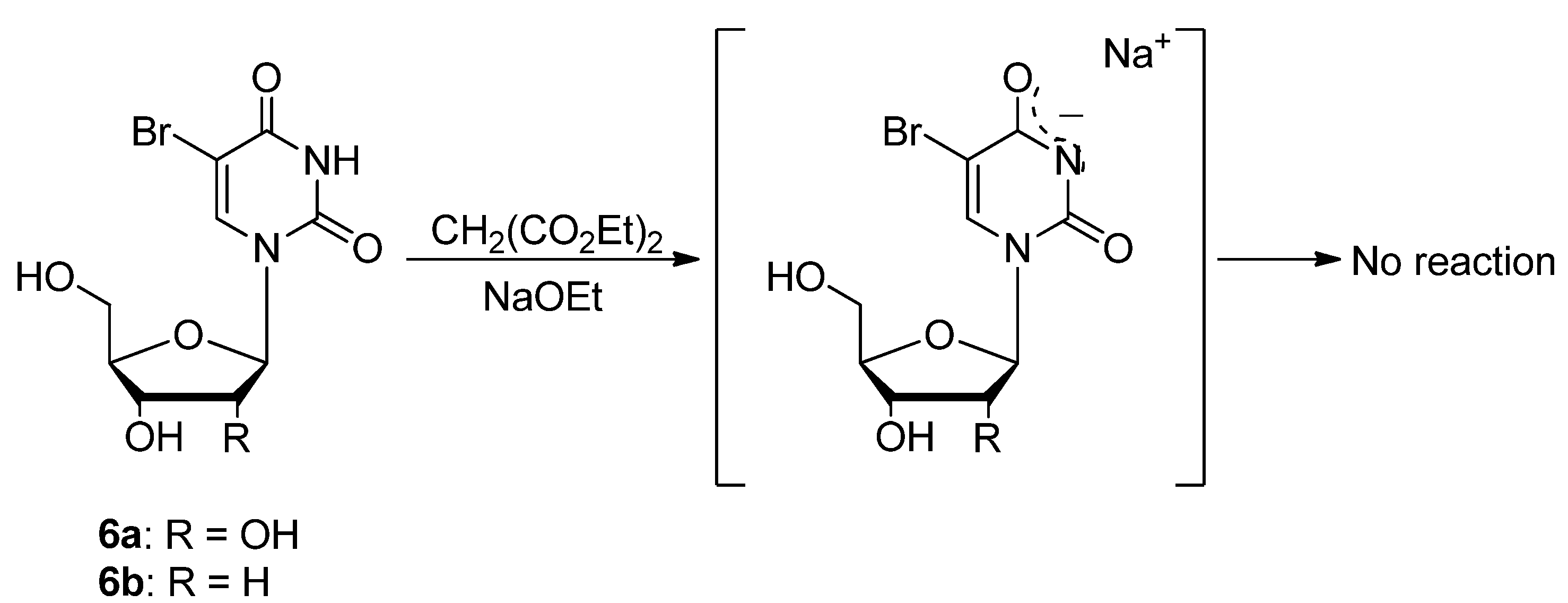
3.1. General
All reagents were obtained from commercial sources and used without further purification. Analytical thin-layer chromatography (TLC) was carried out on pre-coated Silica gel 60 F-254 plates (32–63 µm particle size) and visualized with UV light (254 nm). The 10% Pd/C was obtained from Merck KGaA or Aldrich. Flash column chromatography was performed with Silica gel 60 (40–63 µm particle size, Merck KGaA) or Silica gel 60N (100–210 µm, Kanto Chemical). The 1H and 13C-NMR spectra were recorded by a JEOL AL 400 spectrometer (Tokyo, Japan), JEOL EX 400 spectrometer (400 MHz for 1H-NMR and 100 MHz for 13C-NMR) or JEOL TNG-GX270 spectrometer (270 MHz for 1H-NMR) with tetramethylsilane or residual protonated solvent used as a reference. Elemental analyses were carried out at the Microanalytical Laboratory of our university (YANACO CHN CORDER MT-5 instrument, Tokyo, Japan). The EI and FAB Mass spectra were obtained using a JEOL JMS-SX102A instrument (Tokyo, Japan). The UV spectra were obtained in ethanol using a Shimadzu UV-260 spectrophotometer (Kyoto, Japan).
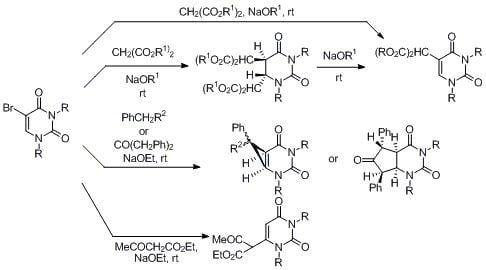
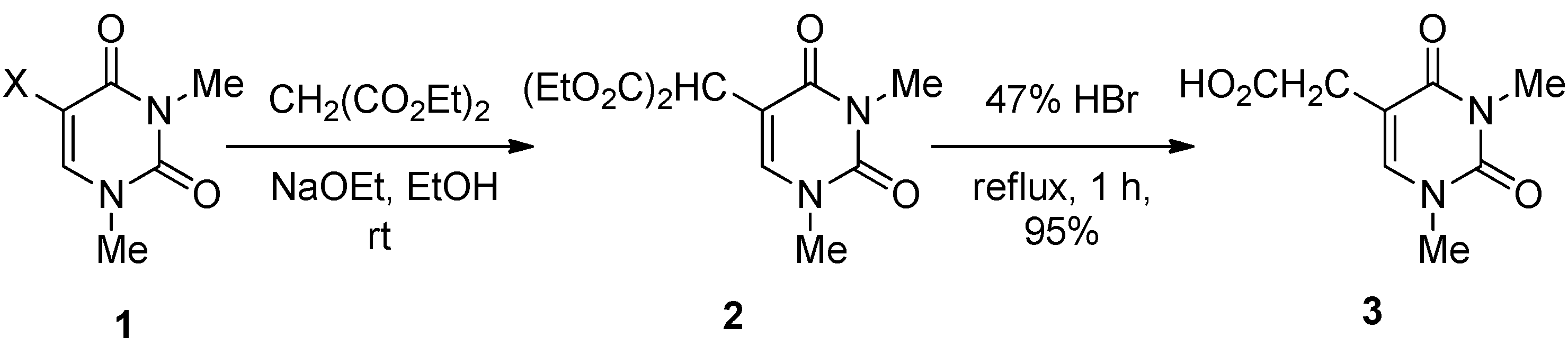
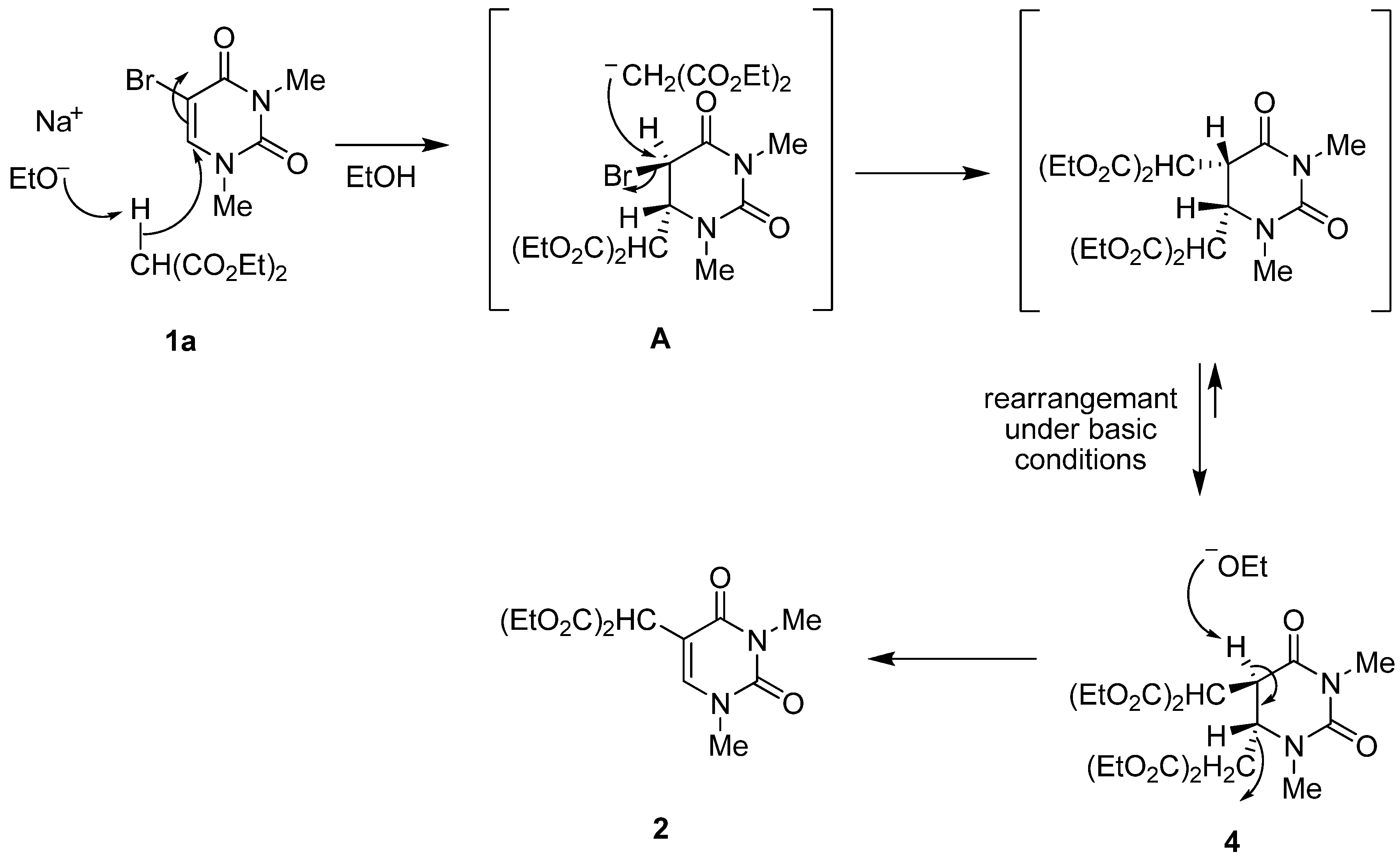
5-Bis(ethoxycarbonyl)methyl-1,3-dimethyluracil (
2) (
Table 2, Entries 1–3). (a) A solution of 5-bromo-1,3-dimethyluracil (
1a) (1.85 g, 8.45 mmol) and diethyl malonate (4.48 g, 27.9 mmol) in ethanolic NaOEt [prepared from Na (584 mg, 25.4 mmol) in absolute EtOH (85 mL)] was stirred for 8 h at room temperature. The mixture was evaporated under reduced pressure and the residue was dissolved in H
2O (20 mL). The mixture was neutralized with conc. HCl and extracted with CHCl
3. The extract was dried over MgSO
4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl
3 as the eluant to give compound
2 (1.51 g, 60%) as a colorless oil.
1H-NMR (CDCl
3) 7.54 (1H, s, 6-H), 4.87 (s, 1H, CH), 4.21 (q,
J = 7.0 Hz, 4H, CH
2), 3.42 and 3.31 (each s, each 3H, NMe), 1.25 (t,
J = 7.0 Hz, 6H, CMe);
13C-NMR (CDCl
3) 167.9, 162.5, 151.2, 142.9, 105.6, 62.3, 48.0, 37.4, 28.3, 14.0; HRMS (EI) calcd. for C
13H
18N
2O
6 (M
+):
m/z 298.1165; found 298.1174; (b) A solution of 5-chloro-1,3-dimethyluracil (
1b, 1.75 g, 10.0 mmol) and diethyl malonate (5.29 g, 33.0 mmol) in ethanolic NaOEt [prepared from Na (690 mg, 30.0 mmol) in absolute EtOH (100 mL)] was stirred for 16 h at room temperature and then treated as described above to give
2 (2.00 g, 67%); (c) A solution of 5-fluoro-1,3-dimethyluracil (
1c, 474 mg, 3.00 mmol) and diethyl malonate (1.59 g, 9.90 mmol) in ethanolic NaOEt [prepared from Na (207 mg, 9.00 mmol) in absolute EtOH (30 mL)] was stirred for 24 h at room temperature and then treated as described above to give
2 (582 mg, 65%).
1,
3-Dimethyluracil-5-acetic acid (
3) [
23]. A mixture of 5-bis(ethoxycarbonyl)methyl-1,3-dimethyluracil (
2) (349 mg, 1.17 mmol) in a 47% HBr solution was refluxed for 1 h. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel with CHCl
3–MeOH (5:1) as the eluant to give
3 (220 mg, 95%), which was identical to the authentic sample.
