3.1. General
All reagents and solvents were used as obtained from commercial suppliers without further purification. Monitoring of reactions was carried out using Merck 60 F254 silica gel, glass-supported TLC plates, and visualization with UV light (254 nm). NMR spectra were recorded on a JEOL JNM-AL400 spectrometer (1H at 400 MHz and 13C at 100 MHz) at room temperature. Chemical shifts were given in δ values (ppm), and following abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet and m = multiplet. IR spectra were recorded on a PerkinElmer Spectrum One using the attenuated total reflection (ATR) technology. Optical rotations were recorded on a JASCO Polarimeter P-1020. Melting points were recorded on a Yanako MP-J3 melting point apparatus without correction. Low-resolution mass spectra were recorded on a Shimadzu LCMS-2010EV instrument under electron spray ionization (ESI) conditions. Elemental analyses were obtained on a CE Instruments EA1110. [14C] Zn(CN)2 (5.0 Ci, 115 mCi/mmol) was purchased from American Radiolabeled Chemicals, Inc. Radio-TLC was scanned on a raytest Rita Star. Quantitation of radioactivity was recorded on an AB Sciex 4000QTrap MS instrument. The following abbreviations are used for reagents and solvents: TFA (trifluoroacetic acid), Boc2O (di-tert-butyl dicarbonate), DMF (N,N-dimethylformamide), EtOAc (ethyl acetate), THF (tetrahydrofuran), IPE (diisopropyl ether).
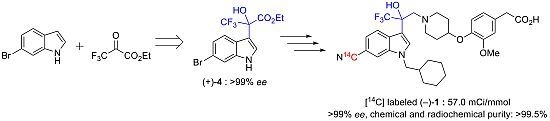
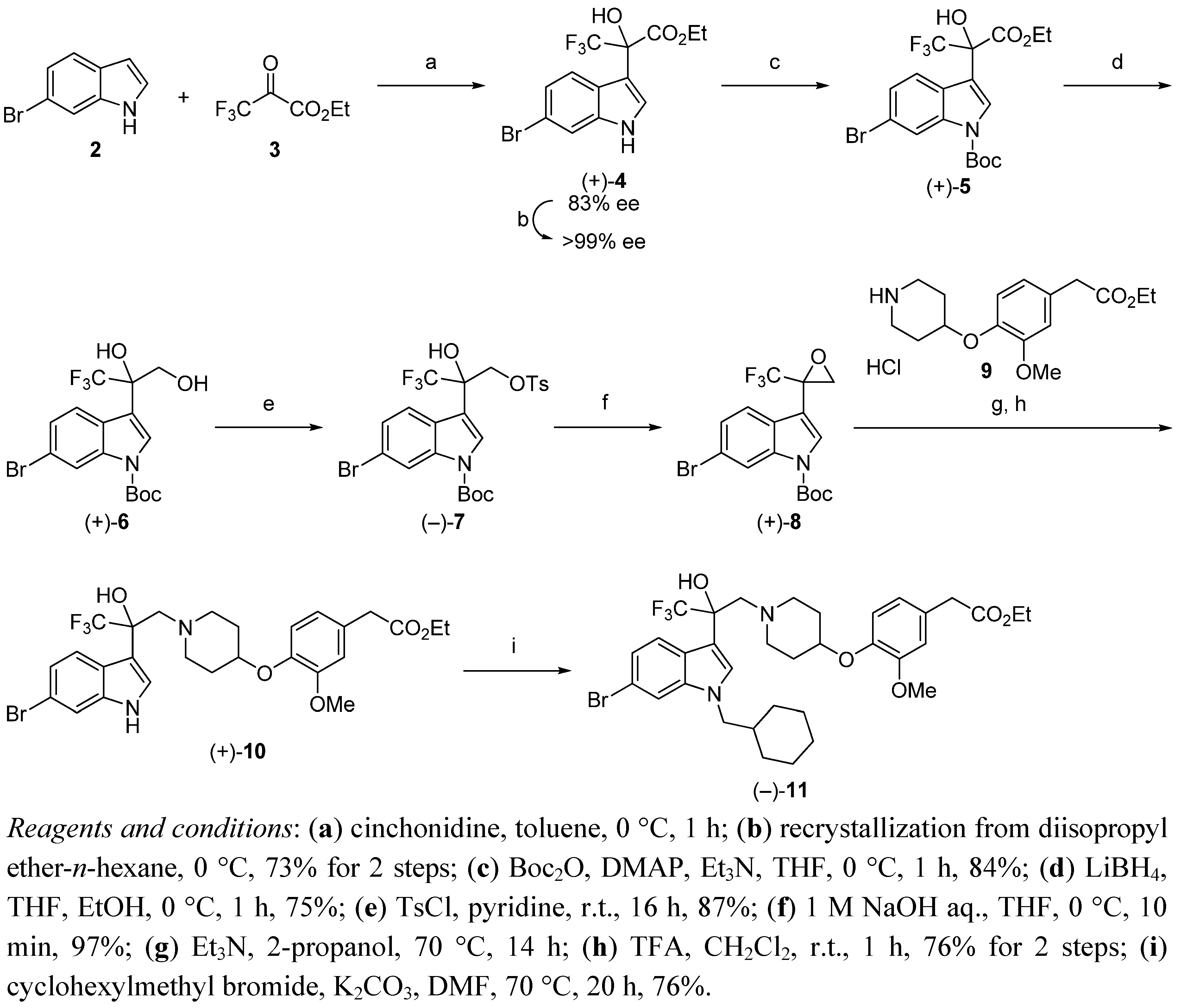
