 1,2-phenylene-capped α-cyclodextrins. C.R. Chim. 2002, 5, 359–372. [Google Scholar]
- Reetz, M.T.; Kostas, I.D.; Waldvogel, S.R. Synthesis of a gold(I) complex with a (thio)phosphine-modified β-cyclodextrin. Inorg. Chem. Commun. 2002, 5, 252–254. [Google Scholar] [CrossRef]
- Engeldinger, E.; Armspach, D.; Matt, D.; Jones, P.G.; Welter, R. A cyclodextrin diphosphane as a first and second coordination sphere cavitand: evidence for weak C-H⋅⋅⋅Cl-M hydrogen bonds within metal-capped cavities. Angew. Chem. Int. Ed. 2002, 41, 2593–2596. [Google Scholar] [CrossRef]
- Wong, Y.T.; Yang, C.; Ying, K.C.; Jia, G. Synthesis of a novel beta-cyclodextrin-functionalized diphosphine ligand and its catalytic properties for asymmetric hydrogenation. Organometallics 2002, 21, 1782–1787. [Google Scholar]
- Yang, C.; Cheung, Y.K.; Yao, J.; Wong, Y.T.; Jia, G. Palladium and platinum complexes with a beta-cyclodextrin-functionalized phosphine ligand. Organometallics 2001, 20, 424–429. [Google Scholar]
- Yang, C.; Wong, Y.T.; Li, Z.; Krepinsky, J.J.; Jia, G. Synthesis of beta-cyclodextrin-functionalized (2S,4S)-(-)-4-(diphenylphosphino)-2-(diphenylphosphinomethyl) ligands and their rhodium and platinum complexes. Organometallics 2001, 20, 5220–5224. [Google Scholar] [CrossRef]
- Engeldinger, E.; Armspach, D.; Matt, D. Cyclodextrin cavities as probes for ligand-exchange processes. Angew. Chem. Int. Ed. 2001, 40, 2526–2529. [Google Scholar] [CrossRef]
- Deshpande, R.M.; Fukuoka, A.; Ichikawa, M. Novel phosphinite capped cyclodextrin-rhodium catalysts in substrate selective hydroformylation. Chem. Lett. 1999, 1, 13–14. [Google Scholar]
- Sawamura, M.; Kitayama, K.; Ito, Y. Synthesis and properties of a new chiral diphosphine ligand bearing a cyclodextrin-based molecular recognition site and its palladium(II) complex. Tetrahedron: Asymmetry 1993, 4, 1829–1832. [Google Scholar]
- Reetz, M.T.; Rudolph, J. Synthesis of a phosphine-modified cyclodextrin and its rhodium complex. Tetrahedron: Asymmetry 1993, 4, 2405–2406. [Google Scholar]
- Hapiot, F.; Bricout, H.; Tilloy, S.; Monflier, E. Functionalized Cyclodextrins as First and Second Coordination Sphere Ligands for Aqueous Organometallic Catalysis. Eur. J. Inorg. Chem. 2012, 2002, 1571–1578. [Google Scholar]
- Tran, D.N.; Legrand, F.X.; Menuel, S.; Bricout, H.; Tilloy, S.; Monflier, E. Cyclodextrin-phosphane possessing a guest-tunable conformation for aqueous rhodium-catalyzed hydroformylation. Chem. Commun. 2012, 48, 753–755. [Google Scholar]
- Legrand, F.X.; Six, N.; Slomianny, C.; Bricout, H.; Tilloy, S.; Monflier, E. Synthesis, Rhodium complexes and catalytic applications of a new water-soluble triphenylphosphane-modified β-cyclodextrin. Adv. Synth. Catal. 2011, 353, 1325–1334. [Google Scholar] [CrossRef]
- Machut-Binkowski, C.; Legrand, F.X.; Azaroual, N.; Tilloy, S.; Monflier, E. New phosphane based on a β-cyclodextrin exhibiting a solvent-tunable conformation and its catalytic properties. Chem. Eur. J. 2010, 16, 10195–10201. [Google Scholar]
- Armspach, D.; Matt, D. Metal-capped β-cyclodextrins: The crowing of the oligosaccharide torus with precious metals. Chem. Commun. 1999, 12, 1073–1074. [Google Scholar] [CrossRef]
- Reetz, M.T.; Frömbgen, C. Chemoselective reduction of halo-nitro aromatic compounds by β-cyclodextrin-modified transition metal catalysts in a biphasic system. Synthesis 1999, 1999, 1555–1557. [Google Scholar]
- Reetz, M.T. Supramolecular transition metal catalysts in two-phase systems. Catal. Today 1998, 42, 399–411. [Google Scholar]
- Reetz, M.T. New supramolecular transition metal catalysis. J. Heterocycl. Chem. 1998, 35, 1065–1073. [Google Scholar] [CrossRef]
- Reetz, M.T.; Waldvogel, S.R. Diphosphanes as ligands for supramolecular rhodium catalysts. Angew. Chem. Int. Ed. Engl. 1997, 36, 865–867. [Google Scholar] [CrossRef]
- Feliciano, C.E. Quiñones, E. The association of 4-(N,N-dimethylamino)benzonitrile and beta-cyclodextrin in dimethyl sulfoxide and N,N-dimethylformamide. J. Photochem. Photobiol. A Chem. 1999, 120, 23–28. [Google Scholar]
- Reetz, M.T.; Rudolph, J.; Goddard, R. Diastereotopic group recognition in the solid state- A unique intramolecular β-cyclodextrin inclusion complex. Can. J. Chem. 2001, 79, 1806–1811. [Google Scholar]
- Loftsson, T.; Jahro, P.; Masson, M.; Jarvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef]
- Rekharsky, M.V.; Inoue, Y. Complexation thermodynamics of cyclodextrins. Chem. Rev. 1998, 98, 1875–1910. [Google Scholar] [CrossRef]
- Legrand, F.X. New mass transfer agents and ligands based on cyclodextrin for aqueous organometallic catalysis. Ph.D. Thesis, University of Artois, Lens, France, 23 November 2010. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tilloy, S.; Binkowski-Machut, C.; Menuel, S.; Bricout, H.; Monflier, E. Phosphane-Based Cyclodextrins as Mass Transfer Agents and Ligands for Aqueous Organometallic Catalysis. Molecules 2012, 17, 13062-13072. https://doi.org/10.3390/molecules171113062
Tilloy S, Binkowski-Machut C, Menuel S, Bricout H, Monflier E. Phosphane-Based Cyclodextrins as Mass Transfer Agents and Ligands for Aqueous Organometallic Catalysis. Molecules. 2012; 17(11):13062-13072. https://doi.org/10.3390/molecules171113062
Chicago/Turabian StyleTilloy, Sébastien, Cécile Binkowski-Machut, Stéphane Menuel, Hervé Bricout, and Eric Monflier. 2012. "Phosphane-Based Cyclodextrins as Mass Transfer Agents and Ligands for Aqueous Organometallic Catalysis" Molecules 17, no. 11: 13062-13072. https://doi.org/10.3390/molecules171113062
APA StyleTilloy, S., Binkowski-Machut, C., Menuel, S., Bricout, H., & Monflier, E. (2012). Phosphane-Based Cyclodextrins as Mass Transfer Agents and Ligands for Aqueous Organometallic Catalysis. Molecules, 17(11), 13062-13072. https://doi.org/10.3390/molecules171113062