5,6-Di-[bis(ethoxycarbonyl)methyl]-5,6-dihydro-1,3-dimethyluracil (4). A solution of 5-bromo-1,3-dimethyluracil (1a) (657 mg, 3.00 mmol) and diethyl malonate (1.59 g, 9.90 mmol) in ethanolic NaOEt [prepared from Na (207 mg, 9.00 mmol) in absolute EtOH (30 mL)] was stirred at room temperature for 2 h. The mixture was neutralized with Amberlite CG-50 (H+) and filtered. The ion exchange resin was washed with ethanol, and the combined filtrates were concentrated under reduced pressure. The residue was treated with H2O (30 mL). The aqueous solution was extracted with CHCl3 and the extract was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel with benzene–EtOAc (6:1) as the eluant to give the starting material 1a (217 mg, 33%) and the 5,6-dihydrouracil 4 (564 mg, 41%), which was recrystallized from EtOH. m.p. 85–86 °C; UV λmax. (EtOH) only end absorption; 1H-NMR (CDCl3) 4.29 (brd, J = 7.3 Hz, 1H, CH), 4.27–4.15 (m, 8H, CH2), 3.75 (d, J = 5.6 Hz, 1H, CH), 3.66 (d, J = 7.3 Hz, 1H, CH), 3.50 (brd, J = 5.6 Hz, 1H, CH), 2.96 and 3.16 (each s, each 3H, NMe), 1.35–1.20 (m, 12H, CMe); 13C-NMR (CDCl3) 168.3, 166.8, 166.8, 166.2, 166.1, and 152.4 (each C=O), 62.5, 62.3, 62.3, and 62.2 (each CH2), 55.6, 54.5, 52.0, and 44.6 (C-5, C-6 and CH × 2), 36.5 and 27.8 (each NMe), 13.9, 13.9, 13.9, and 13.8 (each CMe); MS (EI) m/z 459 (M++H, 3%), 413 (25), 299 (100), 207 (47); Anal calcd. for C20H30N2O10: C, 52.39; H, 6.60; N, 6.11%; found: C, 52.14; H, 6.65; N, 6.11.
Reaction of 4 with sodium ethoxide: A solution of 4 (101 mg, 0.220 mmol) in ethanolic NaOEt [prepared from Na (14.9 mg, 0.650 mmol) in absolute EtOH (5 mL)] was stirred at room temperature for 8 h. The solvent was removed under reduced pressure and the residue was treated with H2O (20 mL). The solution was neutralized with c.HCl and the aqueous solution was extracted with CHCl3. The extract was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel with benzene–EtOAc (6:1) as the eluant to give 1,3-dimethyluracil-5-malonic acid diethyl ester (2, 44.0 mg, 67%).
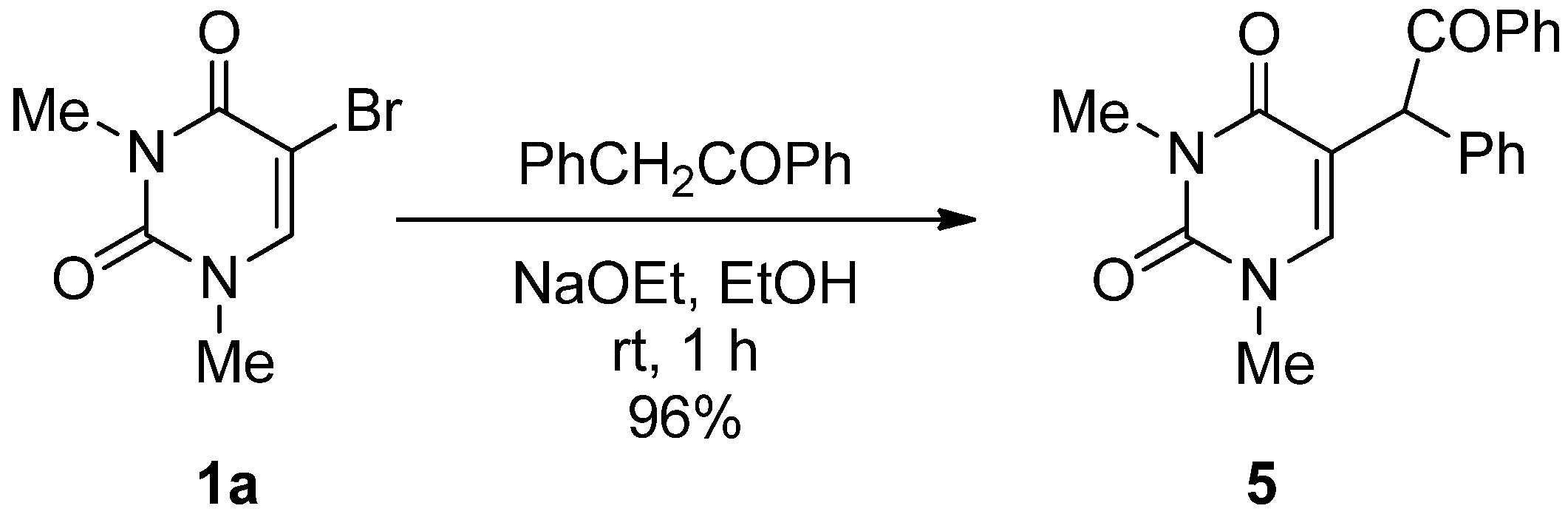
5-(α-Benzoyl)benzyl-1,3-dimethyluracil (5). A mixture of 5-bromo-1,3-dimethyluracil (1a) (1.32 g, 6.00 mmol) and benzylphenylketone (3.69 g, 19.8 mmol) in ethanolic NaOEt [prepared from Na (414 mg, 18.0 mmol) in absolute EtOH (60 mL)] was stirred at room temperature for 1 h. The solvent was removed under reduced pressure and the residue was treated with H2O (40 mL). The solution was neutralized with conc. HCl and the aqueous solution was extracted with CHCl3. The extract was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3 as the eluant to give 5 (1.94 g, 96%), which was recrystallized from CHCl3. m.p. 128.5–130 °C; UV λmax. (EtOH) 249 nm (ε 16,300 dm3mol–1cm–1); 1H-NMR (CDCl3) 8.27–7.88 (m, 2H, COPh), 7.74–7.15 (m, 8H, Ph and COPh), 6.73 (brs, 1H, 6-H), 6.09 (brs, 1H, CH), 3.34 and 3.27 (each s, each 3H, NMe); 13C-NMR (CDCl3) 197.5, 163.2, 151.4, 142.1, 136.1, 135.3, 133.2, 129.5, 129.1, 128.9, 128.6, 128.0, 113.8, 50.8, 37.3, 28.1; MS (EI) m/z 334 (M+, 13%), 229 (100), 172 (25), 131 (38), 105 (70); Anal calcd. for C20H18N2O3: C, 71.84; H, 5.43; N, 8.38%; found: C, 71.79; H, 5.47; N, 8.37.
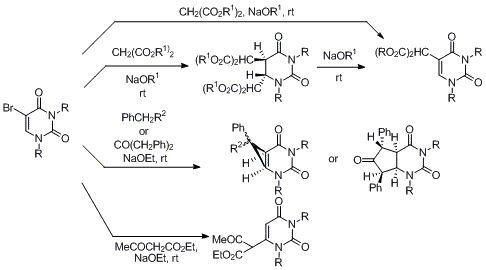
5-Bromo-3-p-methoxybenzyluridine (7a). p-Methoxybenzyl chloride (0.93 mL, 6.80 mmol) was added dropwise to a mixture of 5-bromouridine (2.00 g, 6.20 mmol) and K2CO3 (1.11 g, 8.05 mmol) in DMF (10 mL) at room temperature. The mixture was stirred for 24 h and filtered. The filtrate was concentrated in vacuo, and the residue was purified by recrystallization from MeOH to give 7a (2.42 g, 88%) as a colorless powder. 1H-NMR (DMSO-d6) 8.60 (s, 1H, 6-H), 7.25 and 7.05 (each d, each J = 8.8 Hz, each 2H, C6H4), 5.75 (d, J = 4.9 Hz, 1H, 1'-H), 5.46 (brs, 1H, OH), 5.30 (brs, 1H, OH), 5.05 (brd, J = 5.9 Hz, 1H, OH), 4.93 (s, 2H, CH2), 4.03–3.99 (m, 2H, 2'-H and 3'-H), 3.87–3.82 (m, 1H, 4'-H), 3.77 (s, 3H, CH3), 3.62–3.57 (m, 2H, 5'-H); 13C-NMR (DMSO-d6) 158.5, 158.3, 149.9, 139.1, 129.5, 128.5, 113.7, 94.9, 89.7, 84.5, 73.9, 68.8, 59.7, 55.0, 44.0; MS (EI) m/z 442 (M+, 8%), 444 (8), 310 (25), 312 (25), 162 (12), 121 (100); HRMS (EI) calcd. for C17H19BrN2O7 (M+): 442.03756; found: 442.03681; Anal calcd. for C17H19BrN2O7: C, 46.07; H, 4.32; N, 6.32%; found: C, 45.96; H, 4.46; N, 6.33.
5-Bromo-3-benzyloxymethyluridine (7b). Benzyloxymethyl chloride (0.930 mL, 6.80 mmol) was dropwise added to a mixture of 5-bromouridine (2.00 g, 6.20 mmol) and K2CO3 (1.11 g, 8.05 mmol) in DMF (10 mL) at room temperature. The mixture was stirred for 24 h and filtered. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography on silica gel with CHCl3-MeOH (50:1) as the eluant to give 7b (1.51 g, 55%) as a colorless powder. 1H-NMR (DMSO-d6) 8.60 (s, 1H, 6-H), 7.34–7.25 (m, 5H, C6H5), 5.72 (d, J = 3.7 Hz, 1H, 1'-H), 5.50–5.47 (m, 1H, OH), 5.37–5.32 (m, 3H, OH and CH2), 5.09–5.05 (m, 1H, OH), 4.58 (s, 2H, CH2) 4.09–4.00 (m, 1H, 2'-H), 3.96–3.95 (m, 1H, 3'-H), 3.81–3.74 (m, 1H, 4'-H), 3.61–3.55 (m, 2H, 5'-H); 13C-NMR (DMSO-d6) 158.6, 150.0, 139.7, 128.1, 127.5, 127.4, 94.8, 89.7, 84.4, 74.0, 71.3, 71.1, 68.6, 59.6; MS (FAB, Gly) m/z 443 (M++H, 8%), 445 (8%), 365 (5), 277 (12) 185 (100); HRMS (FAB, Gly) calcd. for C17H19BrN2O7 (M+): 442.0376; found: 442.0447; Anal calcd. for C17H19BrN2O7: C, 46.07; H, 4.32; N, 6.32; found: C, 46.07; H, 4.48; N, 6.28.
5-Bromo-3-benzyloxymethyl-2'-deoxyuridine (7c). Benzyloxymethyl chloride (0.460 mL, 2.59 mmol) was dropwise added to a mixture of 5-bromo-2'-deoxyuridine (1.00 g, 2.36 mmol) and K2CO3 (561 mg, 3.07 mmol) in DMF (10 mL) at room temperature. The mixture was stirred for 24 h and filtered. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (100:1) as the eluant to give 7c (549 mg, 55%) as colorless oil. 1H-NMR (DMSO-d6) 8.49 (s, 1H, 6-H), 7.34-7.23 (m, 5H, C6H5), 6.10 (t, J = 6.3 Hz, 1H, 1'-H), 5.34 (s, 2H, CH2), 5.25 (brd, J = 4.4 Hz, 1H, OH), 5.21-5.18 (m, 1H, OH), 4.58 (s, 2H, CH2), 4.25-4.20 (m, 1H, 3'-H), 3.83-3.79 (m, 1H, 4'-H), 3.67-3.54 (m, 2H, 5'-H), 2.19-2.12 (m, 2H, 2'-H); MS (FAB, NBA) m/z 427 (M++H, 5%), 429 (5), 154 (100), 146 (64); HRMS (FAB, NBA) calcd. for C17H19BrN2O6 (M++H): 427.04265; found: 427.04935.