(+)-Ethyl 2-(6-bromo-1H-indol-3-yl)-3,3,3-trifluoro-2-hydroxypropanoate (+)-4. To a mixture of 6-bromoindole 2 (294 mg, 1.5 mmol) and cinchonidine (11.0 mg, 37.5 μmol) in dry toluene (7 mL) was added ethyl trifluoropyruvate 3 (298 μL, 2.25 mmol) in dry toluene (2 mL) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 1 h. The mixture was quenched with MeOH (20 mL) and DMF (2 mL) and stirred at room temperature over 30 min. The mixture was concentrated and the residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/2) to afford (+)-4 (495 mg, 90%, 83% ee) as a pale brown oil; Anal. Calcd for C13H11BrF3NO3: C, 42.65; H, 3.03; N, 3.83. Found: C, 42.87; H, 3.04; N, 3.93.; 1H-NMR (DMSO-d6) δ 11.49 (s, 1H, NH), 7.64 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 1.8 Hz, 1H), 7.51 (s, 1H, OH), 7.47 (d, J = 2.2 Hz, 1H), 7.17 (dd, J = 8.7, 1.8 Hz, 1H), 4.42–4.16 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6) δ 167.8, 137.3, 126.1, 124.2 (q, 1JC-F = 287 Hz), 124.0, 122.30, 122.26, 114.4, 114.3, 108.7, 76.6 (q, 2JC-F = 29.8 Hz), 62.1, 13.9; IR (ATR) ν 3469, 3391, 1729, 1615, 1540, 1455, 1390, 1370, 1335, 1300, 1256, 1275, 1222, 1171, 1137, 1110, 1095, 1074, 1006 cm−1; MS (ESI) m/z 388 (M+Na)+; Reaction progress was monitored by UFLC at room temperature using a Shimadzu LC-20AD pump equipped with a Shimadzu SPD-M20A detector and a Phenomenex Kinetex C18 100A column (3 mm × 75 mm, 2.6 μm), eluted at 0.8 mL/min with a 20 min gradient (from 10% B to 90% B), where solvent A is water (0.05% TFA solution) and solvent B is acetonitrile (0.05% TFA solution). Enantiomeric excess was measured by HPLC at room temperature using a Shimadzu LC-10AT pump equipped with a Shimadzu SPD-10A UV detector and a Chiralpak AD-H column (4.6 mm × 250 mm, 5 μm), eluted with EtOH/n-hexane = 10/90, flow rate 1.0 mL/min, λ = 254 nm, retention times: (+)-isomer 14.2 min, (−)-isomer 23.3 min.
10 g-Scale Synthesis of (+)-Ethyl 2-(6-bromo-1H-indol-3-yl)-3,3,3-trifluoro-2-hydroxypropanoate (+)-4. To a mixture of 6-bromoindole 2 (10.0 g, 51.0 mmol) and cinchonidine (375 mg, 1.28 mmol) in dry toluene (80 mL) was added ethyl trifluoropyruvate 3 (10.4 g, 61.2 mmol) in dry toluene (20 mL) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 1 h. The mixture was concentrated and the residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/2) to afford (+)-4 (83% ee) as a pale brown oil. The residue was triturated with IPE (20 mL) and n-hexane (60 mL) at 0 °C. The mixture was filtered and the filtrate was concentrated to give (+)-4 (13.6 g, 73%, 99% ee) as a pale brown oil; [α]D25 +12.4 (c 2.01, CHCl3).
(+)-tert-Butyl 6-bromo-3-(3-ethoxy-1,1,1-trifluoro-2-hydroxy-3-oxopropan-2-yl)-1H-indole-1-carboxylate (+)-5. To a mixture of (+)-4 (9.3 g, 25.4 mmol, 99% ee), triethylamine (4.24 ml, 30.5 mmol) and 4-dimethylaminopyridine (310 mg, 2.54 mmol) in dry THF (38 mL) was added Boc2O (6.1 g, 27.9 mmol) in dry THF (13 mL) dropwise at 0 °C. The mixture was stirred at 0 °C for 1 h. The reaction mixture was neutralized with 5% KHSO4 solution. The mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/5) to afford (+)-5 (9.9 g, 84%) as a pale brown solid; Anal. Calcd for C18H19BrF3NO5: C, 46.37; H, 4.11; N, 3.00. Found: 46.53; H, 4.04; N, 3.06; [α]D25 +15.2 (c 2.03, CHCl3); m.p. 54–55 °C; 1H-NMR (DMSO-d6) δ 8.27 (s, 1H), 7.97 (s, 1H, OH), 7.75 (s, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.46 (d, J = 8.5 Hz, 1H), 4.42–4.21 (m, 2H), 1.61 (s, 9H), 1.20 (t, J = 7.1 Hz, 3H); 13C-NMR (DMSO-d6) δ 166.9, 148.2, 135.7, 126.3, 126.02, 125.99, 123.7 (q, 1JC-F = 287 Hz), 123.1, 117.7, 117.5, 114.4, 85.2, 76.2 (q, 2JC-F = 30.1 Hz), 62.7, 27.4, 13.8; IR (ATR) ν 3437, 1738, 1603, 1555, 1455, 1433, 1368, 1290, 1253, 1239, 1220, 1152, 1126, 1088, 1009 cm−1; MS (ESI) m/z 488 (M+Na)+.
(+)-tert-Butyl 6-bromo-3-(1,1,1-trifluoro-2,3-dihydroxypropan-2-yl)-1H-indole-1-carboxylate (+)-6. To a mixture of LiBH4 (1.68 g, 77.2 mmol) in dry THF (72 mL) and EtOH (9 mL) was added (+)-5 (9.0 g, 19.3 mmol) in dry THF (18 mL) dropwise at 0 °C. The mixture was stirred at 0 °C for 1 h. The reaction mixture was quenched with 5% KHSO4 solution. The mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/2) to afford (+)-6 (6.16 g, 75%) as a white solid; Anal. Calcd for C16H17BrF3NO4: C, 45.30; H, 4.04; N, 3.30. Found: 45.41; H, 3.98; N, 3.34; [α]D25 +12.1 (c 2.00, CHCl3); m.p. 120–121 °C; 1H-NMR (DMSO-d6) δ 8.24 (d, J = 1.7 Hz, 1H), 7.83 (d, J = 8.6 Hz, 1H), 7.74 (s, 1H), 7.42 (dd, J = 8.6, 1.7 Hz, 1H), 6.67 (s, 1H, OH), 5.28 (t, J = 5.8 Hz, 1H, OH), 4.00 (dd, J = 11.5, 5.8 Hz, 1H), 3.89 (dd, J = 11.5, 5.8 Hz, 1H), 1.62 (s, 9H); 13C-NMR (DMSO-d6) δ 148.5, 135.6, 127.6, 125.70, 125.66 (q, 1JC-F = 288 Hz), 125.5, 124.0, 117.266, 117.258, 117.1, 84.9, 75.2 (q, 2JC-F = 27.3 Hz), 63.4, 27.5; IR (ATR) ν 3410, 3294, 1707, 1606, 1557, 1465, 1454, 1430, 1383, 1371, 1331, 1310, 1289, 1271, 1252, 1189, 1156, 1135, 1109, 1083 cm−1; MS (ESI) m/z 446 (M+Na)+.