5-Bis(ethoxycarbonyl)methyl-3-p-methoxybenzyluridine (8a). A solution of 5-bromo-3-p-methoxybenzyluridine (7a) (500 mg, 1.13 mmol) and diethyl malonate (597 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8a (301 mg, 51%) as a colorless foam. 1H-NMR (DMSO-d6) 8.00 (s, 1H, 6-H), 7.26 and 6.88 (each d, each J = 8.7 Hz, each 2H, C6H4), 5.88 (d, J = 5.1 Hz, 1H, 1'-H), 5.46 (brd, J = 5.9 Hz, 1H, OH), 5.13–5.09 (m, 1H, OH), 4.94 (brd, J = 5.0 Hz, 1H, OH), 4.94 (s, 2H, CH2), 4.16 (q, J = 7.2 Hz, 4H, CH2 × 2), 4.03–3.98 (m, 1H, 2’-H), 3.95–3.90 (m, 1H, 3'-H), 3.90–3.89 (m, 1H, 4’-H), 3.74 (s, 3H, CH3), 3.62–3.45 (m, 2H, 5'-H), 1.19 (t, J = 7.1 Hz, 6H, CH3 × 2); 13C-NMR (DMSO-d6) 166.9, 161.1, 158.6, 150.2, 137.9, 129.5, 128.7, 113.6, 106.9, 88.9, 85.1, 73.8, 69.8, 61.5, 60.8, 55.0, 49.7, 43.3, 13.8; MS (FAB, NBA) m/z 523 (M++H, 29%), 522 (7), 391 (9), 154 (100), 121 (76); HRMS (FAB, NBA) calcd. for C24H31N2O11 (M++H) 523.185; found: 523.1935.
5-Bis(methoxycarbonyl)methyl-3-p-methoxybenzyluridine (8b). A solution of 5-bromo-3-p-methoxybenzyluridine (7a) (500 mg, 1.13 mmol) and dimethyl malonate (493 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8b (262 mg, 47%) as a colorless foam. 1H-NMR (DMSO-d6) 7.99 (s, 1H, 6-H), 7.24 and 6.86 (each d, each J = 8.5 Hz, each 2H, C6H4), 5.84 (d, J = 3.9 Hz, 1H, 1'-H), 5.45 (brd, J = 5.8 Hz, 1H, OH), 5.13–5.11 (m, 1H, OH), 5.05 (brd, J = 4.3 Hz, 1H, OH), 4.85 (s, 2H, CH2), 4.64 (s, 1H, CH), 4.01–3.99 (m, 1H, 2'-H), 3.98–3.97 (m, 1H, 3'-H), 3.92–3.87 (m, 1H, 4'-H), 3.17 (s, 6H, CH3 × 2), 3.66 (s, 3H, CH3), 3.57–3.50 (m, 2H, 5'-H); MS (EI) m/z 494 (M+, 5%), 462 (17), 304 (18), 162 (17), 121 (100); HRMS (EI) calcd. for C22H28N2O11 (M+): 494.15366; found: 494.15423.
5-(α-Benzoyl)benzyl-3-p-methoxybenzyluridine (8c). A solution of 5-bromo-3-p-methoxybenzyluridine (7a) (500 mg, 1.13 mmol) and benzylphenylketone (732 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute ethanol (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8c (379 mg, 60%) as a colorless foam. 1H-NMR (DMSO-d6) 7.99 (d, J = 7.8 Hz, 2H, o-Bz), 7.55 (t, J = 7.3 Hz, 1H, p-Bz), 7.46 (t, J = 6.8 Hz, m-Bz), 7.35–7.31 (m, 5H, C6H5), 7.28 and 6.83 (each d, each J = 6.3 Hz, each 2H, C6H4), 7.01 and 7.04 (each s, total 1H, 6-H), 6.10 (s, 1H, CH), 5.79 (d, J = 5.4 Hz, 1H, 1'-H), 5.41 (brd, J = 5.9 Hz, 1H, OH), 5.33–5.32 (m, J = 5.8 Hz, 1H, OH), 5.10–5.06 (m, 1H, OH), 4.89 (s, 2H, CH2) 4.66–4.63 (m, 1H, 2'-H), 4.59–4.56 (m, 1H, 3'-H), 3.87–3.84 (m, 1H, 4'-H), 3.76 (s, 3H, CH3), 3.68–3.58 (m, 2H, 5'-H); 13C-NMR (DMSO-d6) 196.5, 161.5, 158.2, 150.3, 136.9, 136.8, 136.7, 135.8, 135.0, 134.8, 133.2, 129.4, 129.1, 128.6, 127.7, 114.2, 113.6, 88.7, 84.8, 73.4, 70.2, 61.2, 55.0, 50.5, 43.4; MS (FAB, Gly) m/z 559 (M++H, 29%), 427 (7), 321 (8), 185 (100), 121 (78); HRMS (FAB, Gly) calcd. for C31H30N2O8 (M++H): 559.2002; found: 559.2090.
5-Bis(ethoxycarbonyl)methyl-3-benzyloxymethyluridine (8d): A solution of 5-bromo-3-benzyloxy-methyluridine (7b) (500 mg, 1.13 mmol) and diethyl malonate (597 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute ethanol (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with CHCl3 and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8d (307 mg, 52%) as a colorless oil. 1H-NMR (DMSO-d6) 8.00 (s, 1H, 6-H), 7.34–7.24 (m, 5H, C6H5), 5.83 (d, J = 5.4 Hz, 1H, 1'-H), 5.48 (brd, J = 5.4 Hz, 1H, OH), 5.39–5.26 (m, 2H, CH2), 5.16–5.14 (m, 1H, OH), 5.04 (brd, J = 4.9 Hz, 1H, OH), 4.59 (s, 2H, CH2), 4.20–4.10 (m, 5H, CH2 × 2 and 2'-H), 4.00–3.98 (m, 1H, 3'-H), 3.89–3.82 (m, 1H, 4'-H), 3.65–3.50 (m, 2H, 5'-H), 1.17 (J = 6.9 Hz, 6H, CH3 × 2); MS (FAB, NBA) m/z 523 (M++H, 32%), 416 (8%), 238 (9), 154 (100), 107 (24); HRMS (FAB, NBA) calcd. for C24H30N2O11 (M++H): 523.19343; found: 523.19283.
5-Bis(methoxycarbonyl)methyl-3-benzyloxymethyl-2'-deoxyuridine (8e). A solution of 5-bromo-3-benzyloxymethyluridine (7b) (500 mg, 1.13 mmol) and dimethyl malonate (493 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8e (263 mg, 47%) as a colorless oil. 1H-NMR (DMSO-d6) 8.01 (s, 1H, 6-H), 7.35–7.27 (m, 5H, -C6H5), 5.83 (d, J = 4.6 Hz, 1H, 1'-H), 5.47 (brd, J = 5.9 Hz, 1H, OH), 5.32 (s, 2H, CH2), 5.14–5.12 (m, 1H, OH), 5.05 (brd, J = 4.8 Hz, 1H, OH), 4.70 (s, 1H, CH), 4.57 (s, 2H, CH2), 4.04–4.02 (m, 1H, 2'-H), 4.01–3.98 (m, 1H, 3'-H), 3.94–3.92 (m, 1H, 4'-H), 3.69 (s, 6H, CH3 × 2), 3.68–3.65 (m, 2H, 5'-H); MS (FAB, NBA) m/z 495 (M++H, 5%), 238 (10), 176 (8), 154 (100), 85 (47); HRMS (FAB, NBA) calcd. for C22H28N2O11 (M++H): 495.15366; found: 494.16221.
5-(α-Benzoyl)benzyl-3-p-methoxybenzyluridine (8f): A solution of 5-bromo-3-benzyloxymethyluridine (7b) (500 mg, 1.13 mmol) and benzylphenylketone (732 mg, 3.73 mmol) in ethanolic NaOEt [prepared from Na (77.8 mg, 3.38 mmol) in absolute ethanol (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 8f (297 mg, 47%) as a colorless oil. 1H-NMR (DMSO-d6) 8.01 (d, J = 7.7 Hz, 2H, o-Bz), 7.57 (t, J = 7.7 Hz, 1H, p-Bz), 7.48 (t, J = 7.2 Hz, m-Bz),7.49–7.24 (m, 10H, C6H5 × 2), 7.10 and 7.04 (each s, total 1H, 6-H), 6.08 (s, 1H, CH), 5.80 (d, J = 5.8 Hz, 1H, 1'-H), 5.45 (brd, J = 5.8 Hz, 1H, OH), 5.36–5.32 (m, 3H, OH and CH2), 5.08 (brd, J = 4.8 Hz, 1H, OH), 4.55 (s, 2H, CH2), 4.04–3.98 (m, 1H, 2'-H), 3.90–3.88 (m, 1H, 3'-H), 3.76–3.73 (m, 1H, 4'-H), 3.68–3.58 (m, 2H, 5′-H); MS (FAB, NBA) m/z 559 (M++H, 5%), 238 (21), 176 (6), 154 (100), 85 (67); HRMS (FAB, NBA) calcd. for C31H30N2O8 (M++H): 559.20803; found: 559.20701.
5-Bis(ethoxycarbonyl)methyl-3-benzyloxymethyl-2'-deoxyuridine (8g). A solution of 5-bromo-3-benzyloxymethyl-2’-deoxyuridine (7c) (299 mg, 0.700 mmol) and diethyl malonate (370 mg, 2.31 mmol) in ethanolic NaOEt [prepared from Na (48.3 mg, 2.10 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (200:1) as the eluant to give 8g (170 mg, 48%) as a colorless oil. 1H-NMR (CDCl3) 8.09 (s, 1H, 6-H), 7.36–7.23 (m, 5H, C6H5), 6.26 (t, J = 6.5 Hz, 1H, 1'-H), 5.47 (s, 2H, CH2), 5.09–5.02 (brs, 1H, OH), 4.99–4.93 (brs, 1H, OH), 4.87 (s, 1H, CH), 4.68 (s, 2H, CH2), 4.53–4.48 (m, 1H, 3'-H), 4.28–4.16 (m, 4H, CH2 × 2), 3.82–3.75 (m, 1H, 4'-H), 3.75–3.54 (m, 2H, 5'-H), 2.47–2.25 (m, 2H, 2'-H), 1.28 (t, J = 7.2 Hz, 6H, CH3 × 2); MS (FAB, NBA) m/z 507 (M++H, 36%), 391 (18), 284 (11), 154 (100), 91 (50); HRMS (FAB, NBA) calcd. for C24H30N2O10 (M++H): 507.19783; found: 507.19747.
5-Bis(methoxycarbonyl)methyl-3-benzyloxymethyl-2'-deoxyuridine (8h). A solution of 5-bromo-3-benzyloxy-2′-deoxymethyluridine (7c) (299 mg, 0.700 mmol) and dimethyl malonate (305 mg, 2.31 mmol) in ethanolic NaOEt [prepared from Na (48.3 mg, 2.10 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (200:1) as the eluant to give 8h (141 mg, 42%) as a colorless oil. 1H-NMR (CDCl3) 8.10 (s, 1H, 6-H), 7.36–7.23 (m, 5H, C6H5), 6.26 (t, J = 6.2 Hz, 1H, 1'-H), 5.47 (s, 2H, CH2), 5.10–5.06 (m, 1H, OH), 4.93 (brs, 1H, OH), 4.90 (s, 1H, CH), 4.68 (s, 2H, CH2), 4.03–3.98 (m, 1H, 3′-H), 3.93–3.86 (d, J = 3.6 Hz, 1H, 4'-H), 3.77 (s, 4H, CH2 × 2), 3.58–3.46 (m, 2H, 5'-H), 2.44–2.24 (m, 2H, 2'-H); MS (FAB, NBA) m/z 479 (M++H, 5%), 391 (5%), 176 (10), 154 (100), 89 (64); HRMS (FAB, NBA) calcd. for C22H26N2O10 (M++H): 479.15875; found: 479.16750.
5-(α-Benzoyl)benzyl-3-benzyloxymethyl-2'-deoxyuridine (8i). A solution of 5-bromo-3-benzyloxy-2'-deoxymethyl uridine (7c) (299 mg, 0.700 mmol) and dimethyl malonate (305 mg, 2.31 mmol) in ethanolic NaOEt [prepared from Na (48.3 mg, 2.10 mmol) in absolute EtOH (10 mL)] was stirred for 24 h at room temperature. The mixture was evaporated under reduced pressure, the residue was dissolved in H2O (30 mL), and the mixture was neutralized with NaHSO4. The solution was extracted with EtOAc and the extract was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel with CHCl3–MeOH (200:1) as the eluant to give 8i (163 mg, 43%) as a colorless oil. 1H-NMR (DMSO-d6) 8.02 (d, J = 7.6 Hz, 2H, O-Bz), 7.58 (t, J = 7.3 Hz, 1H, p-Bz), 7.47 (t, J = 7.6 Hz, m-Bz), 7.40–7.26 (m, 10H, C6H5 × 2), 7.00 and 6.93 (each s, total 1H, 6-H), 6.12–6.06 (m, 2H, CH and 1′-H), 5.31 (s, 2H, CH2), 5.24–5.20 (m, 1H, OH), 4.81-4.77 (m, 1H, OH), 4.56 (s, 2H, CH2), 4.05–3.92 (m, 1H, 3'-H), 3.73–3.67 (m, 1H, 4'-H), 3.52–3.35 (m, 2H, 5'-H), 2.17–2.10 (m, 2H, 2'-H); MS (FAB, NBA) m/z 543 (M++H, 5%), 282 (21), 238 (11), 154 (100), 107 (20), 85 (44); HRMS (FAB, NBA) calcd. for C31H30N2O7 (M++H): 543.20530, found: 543.21377.