(−)-tert-Butyl 6-bromo-3-(1,1,1-trifluoro-2-hydroxy-3-{[(4-methylphenyl)sulfonyl]oxy}propan-2-yl)-1H-indole-1-carboxylate (−)-7. To a mixture of (+)-6 (5.9 g, 13.9 mmol) in pyridine (47 mL) was added p-toluenesulfonyl chloride (13.3 g, 69.5 mmol) at room temperature. The mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated, CHCl3 was added to the residue and washed with 2 M HCl (×2). The organic layer was washed with water (×2) and brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/4) to afford (−)-7 (6.99 g, 87%) as a white solid; Anal. Calcd for C23H23BrF3NO6S: C, 47.76; H, 4.01; N, 2.42. Found: 47.86; H, 3.93; N, 2.48; [α]D25 −10.8 (c 2.08, CHCl3); m.p. 118–119 °C; 1H-NMR (DMSO-d6) δ 8.20 (d, J = 1.7 Hz, 1H), 7.66 (s, 1H), 7.64–7.54 (m, 3H), 7.52 (s, 1H, OH), 7.33 (dd, J = 8.7, 1.7 Hz, 1H), 7.28 (d, J = 8.3 Hz, 2H), 4.57 (d, J = 10.6 Hz, 1H), 4.47 (d, J = 10.6 Hz, 1H), 2.38 (s, 3H), 1.63 (s, 9H); 13C-NMR (DMSO-d6) δ 148.2, 145.1, 135.4, 131.3, 129.8, 127.5, 126.4, 125.8, 125.7, 124.5 (q, 1JC-F = 288 Hz), 123.1, 117.41, 117.36, 114.6, 85.2, 73.7 (q, 2JC-F = 29.2 Hz), 69.3, 27.5, 21.1; IR (ATR) ν 3404, 1735, 1660, 1555, 1454, 1436, 1364, 1337, 1285, 1252, 1232, 1189, 1171, 1134, 1094, 1054, 1027 cm−1; MS (ESI) m/z 602 (M+Na)+.
(+)-tert-Butyl 6-bromo-3-[2-(trifluoromethyl)oxiran-2-yl]-1H-indole-1-carboxylate (+)-8. To a mixture of (−)-7 (6.6 g, 11.4 mmol) in THF (57 mL) was added 1 M NaOH (11.4 mL) at 0 °C. The mixture was stirred at 0 °C for 10 min. The reaction mixture was neutralized with saturated NH4Cl solution. The mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4 and concentrated to afford (+)-8 (4.51 g, 97%) as a white solid; Anal. Calcd for C16H15BrF3NO3: C, 47.31; H, 3.72; N, 3.45. Found: 47.42; H, 3.66; N, 3.49; [α]D25 +13.4 (c 2.08, CHCl3); m.p. 81–82 °C; 1H-NMR (CDCl3) δ 8.26 (d, J = 1.8 Hz, 1H), 7.99 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.47 (dd, J = 8.5, 1.8 Hz, 1H), 3.57 (d, J = 4.5 Hz, 1H), 3.37–3.32 (m, 1H), 1.61 (s, 9H); 13C-NMR (CDCl3) δ 148.1, 135.1, 128.3, 127.0, 126.3, 123.4 (q, 1JC-F = 278 Hz), 121.6, 117.9, 117.7, 110.5, 85.3, 53.0 (q, 2JC-F = 38.1 Hz), 50.0, 27.4; IR (ATR) ν 1732, 1609, 1586, 1562, 1457, 1435, 1390, 1370, 1312, 1252, 1224, 1163, 1146, 1094, 1068, 1050 cm−1; MS (ESI) m/z 306 (M−Boc)+.