5-Bis(ethoxycarbonyl)methyluridine (
9a). (a) A solution of 5-bis(ethoxycarbonyl)methyl-3-
p-methoxy-benzyluridine (
8a) (100 mg, 0.191 mmol) and AlCl
3 (204 mg, 1.53 mmol) in absolute anisole (1.0 mL) was stirred for 12 h at room temperature. To the mixture was added MeOH, and the mixture was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl
3–MeOH (30:1) as the eluant to give
9a (35.4 mg, 46%) as a colorless oil (
Table 3, Entry 1).
1H-NMR (DMSO-
d6) 11.58 (s, 1H, 3-NH), 7.87 (s, 1H, 6-H), 5.80 (d,
J = 5.4 Hz, 1H, 1'-H), 5.40 (brd,
J = 5.4 Hz, 1H, OH), 5.13–5.09 (m, 1H, OH), 5.02–4.92 (m, 1H, OH), 4.57 (s, 1H, CH), 4.13 (q,
J = 7.0 Hz, 4H, CH
2 × 2), 4.01–3.97 (m, 1H, 2'-H), 3.95–3.90 (m, 1H, 3'-H), 3.88–3.82 (m, 1H, 4'-H), 3.60–3.50 (m, 2H, 5'-H), 1.17 (
J = 7.0 Hz, 6H, CH
3 × 2). MS (EI)
m/z 402 (M
+, 7%), 152 (100); HRMS (EI) calcd. for C
16H
22N
2O
10 (M
+) 402.1274; found: 402.1291; (b) A mixture of
8d (136 mg, 0.260 mmol) and Pd/C (40.8 mg) was stirred at room temperature under an H
2 atmosphere. After 72 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated
in vacuo. The residue was purified by column chromatography on silica gel with CHCl
3–MeOH (30:1) to give
9a (72.2 mg, 69%) (
Table 3, Entry 4).
5-Bis(methoxycarbonyl)methyluridine (
9b). (a) A solution of 5-bis(methoxycarbonyl)methyl-3-
p-methoxybenzyluridine (
8b) (100 mg, 0.202 mmol) and AlCl
3 (216 mg, 1.62 mmol) in absolute anisole (1.0 mL) was stirred for 24 h at room temperature. To the mixture was MeOH, and the mixture was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl
3–MeOH (30:1) as the eluant to give
9b (31.8 mg, 42%) as a colorless oil (
Table 3, Entry 2).
1H-NMR (DMSO-
d6) 11.60 (s, 1H, 3-NH), 7.87 (s, 1H, 6-H), 5.79 (d,
J = 5.6Hz, 1H, 1'-H), 5.39 (brs, 1H, OH), 5.01 (brs, 1H, OH), 5.00 (brs, 1H, OH), 4.62 (s, 1H, CH), 4.03–3.96 (m, 1H, 2'-H), 3.95–3.90 (m, 1H, 3'-H), 3.93–3.84 (m, 1H, 4′-H), 3.66 (s, 6H, CH
3 × 2), 3.61–3.49 (m, 2H, 5'-H); MS (FAB, Gly)
m/z 375 (M
++H, 3%), 307 (20), 289 (15), 238 (16), 154 (100), 136 (65), 107 (19), 85 (56); HRMS (FAB, Gly) calcd. for C
14H
19N
2O
10 (M
++H): 375.0961; found: 375.1042; (b) A mixture of
8e (129 mg, 0.26 mmol) and Pd/C (38.7 mg) was stirred at room temperature under an H
2 atmosphere. After 72 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated
in vacuo. The residue was purified by column chromatography on silica gel with CHCl
3/MeOH (30:1) to give
9b (65.2 mg, 67%) (
Table 3, Entry 5).
5-(α-Benzoyl)benzyluridine (9c). A solution of 5-(α-benzoyl)benzyl-3-p-methoxybenzyluridine (8c) (100 mg, 0.179 mmol) and AlCl3 (191 mg, 1.43 mmol) in absolute anisole (1.0 mL) was stirred for 4 h at room temperature. To the mixture was added MeOH, and the mixture was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (30:1) as the eluant to give 9c (64 mg, 82%) as a pale yellow foam. 1H-NMR (DMSO-d6) 11.50 (s, 1H, 3-NH), 7.97 (d, J = 7.8 Hz, 2H, o-Bz), 7.55 (t, J = 7.3 Hz, 1H, p-Bz), 7.44 (t, J = 7.3 Hz, m-Bz), 7.33–7.27 (m, 5H, C6H5), 7.01 and 6.95 (each s, total 1H, 6-H), 6.10 (s, 1H, CH), 5.78–5.72 (m, 1H, 1'-H), 5.37 (brs, 1H, OH), 5.28 (brs, 1H, OH), 5.10–5.06 (m, 1H, OH), 4.68–4.63 (m, 2H. CH2), 3.86–3.78 (m, 1H, 2'-H), 3.77–3.71 (m, 1H, 3'-H), 3.66–3.60 (m, 1H, 4'-H), 3.53–3.29 (m, 2H, 5'-H); MS (EI) m/z 438 (M+, 6%), 306 (17), 201 (55), 158 (13), 115 (10), 105 (100), 77 (25); HRMS (EI) calcd. for C23H22N2O7 (M+): 438.1427; found: 438.1418.
5-Bis(ethoxycarbonyl)methyl-2'-deoxyuridine (9d). A mixture of 5-bis(ethoxycarbonyl)methyl-3-benzyloxymethyl-2’-deoxyuridine (8g) (50.7 mg 0.100 mmol) and Pd/C (Merck) (15.0 mg) in MeOH (1.0 mL) was stirred under H2 atmosphere at room temperature. After 24 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (30:1) as the eluant to give 9d (24.7 mg, 64%) as a colorless oil. 1H-NMR (DMSO-d6) 11.55 (s, 1H, 3-NH), 7.84 (s, 1H, 6-H), 6.16 (t, J = 6.8 Hz, 1H, 1'-H), 5.24 (brd, J = 4.1 Hz, 1H, OH), 4.95–4.91 (m, 1H, OH), 4.58 (s, 1H, CH), 4.22–4.18 (m, 1H, 3'-H), 4.06 (q, J = 7.2 Hz, 4H, CH2 × 2), 3.80–3.78 (m, 1H, 4'-H), 3.55–3.40 (d, J = 4.6 Hz, 2H, 5'-H), 2.17–1.97 (m, 2H, 2’-H), 1.17 (t, J = 7.1 Hz, 6H, CH3 × 2); MS (FAB, NBA) m/z 387 (M++H, 9%), 271 (12), 176 (8), 154 (100), 107 (19), 89 (17); HRMS (FAB, NBA) calcd. for C16H22N2O9 (M++H): 387.13253; found: 387.14105.
5-Bis(methoxycarbonyl)methyl-2'-deoxyuridine (9e). A mixture of 5-bis(methoxycarbonyl)methyl-3-benzyloxymethyl-2′-deoxyuridine (8h) (50 mg 0.105 mmol) and Pd/C (Merck) (15.0 mg) in MeOH (1.0 mL) was stirred under an H2 atmosphere at room temperature. After 72 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (30:1) as the eluant to give 9e (22.2 mg, 59%) as a colorless oil. 1H-NMR (DMSO-d6) 11.56 (s, 1H, 3-NH), 7.86 (s, 1H, 6-H), 5.81–5.78 (m, 1H, 1'-H), 5.24 (brd, J = 4.1 Hz, 1H, OH), 4.95–4.90 (m, 1H, OH), 4.64 (s, 1H, CH), 4.09–4.02 (m, 1H, 3'-H), 3.81–3.76 (m, 1H, 4'-H), 3.66 (s, 4H, CH2 × 2), 3.55–3.49 (m, 2H, 5'-H), 2.17–1.99 (m, 2H, 2'-H); MS (FAB, NBA) m/z 359 (M++H, 32%), 243 (33%), 154 (100), 107 (10), 85 (62); HRMS (FAB, NBA) calcd. for C14H18N2O9 (M++H): 359.10123; found: 359.10827.
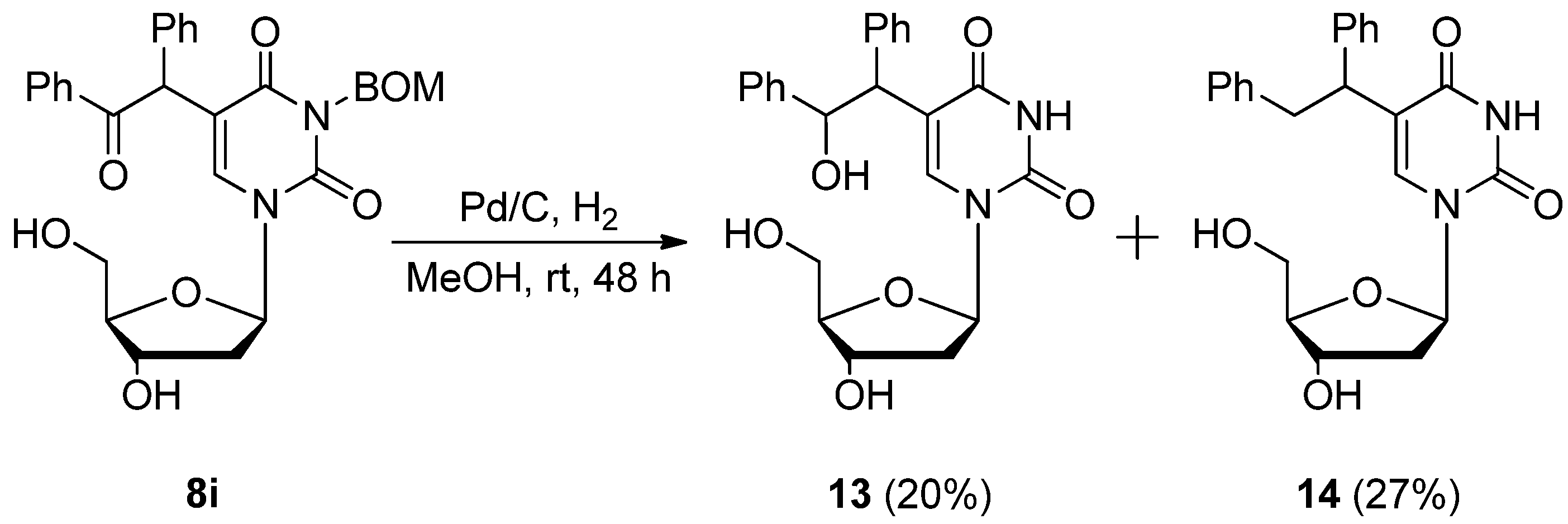
5-(2-Hydroxy-1,2-diphenylethyl)-2'-deoxyuridine (13) and 5-(1,2-diphenylethyl)-2'-deoxyuridine (14). A mixture of 5-α-benzoylbenzyl-3-benzyloxymethyl-2’-deoxyuridine (8i) (81.4 mg 0.150 mmol) and Pd/C (20.3 mg) in MeOH (1.0 mL) was stirred under H2 atmosphere at room temperature for 48 h. The mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated in vacuo. The residue was purified by PTLC with CHCl3-MeOH (5:1) as the eluant to give 13 (12.7 mg, 20%) as a light brown oil and 14 (16.5 mg, 27%) as a colorless oil.
13: 1H-NMR (DMSO-d6) 11.11 (d, J = 10.6 Hz, 1H, 3-NH), 7.97 and 7.90 (each s, 1H, 6-H), 7.25–7.10 (m, 10H, C6H5 × 2), 6.11–6.05 (m, 1H, 1'-H), 5.32–5.09 (m, 4H, CH × 2 and OH × 2), 4.53 (s, 1H, OH), 4.30–4.22 (m, 1H, 3'-H), 3.82–3.78 (m, 1H, 4'-H), 3.70–3.59 (m, 2H, 5'-H), 2.11–1.90 (m, 2H, 2'-H); MS (FAB, NBA) m/z 425 (M++H, 19%), 291 (65), 202 (17), 176 (8), 154 (100), 107 (26), 77 (21); HRMS (FAB, NBA) calcd. for C23H24N2O6 (M++H): 425.16344; found: 425.17190.