(+)-Ethyl [4-({1-[2-(6-bromo-1H-indol-3-yl)-3,3,3-trifluoro-2-hydroxypropyl]piperidin-4-yl}oxy)-3-methoxyphenyl]acetate (+)-10. To a mixture of (+)-8 (4.3 g, 10.6 mmol) and Et3N (3.68 mL, 26.5 mmol) in 2-propanol (35 mL) was added 9 (3.84 g, 11.6 mmol) at room temperature. The mixture was stirred at 70 °C for 14 h. After cooling to room temperature, the reaction mixture was neutralized with 5% KHSO4 solution. The mixture was extracted with EtOAc. The organic layer was washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. To a mixture of the residue in CH2Cl2 (17 mL) was added TFA (34 mL) at room temperature. The mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated, EtOAc was added to the residue and the solution washed with saturated NaHCO3 solution and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/1) to afford (+)-10 (4.81 g, 76%) as an amorphous solid; Anal. Calcd for C27H30BrF3N2O5: C, 54.10; H, 5.04; N, 4.67. Found: 54.26; H, 5.04; N, 4.73; [α]D25 +4.51 (c 2.09, CHCl3); m.p. 55–56 °C; 1H-NMR (400 MHz, DMSO-d6) δ 11.32 (d, J = 2.2 Hz, 1H, NH), 7.73 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 1.7 Hz, 1H), 7.46 (d, J = 2.7 Hz, 1H), 7.12 (dd, J = 8.5, 1.7 Hz, 1H), 6.98–6.79 (m, 2H), 6.70 (dd, J = 8.2, 1.8 Hz, 1H), 6.11 (s, 1H, OH), 4.20–4.09 (m, 1H), 4.05 (q, J = 7.1 Hz, 2H), 3.70 (s, 3H), 3.53 (s, 2H), 3.12 (d, J = 13.8 Hz, 1H), 3.00 (d, J = 13.8 Hz, 1H), 2.78–2.59 (m, 2H), 2.40–2.22 (m, 2H), 1.84–1.65 (m, 2H), 1.61–1.41 (m, 2H), 1.16 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 171.3, 150.0, 145.1, 137.3, 127.6, 126.2 (q, 1JC-F = 287 Hz), 125.4, 124.7, 122.9, 121.7, 121.3, 116.5, 114.1, 113.8, 112.6, 74.0 (q, 2JC-F = 27.0 Hz), 73.3, 60.1, 60.0, 55.6, 51.8, 51.7, 39.9, 30.6, 22.1, 21.9, 14.1; IR (ATR) ν 3336, 1723, 1611, 1590, 1541, 1509, 1453, 1420, 1369, 1334, 1265, 1226, 1149, 1101, 1032 cm−1; MS (ESI) m/z 599 (M+H)+.