14: 1H-NMR (DMSO-d6) 11.19 (s, 1H, 3-NH), 8.04 and 8.00 (each s, 1H, 6-H), 7.25–7.10 (m, 10H, C6H5 × 2), 6.16 (t, J = 6.6 Hz, 1H, 1'-H), 5.32–5.20 (m, 4H, CH2 and OH × 2), 5.24–5.20 (m, 1H, OH), 4.32–4.10 (m, 2H, 3'-H and CH), 3.84–3.80 (m, 1H, 4'-H), 3.68–3.60 (m, 2H, 5'-H), 2.12–2.03 (m, 2H, 2'-H); MS (FAB, NBA) m/z 409 (M++1, 6%), 154 (100), 136 (60), 107 (16), 89 (14); HRMS (FAB, NBA) calcd. for C23H24N2O5 (M++H): 409.16852; found: 409.17779.
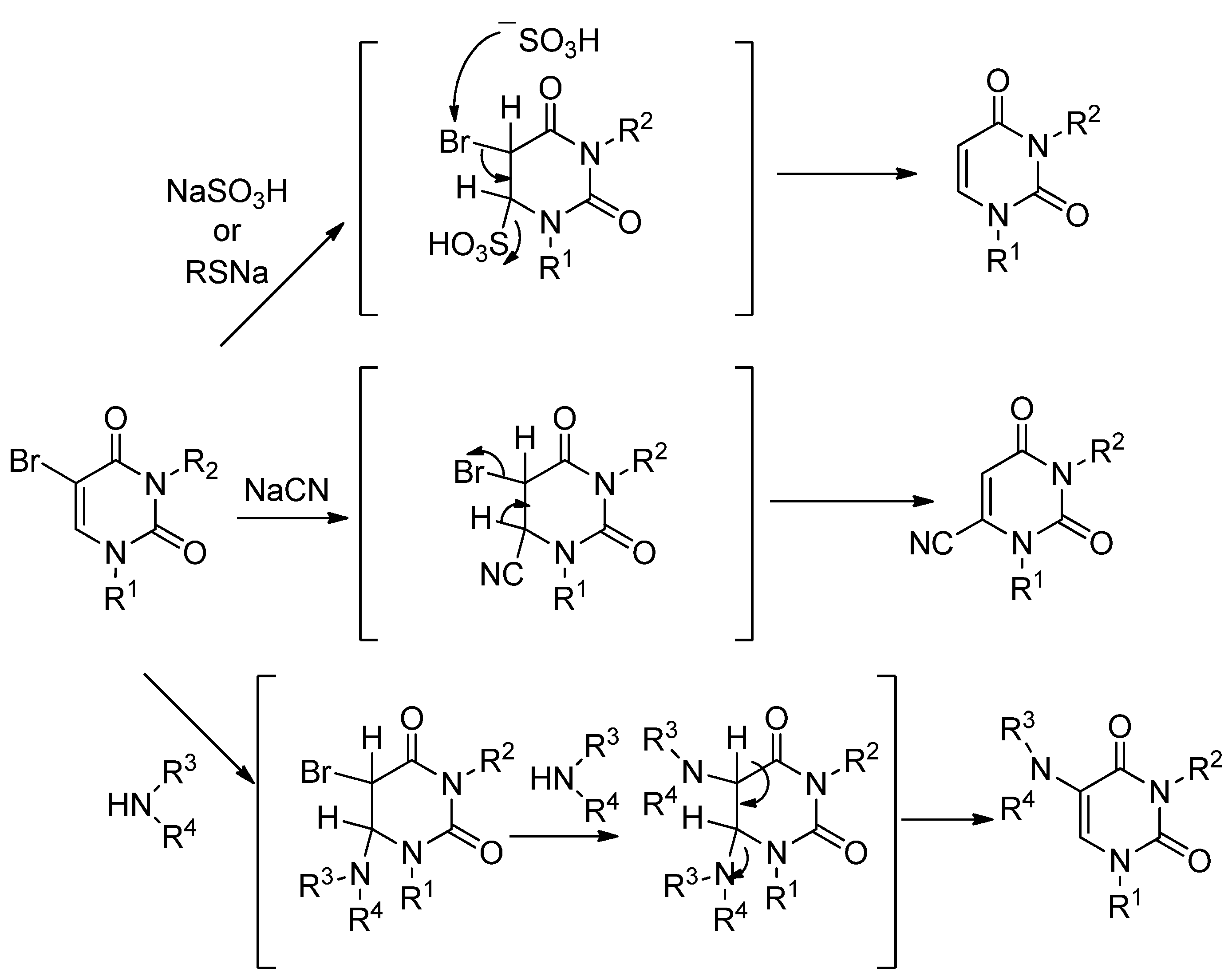
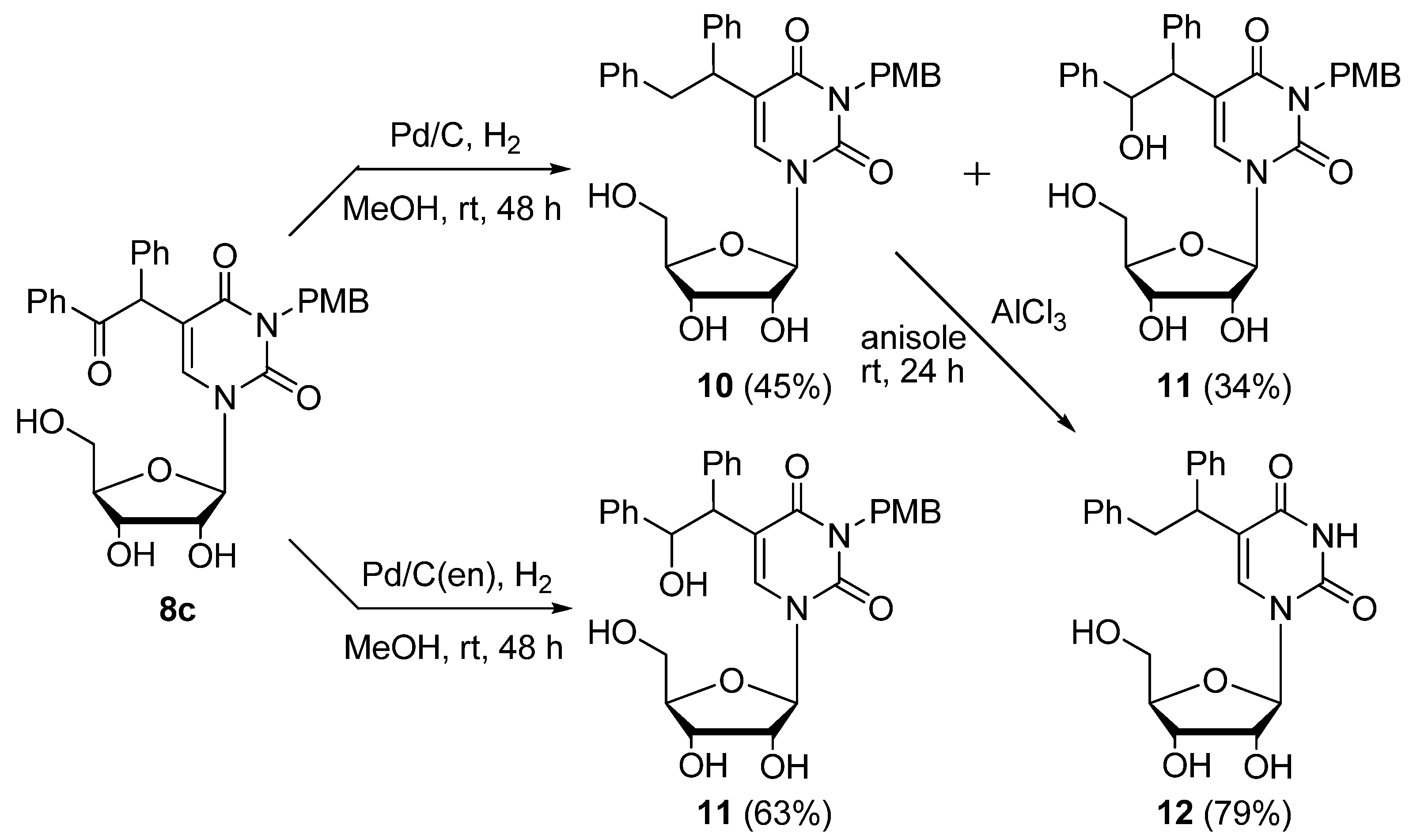
Pd/C-catalyzed hydrogenation of 5-(α-benzoyl)benzyl-3-p-methoxybenzyluridine (
8c) (
Scheme 7). A mixture of
8c (100 mg, 0.179 mmol) and Pd/C (30.0 mg) was stirred at room temperature under an H
2 atmosphere. After 48 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated
in vacuo. The residue was purified by column chromatography on silica gel with CHCl
3/MeOH (100:1 to 50:1) to give 3-
p-methoxybenzyl-5-(1,2-diphenylethyl)uridine (
10, 43.9 mg, 45%) and 3-
p-methoxybenzyl-5-(2-hydroxy-1,2-diphenylethyl)uridine (
11, 34.1 mg, 34%).
3-p-Methoxybenzyl-5-(1,2-diphenylethyl)uridine (10). A colorless foam. 1H-NMR (DMSO-d6) 8.26 and 8.20 (each s, 1H, 6-H), 7.24–7.18 (m, 12H, C6H5 × 2 and o-PMB), 6.80 (d, J = 6.8 Hz, m-PMB), 5.81 (d, J = 4.1 Hz, 1H, 1'-H), 5.48–5.39 (m, 4H, OH × 2 and CH2), 5.12–5.08 (m, 1H, OH), 4.89 (s, 2H, CH2) 4.27–4.21 (m, 1H, 2'-H), 4.09–4.00 (m, 2H, CH and 3'-H), 3.93–3.90 (m, 1H, 4'-H), 3.69 (s, 3H, CH3), 3.40–3.35 (m, 2H, 5'-H); MS (EI) m/z 544 (M+, 2%), 453 (16), 321 (86), 121 (100), 91 (8); HRMS (EI) calcd. for C31H32N2O7 (M+): 544.2210; found: 544.2197.
3-p-Methoxybenzyl-5-(2-hydroxy-1,2-diphenylethyl)uridine (11). Colorless oil. 1H-NMR (DMSO-d6) 8.26 and 8.19 (each s, 1H, 6-H), 7.34–7.04 (m, 12H, C6H5 × 2 and o-PMB), 6.80 (d, J= 6.8 Hz, m-PMB), 5.81 (d, J= 3.9 Hz, 1H, 1'-H), 5.46 (brs, 1H, OH), 5.41 (brs, 1H, OH), 5.14–5.08 (m, 1H, OH), 4.82 (s, 2H, CH2), 4.27–4.21 (m, 1H, 2'-H), 4.09–4.03 (m, 2H, CH and 3'-H), 3.93–3.90 (m, 1H, 4'-H), 3.76–3.60 (m, 5H, CH and OH and CH3), 3.40–3.34 (m, 2H, 5'-H); MS (FAB, Gly) m/z 561 (M++H, 13%), 543 (7), 369 (5), 277 (14), 185 (100), 121 (33); HRMS (FAB, Gly) calcd. for C31H32N2O8 (M++H): 561.2159; found: 561.2228.
Pd/C(en)-catalyzed hydrogenation of 5-(α-Benzoyl)benzyl-3-p-methoxybenzyluridine (
8c) (
Scheme 7). A mixture of
8c (100 mg, 0.179 mmol) and 10% Pd/C(en) (30.0 mg) was stirred at room temperature under an H
2 atmosphere. After 48 h, the mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated
in vacuo. The residue was purified by column chromatography on silica gel with CHCl
3–MeOH (100:1 to 50:1) to give
11 (63.6 mg, 63%) as a colorless oil.
5-(1,2-Diphenylethyl)uridine (12). According to the procedure for the removal of the PMB group of 5-bis(ethoxycarbonyl)methyluridine (8a), 12 (61 mg, 79%) was obtained as a colorless foam. 1H-NMR (DMSO-d6) 11.23 (s, 1H, 3-NH), 8.14 and 8.10 (each s, 1H, 6-H), 7.24–7.18 (m, 10H, C6H5 × 2), 5.77 (d, J = 3.4 Hz, 1H, 1'-H), 5.43 (d, J = 6.2 Hz, 2H, CH2), 5.36 (brs, 1H, OH), 5.36 (brs, 1H, OH), 5.12 (brs, 1H, OH), 4.24–4.10 (m, 3H, 2'-H and 3’-H and CH), 3.72–3.60 (m, 3H, 4'-H and 5'-H); MS (FAB, NBA) m/z 425 (M++H, 21%), 329 (8), 154 (100), 136 (69), 89 (21); HRMS (FAB, NBA) calcd. for C23H24N2O6 (M++H): 425.1634; found: 425.1705.
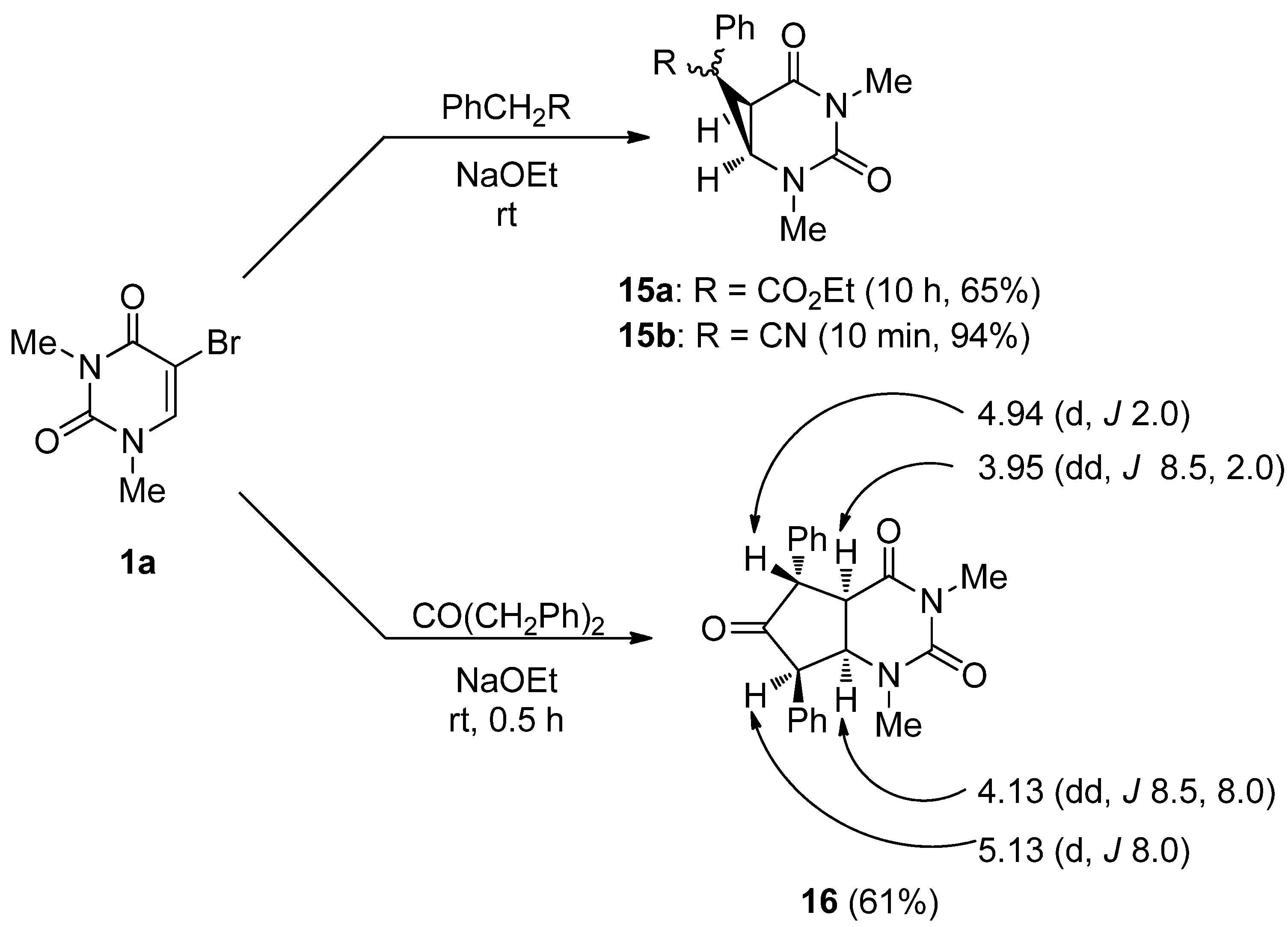
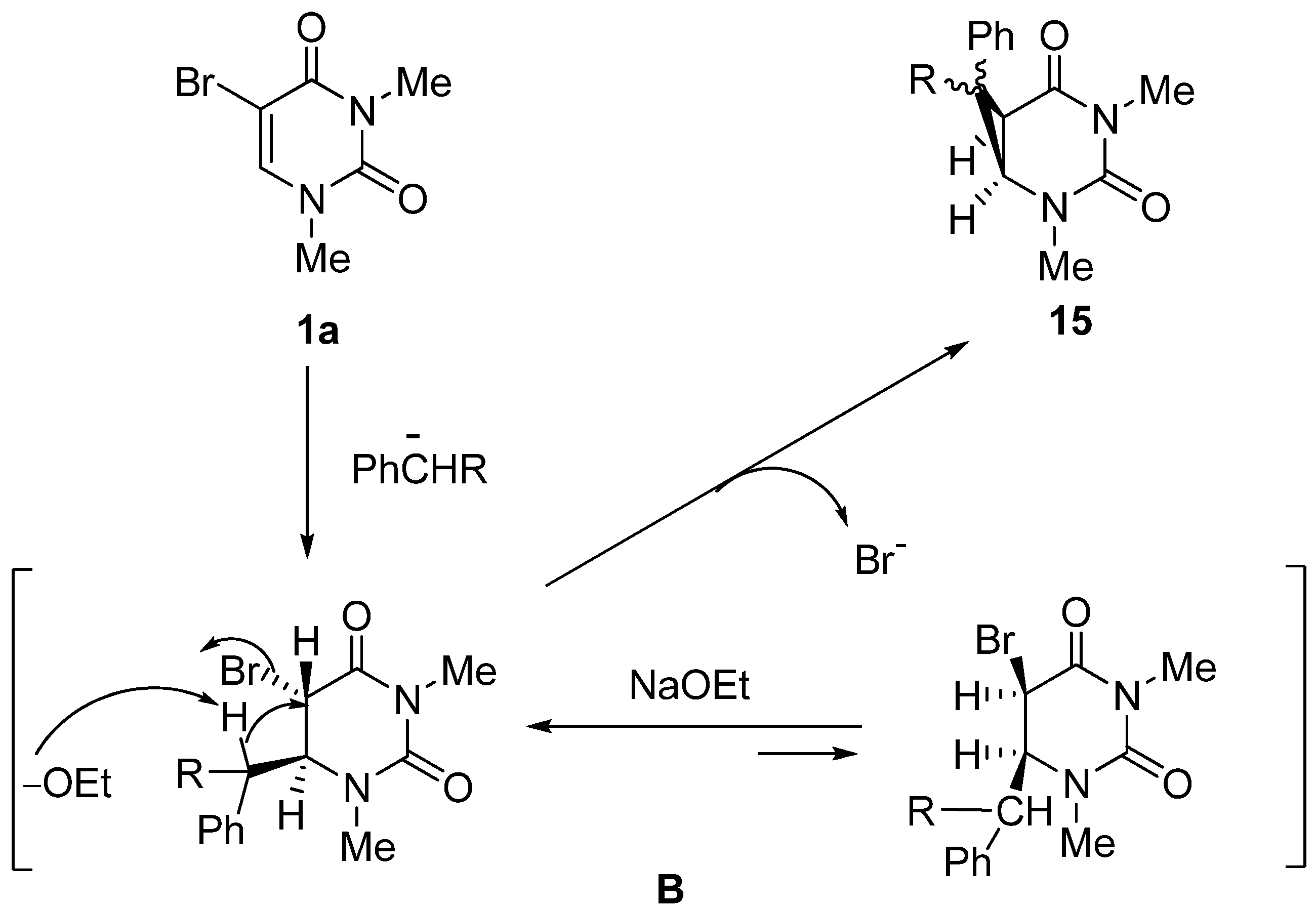
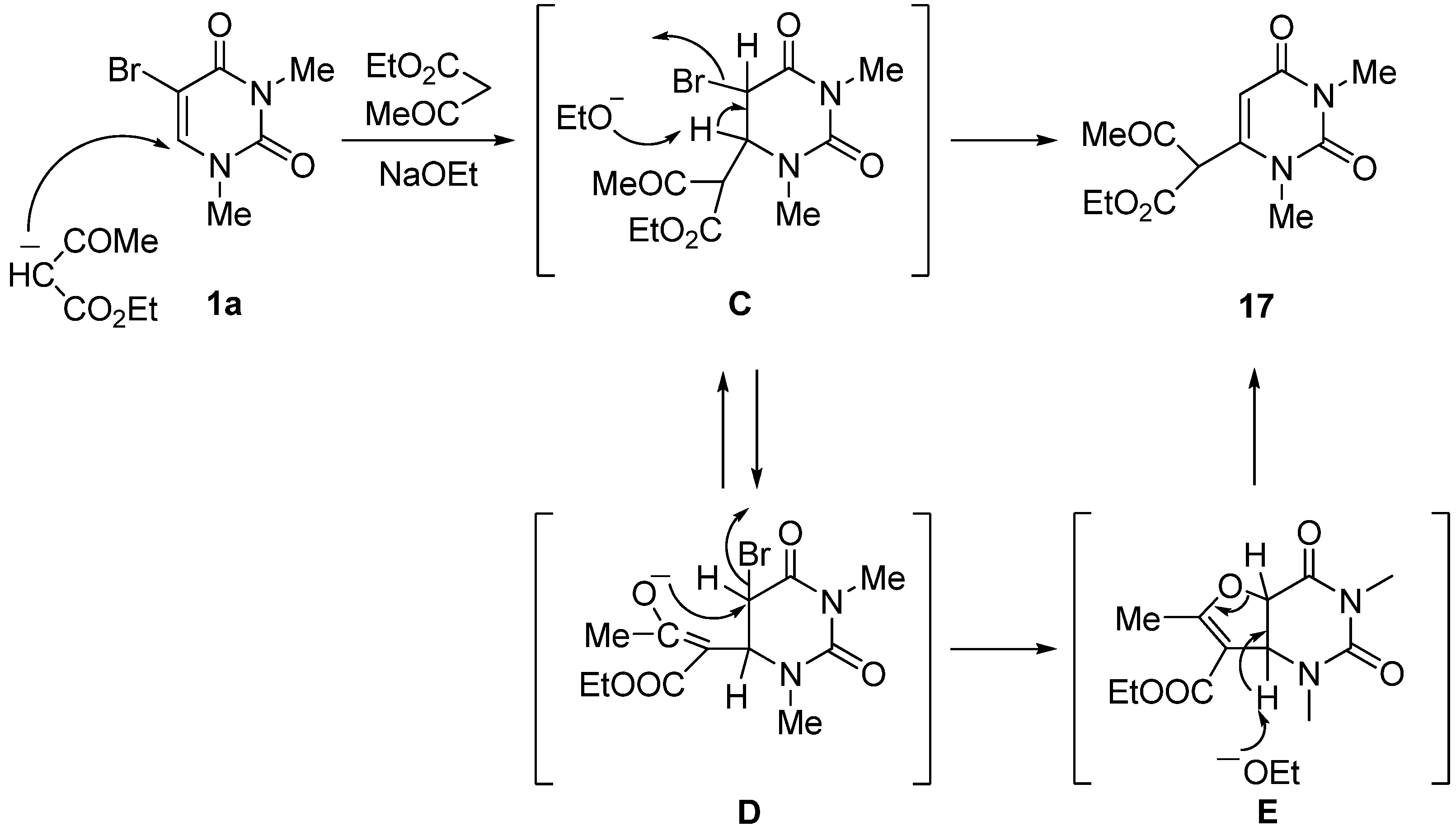
2,4-Dimethyl-7-ethoxycarbonyl-7-phenyl-2,4-diazabicyclo[4,1,0]heptane-3,5-dione (15a). (a) 5-Bromo-1,3-dimethyluracil (1a, 657 mg, 3.00 mmol) was added to a stirred solution of ethyl phenylacetate (1.63 g, 9.90 mmol) in ethanolic NaOEt [prepared from Na (207 mg, 9.00 mmol) in absolute EtOH (30 mL)] and the mixture was stirred at room temperature for 10 h. The solvent was removed under reduced pressure, and the residue was dissolved in H2O (20 mL). The solution was neutralized with c.HCl, and the mixture was extracted with CHCl3. The extract was dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3 as the eluant and recrystallized from Et2O to give 15a (589 mg, 65%). m.p. 123.5–125 °C; UV λmax. (EtOH) only end absorption; 1H-NMR (CDCl3) 7.48–6.98 (m, 5H, Ph), 4.15 (q, J = 7.0 Hz, 2H, CH2), 3.76 and 3.22 (each d, each J = 9.0 Hz, each 1H, 5 and 6-H), 3.25 and 2.76 (each s, each 3H, NMe), 1.18 (t, J = 7 Hz, 3H, CMe); 13C-NMR (CDCl3) 170.5, 165.6, 150.8, 130.7, 129.2, 128.8, 128.8, 62.3, 45.1, 36.3, 30.3, 27.2, 14.0; MS (EI) m/z 302 (M+, 13%), 303 (13), 256 (100), 228 (50), 227 (24), 199 (23) 143 (27); Anal calcd. for C16H18N2O4: C, 63.56; H, 6.00; N, 9.27%; found: C, 63.28; H, 6.00; N, 9.16; (b) 5-Bromo-1,3-dimethyluracil (1a) (657 mg, 3.00 mmol) was dissolved in a solution of ethyl phenylacetate (1.63 g, 9.90 mmol) and DBU (1.37 g, 9.00 mmol) in anhydrous DMF (30 mL). The mixture was stirred at room temperature for 4 days and then the solvent was removed under reduced pressure. The residue was treated as described above to give 15a (635 mg, 70%), which was identical to the product obtained above.
7-Cyano-1,4-dimethyl-7-phenyl-2,4-diazabicyclo[4,1,0]heptane-3,5-dione (15b). 5-Bromo-1,3-dimethyluracil (1a, 657 mg, 3.00 mmol) was added to a stirred solution of benzylcyanide (1.16 g, 9.90 mmol) in ethanolic NaOEt [prepared from Na (207 mg, 9.00 mmol) in absolute EtOH (30 mL)] and the mixture was stirred at room temperature for 10 min. The mixture was neutralized with Amberlite CG-50 (H+) and filtered. The ion exchanger was washed with EtOH. The combined filtrates were concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel with chloroform as the eluant to give 15b (720 mg, 94%), which was recrystallized from CHCl3–Et2O. m.p. 134–135 °C; UV λmax. (EtOH) only end absorption; νmax 2240 cm–1 (CN); 1H-NMR (CDCl3) 7.68–7.06 (5H, m, Ph), 3.57 (1H, d, J = 8.5 Hz, 6-H), 3.27 and 3.24 (each 3H, each s, NMe), 3.02 (d, J = 8.5 Hz, 1H, 5-H); 13C-NMR (CDCl3) 162.9, 150.7, 131.3, 129.5, 129.1, 125.9, 114.7, 46.5, 35.6, 31.4, 28.0, 25.0; MS (EI), m/z 255 (M+, 70%), 198 (36), 170 (100), 169 (85); Anal calcd. for C14H13N3O4: C, 65.87; H, 5.13; N, 16.46%; found: C, 65.88; H, 5.12; N, 16.51.
2,4-Dimethyl-7,9-diphenyl-2,4-diazabicyclo[4,3,0]nonane-3,5,8-trione (16). 5-Bromo-1,3-dimethyl-uracil (1a) (657 mg, 3.00 mmol) was added to a stirred solution of dibenzylketone (2.08 g, 9.90 mmol) in ethanolic NaOEt [prepared from Na (207 g, 9.00 mmol) in absolute EtOH (30 mL)] and the mixture was stirred at room temperature for 30 min. The mixture was neutralized with Amberlite CG-50 (H+) and filtered. The ion exchange resin was washed with EtOH. The combined filtrates were concentrated under reduced pressure, and the residue was purified by chromatography on silica gel with CHCl3 as the eluant to give 16 (638 mg, 61%), which was recrystallized from chloroform-ether. m.p. 220–221 °C; UV λmax. (EtOH) 266 nm (ε 25,100 dm3mol–1cm–1); m/z 348 (M+, 52%), 208 (28), 180 (100); 1H-NMR (CDCl3) 7.70–7.07 (m, 10H, Ph), 5.06 (d, J = 8.0 Hz, 1H, 4a or 7a-H), 4.93 (d, J = 2.0 Hz, 1H, 5 or 7-H), 4.13 (dd, J = 8.5 Hz and 8.0 Hz, 1H, 4a or 7a-H), 3.95 (dd, J = 8.5, 2.0 Hz, 1H, 5 or 7-H), 2.73 and 3.29 (each s, each 3H, NMe); 13C-NMR (CDCl3) 166.4, 155.7, 151.2, 137.5, 134.7, 129.3, 128.6, 128.3, 128.3, 128.1, 126.1, 103.1, 73.3, 64.0, 54.4, 35.8, 28.2; Anal calcd. for C21H20N2O3: C, 72.39; H, 5.79; N, 8.04%; found: C, 72.14; H, 5.87; N, 8.01.
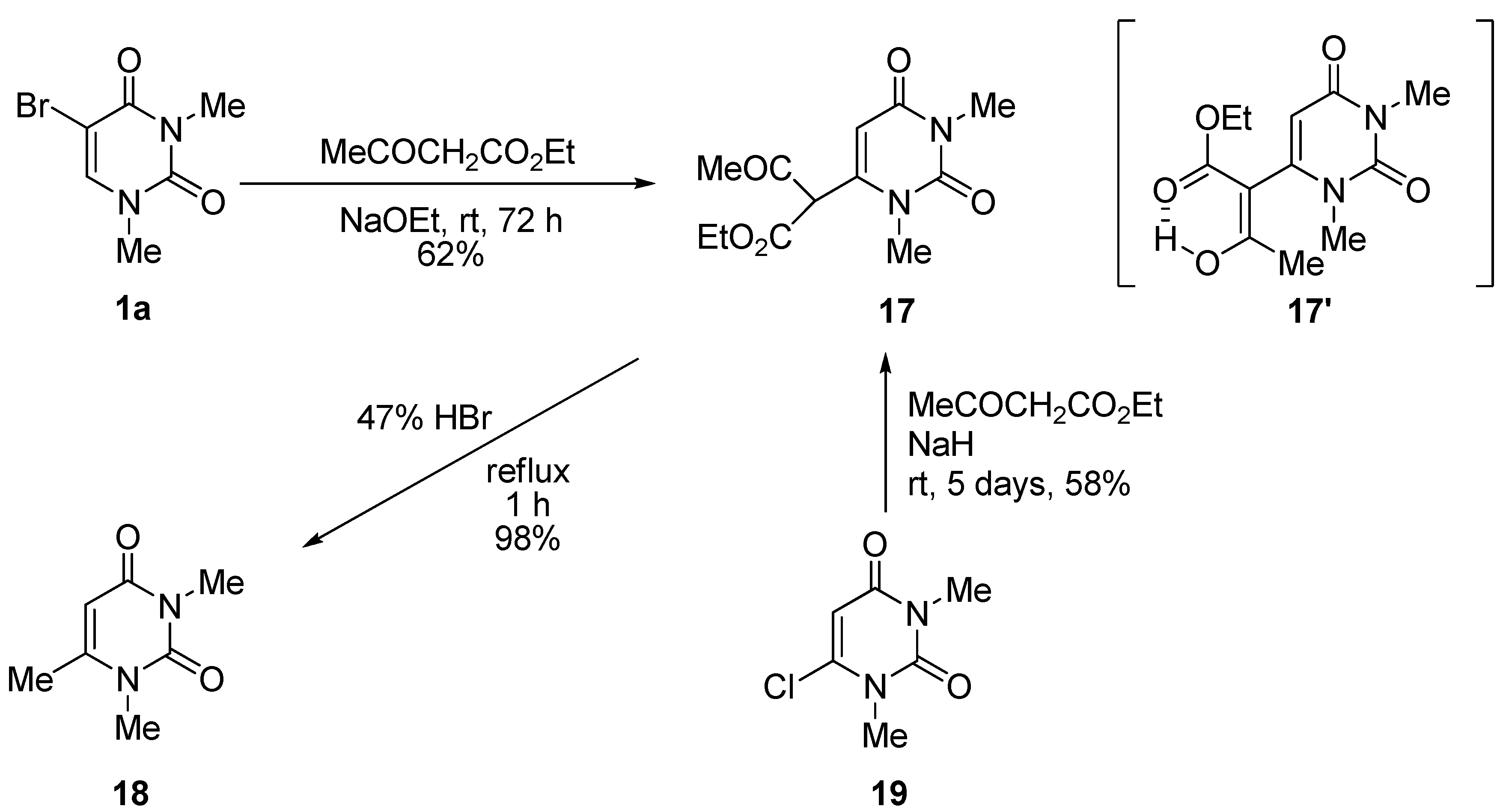
1,3-Dimethyluracil-6-(α-acetyl)acetic acid ethyl ester (17). (a) A solution of 5-bromo-1,3-dimethyluracil (1a, 2.20 g, 10.0 mmol) and ethyl acetoacetate (4.32 g, 33.0 mmol) in ethanolic NaOEt [prepared from Na (690 mg, 30.0 mmol) in absolute EtOH (100 mL)] was stirred for 3 days at room temperature. The mixture was evaporated under reduced pressure, and the residue was treated with H2O. The resulting precipitate was filtered off, and the mother liquor was extracted with CHCl3. The extract was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was treated with Et2O and the resulting precipitate was filtered off. The combined precipitate was washed with Et2O to afford the recovered (1a) (700 mg, 32%), which was identical to the authentic sample. The water layer was neutralized with c.HCl, and the mixture was extracted with CHCl3. The extract was concentrated in vacuo, and the residue was purified by column chromatography on silica gel with CHCl3 as the eluant to give 17 (1.66 g, 62%). m.p. 100–103 °C; UV λmax(EtOH) 268 nm (ε 12,400 dm3mol–1cm–1); 1H-NMR (CDCl3) 13.10 (s, CH, 1H, deuterium exchangeable), 5.68 (s, 1H, 5-H), 4.32 (q, J = 7.5 Hz, 2H, CH2), 3.37 and 3.26 (each s, each 3H, NMe), 2.04 (s, 3H, CMe), 1.28 (t, J = 7.5 Hz, 3H, CMe); MS (EI) m/z 268 (M+, 74%), 222 (32), 207 (54), 82 (32), 43 (100); Anal. calcd. for C12H16N2O5: C, 53.72; H, 6.01; N, 10.44%; found: C, 53.48; H, 6.12; N, 10.52; (b) To a stirred solution of 6-chloro-1,3-dimethyluracil (19) (349 mg, 2.00 mmol) and ethyl acetoacetate (859 mg, 6.60 mmol) in anhydrous DMF (5 mL) was added sodium hydride (60% in mineral oil) (240 mg, 6.00 mmol). The mixture was stirred at room temperature for 5 days, and the solvent was removed under reduced pressure. The residue was dissolved in H2O (20 mL) and then washed with CHCl3. The aqueous layer was neutralized with conc. HCl and extracted with CHCl3. The extract was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl3 as the eluant to give 17 (311 mg, 58%), which was identical to the sample prepared above.
1,3,6-Trimethyluracil (18) (CAS: 13509-52-9). A mixture of the 1,3-dimethyluracil-6-(α-acetyl)acetic acid ethyl ester (2, 1.32 g, 4.92 mmol) and hydrobromic acid (47%) was refluxed for 1 h. The solvent was removed under reduced pressure, and the residue was treated with H2O (30 mL). The suspension was extracted with CHCl3, and the extract was dried over Na2SO4. The solvent was removed under reduced pressure to give 18 (743 mg, 98%), which was identical to the authentic sample.
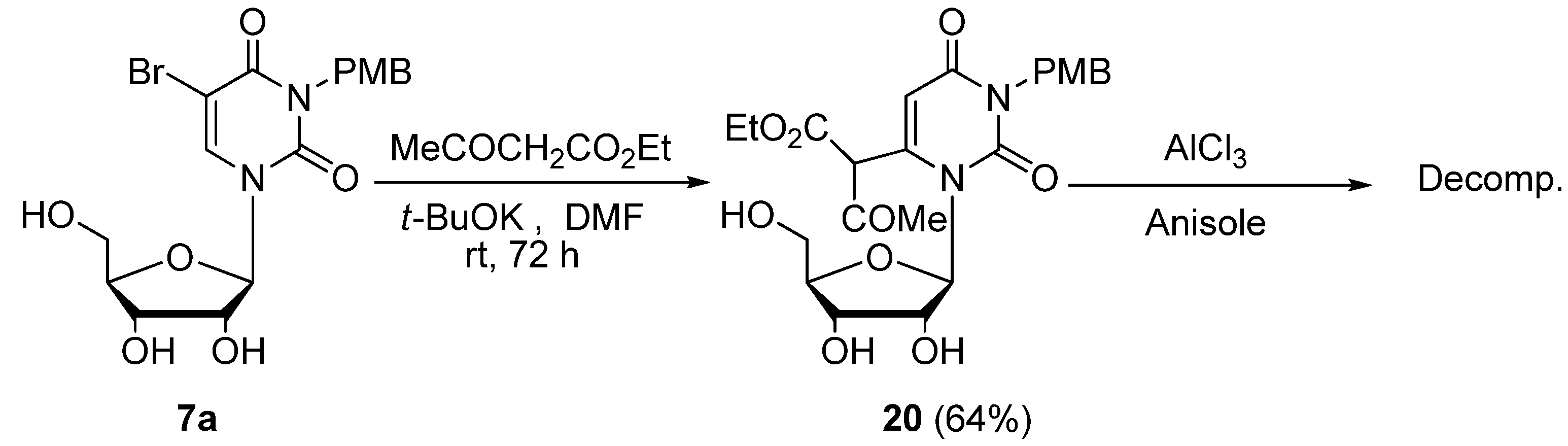
3-p-Methoxybenzyluridine-6-(α-acetyl)acetic acid ethyl ester (20). To a stirred solution of 5-bromo-3-p-methoxybenzyluridine (7a, 1.33 g, 3.00 mmol) and ethyl acetoacetate (1.29 g, 9.90 mmol) in anhydrous DMF (30 mL) was added potassium t-butoxide (1.01 g, 9.00 mmol). The mixture was stirred at room temperature for 3 days, and the solvent was removed under reduced pressure. The residue was dissolved in H2O (10 mL) and then washed with CHCl3. The aqueous layer was neutralized with concentrated NaHSO4 and extracted with CHCl3. The extract was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (150:1) as the eluant to give 20 as a light brown foam (946 mg, 64%). 1H-NMR (CDCl3) 13.19 (s, 1H, CH), 7.43 and 6.84 (each d, each J = 7.8 Hz, each 2H, C6H4), 5.67 (d, 1H, J = 3.4 Hz, 5-H), 5.39 (d, J = 5.4 Hz, 1H, 1′-H), 5.37–5.00 (m, 4H, OH × 2, CH2), 4.90 (brd, J = 3.9 Hz, 1H, OH), 4.21–4.16 (m, 3H, 2'-H, CH2), 3.88 (m, 1H, 3'-H), 3.78 (s, 3H, CH3), 3.72 (m, 1H, 4'-H), 3.48 (m, 2H, 5'-H), 2.07 and 2.00 (each s, total 3H, CH3), 1.22 (brt, J = 7.1 Hz, 3H, CH3). MS (EI) m/z 492 (M+, 8%), 360 (32), 314 (18), 162 (15), 121 (100). HRMS (EI) calcd. for C23H28N2O10 (M+): 492.1744; found: 492.1756.
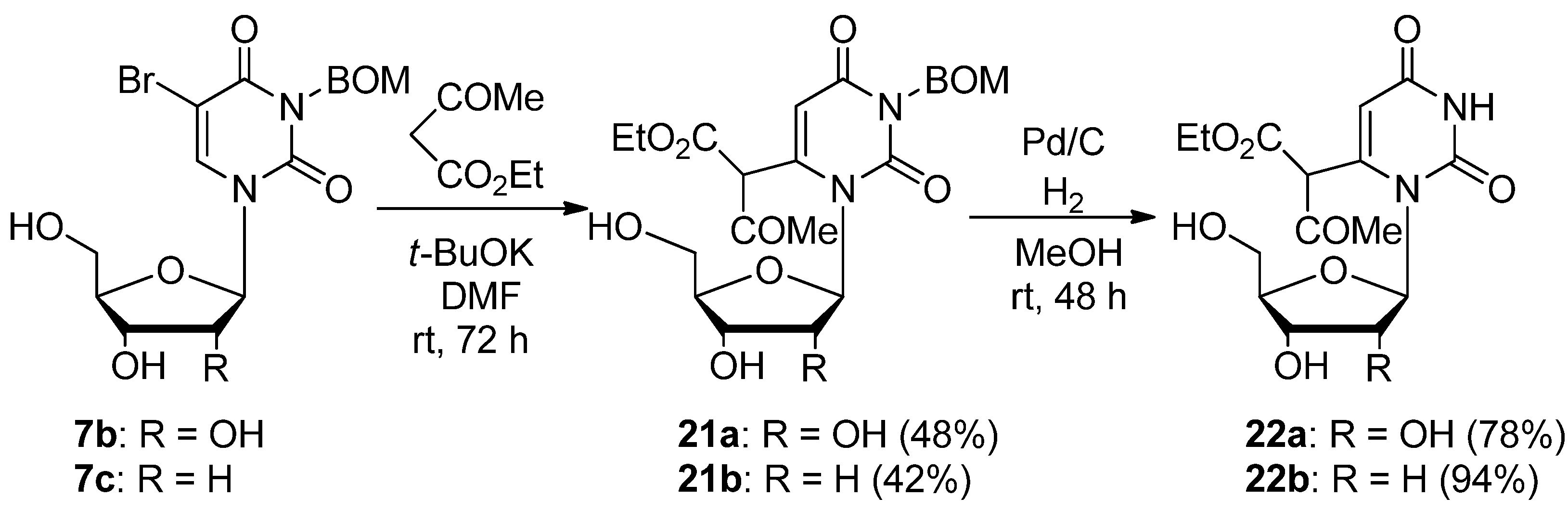
3-Benzyloxymethyluridine-6-(α-acetyl)acetic acid ethyl ester (21a). To a stirred solution of 5-bromo-3-benzyloxymethyluridine (7b, 452 mg, 1.02 mmol) and ethyl acetoacetate (586 mg, 4.50 mmol) in anhydrous DMF (30 mL) was added potassium t-butoxide (337 mg 3.00 mmol). The mixture was stirred at room temperature for 3 days and the solvent was removed under reduced pressure. The residue was dissolved in H2O (10 mL) and then washed with CHCl3. The aqueous layer was neutralized with concentrated NaHSO4 and extracted with CHCl3. The extract was dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (200:1) as the eluant to give 21a (242 mg, 48%) as a colorless oil. 1H-NMR (CDCl3) 12.91 (s, 1H, CH), 7. 31 (m, 5H, C6H5), 5.75 (d, 1H, J = 3.1 Hz, 5-H), 5.54 (d, J = 5.9 Hz, 1H, 1'-H), 5.30 (s, 2H, CH2), 5.22 (brd, J = 4.4 Hz, 1H, OH), 5.10–5.07 (m, 1H, OH), 4.99–4.96 (m, 1H, OH), 4.61 (s, 2H, CH2), 4.21–4.15 (m, 3H, 2’-H and CH2), 4.02-3.99 (m, 1H, 3'-H), 3.62–3.58 (m, 1H, 4'-H), 3.49–3.39 (m, 2H, 5'-H), 2.01 and 1.97 (each s, total 3H, CH3), 1.17 (brt, J = 7.1 Hz, 3H, CH3); 13C-NMR (CDCl3) 177.3, 169.7, 161.9, 151.7, 128.4, 127.8, 127.5, 106.5, 106.2, 97.3, 94.7, 93.6, 84.2, 72.6, 72.5, 70.5, 68.9, 62.4, 61.7, 20.18, 14.0; MS (FAB, NBA) m/z 493 (M++H, 12%), 361 (10), 331 (11), 154 (100), 91 (41); HRMS (FAB, NBA) calcd. for C23H28N2O10 (M++H) 493.18218; found: 493.18141.
3-Benzyloxymethyl-2'-deoxyuridine-6-(α-acetyl)acetic acid ethyl ester (21b). To a stirred solution of 5-bromo-3-p-methoxybenzyl-2'-deoxyuridine (7c, 297 mg, 0.700 mmol) and ethyl acetoacetate (0.290 mL, 2.32 mmol) in anhydrous DMF (5 mL) was added potassium t-butoxide (237 mg, 2.11 mmol). The mixture was stirred at room temperature for 3 days, and the solvent was removed under reduced pressure. The residue was dissolved in H2O (10 mL) and then washed with CHCl3. The aqueous layer was neutralized with concentrated NaHSO4 and extracted with CHCl3. The extract was dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel with CHCl3–MeOH (200:1) as the eluant to give 21b (141 mg, 42%) as a colorless oil. 1H-NMR (CDCl3) 13.09 (s, 1H, CH), 7.39–7.29 (m, 5H, C6H5), 5.82–5.76 (m, 1H, 5-H), 5.63–5.60 (m, 1H, 1'-H), 5.43 (s, 2H, CH2), 5.38 (brs, 1H, OH), 4.96 (brs, 1H, OH), 4.74 (s, 2H, CH2), 4.27 (q, J = 7.1 Hz, 2H, CH3), 3.92–3.86 (m, 1H, 3'-H), 3.80–3.74 (m, 1H, 4'-H), 3.62–3.40 (m, 2H, 5'-H), 2.08–1.96 (m, 5H, CH3 and 2'-H), 1.29 (brt, J = 7.0 Hz, 3H, CH3); MS (FAB, NBA) m/z 477 (M++H, 9%), 361 (29), 256 (6), 154 (100), 91 (31); HRMS (FAB, NBA) calcd. for C23H28N2O9 (M++H): 477.18728; found: 477.18841.
Uridine-6-(α-acetyl)acetic acid ethyl ester (22a). A mixture of 3-benzyloxymethyluridine-6-(α-acetyl)acetic acid ethyl ester (21a, 100 mg, 0.203 mmol) and Pd/C (30.0 mg) in MeOH (1 mL) was stirred under H2 atmosphere at room temperature for 24 h. The mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (40:1) as the eluant to give uridine-6-acetoacetic acid ethyl ester (22a, 59.0 mg, 78%) as a light brown oil. 1H-NMR (CDCl3) 12.89 (s, 1H, CH), 11.42 (s, 1H, 3-NH), 5.66 (d, J = 2.2 Hz, 1H, 5-H), 5.64 (d, J = 5.9 Hz, 1H, 1'-H), 5.24–5.18 (m, 1H, OH), 5.12–5.07 (m, 1H, OH), 5.13–4.97 (m, 1H, OH), 4.17–4.11 (m, 1H, 2'-H), 4.10–3.57 (m, 6H, CH2, 3'-H, 4'-H and 5'-H), 2.02 and 1.98 (each s, total 3H, CH3), 1.18 (brt, J = 7.1 Hz, 3H, CH3); 13C-NMR (CDCl3) 176.5, 175.8, 169.5, 162.5, 105.8, 97.5, 93.8, 92.9, 84.4, 72.2, 70.1, 62.1, 61.2, 19.8, 13.7; MS (FAB, Gly) m/z 373 (M++H, 5%), 277 (10), 270 (33), 184 (100), 115 (57); HRMS (FAB, Gly) calcd. for C15H20N2O9 (M++H): 373.1169; found: 373.1251.
2'-Deoxyuridine-6-(α-acetyl)acetic acid ethyl ester (22b). A mixture of 3-benzyloxymethyl-2'-deoxyuridine-6-(α-acetyl)acetic acid ethyl ester (21b, 100 mg, 0.210 mmol) and Pd/C (30.0 mg) in MeOH (1 mL) was stirred under H2 atmosphere at room temperature for 48 h. The mixture was filtered using a membrane filter (Millex-LH, 0.45 μm), and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel with CHCl3–MeOH (50:1) as the eluant to give 22b (70.3 mg, 94%) as a colorless foam. 1H-NMR (CDCl3) 12.83 (s, 1H, CH), 11.35 (s, 1H, 3-NH), 5.73–5.70 (m, 1H, 5-H), 5.58–5.56 (m, 1H, 1'-H), 5.08–4.90 (m, 1H, OH), 4.54–4.48 (m, 1H, OH), 4.23–4.10 (m, 3H, CH2 and 3'-H), 3.61–3.44 (m, 3H, 4'-H and 5'-H), 2.02–1.88 (m, 3H, CH3), 1.23–1.06 (m, 3H, CH3); MS (FAB, NBA) m/z 356 (M++H, 9%), 241 (39), 195 (17), 154 (100); HRMS (FAB, NBA) calcd. for C15H20N2O8 (M++H): 357.12978; found: 357.13060.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