(−)-Ethyl {4-[(1-{2-[6-bromo-1-(cyclohexylmethyl)-1H-indol-3-yl]-3,3,3-trifluoro-2-hydroxypropyl} piperidin-4-yl)oxy]-3-methoxyphenyl}acetate (−)-11. To a mixture of (+)-10 (4.12 g, 6.87 mmol), K2CO3 (5.70 g, 41.2 mmol) and KI (114 mg, 0.687 mmol) in DMF (69 mL) was added cyclohexylmethyl bromide (3.65 g, 20.6 mmol) at room temperature. The mixture was stirred at 70 °C for 20 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/3) to afford (−)-11 (3.61 g, 76%) as a pale brown oil; Anal. Calcd for C34H42BrF3N2O5·0.25H2O: C, 58.33; H, 6.12; N, 4.00. Found: 58.23; H, 6.01; N, 4.01; [α]D25 −4.65 (c 2.02, CHCl3); 1H-NMR (DMSO-d6) δ 7.81–7.67 (m, 2H), 7.43 (s, 1H), 7.13 (dd, J = 8.7, 1.8 Hz, 1H), 6.92–6.78 (m, 2H), 6.69 (dd, J = 8.2, 1.6 Hz, 1H), 6.13 (s, 1H, OH), 4.18–4.08 (m, 1H), 4.08–3.91 (m, 4H), 3.69 (s, 3H), 3.53 (s, 2H), 3.13 (d, J = 13.7 Hz, 1H), 2.94 (d, J = 13.7 Hz, 1H), 2.78–2.57 (m, 2H), 2.40–2.17 (m, 2H), 1.83–1.34 (m, 10H), 1.23–0.83 (m, 5H), 1.16 (t, J = 7.2 Hz, 3H); 13C-NMR (DMSO-d6) δ 171.2, 150.0, 145.1, 137.4, 129.5, 127.6, 126.1 (q, 1JC-F = 287 Hz), 125.0, 123.1, 121.7, 121.3, 116.6, 114.1, 113.8, 112.9, 101.4, 74.2 (q, 2JC-F = 27.3 Hz), 73.4, 60.1, 59.9, 55.5, 51.71, 51.65, 51.5, 39.9, 38.1, 30.7, 30.6, 29.93, 29.88, 25.9, 25.1, 14.0; IR (ATR) ν 3680, 1731, 1606, 1589, 1541, 1509, 1466, 1450, 1420, 1366, 1265, 1225, 1141, 1032 cm−1; MS (ESI) m/z 695 (M+H)+.
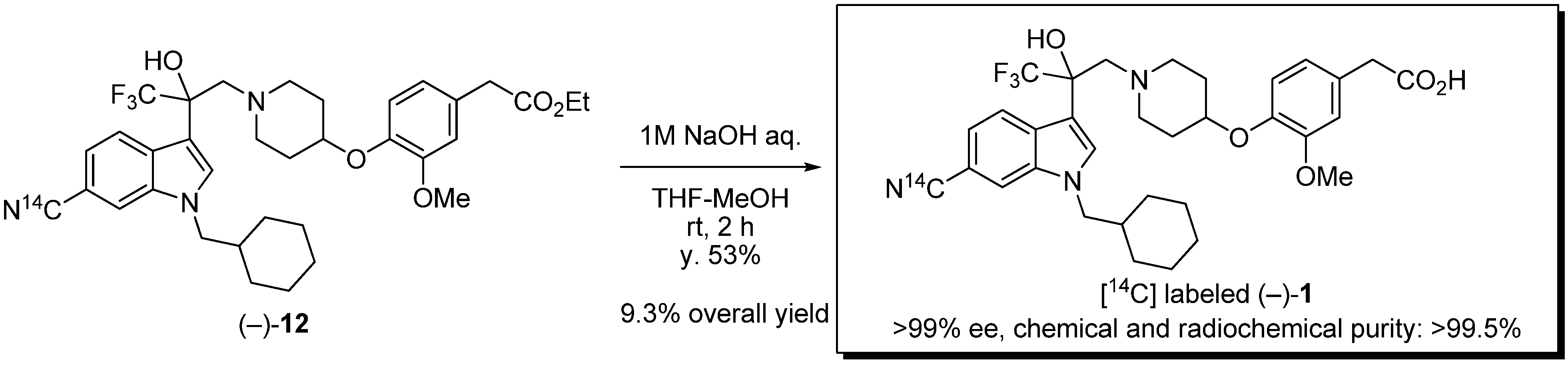
(−)-Ethyl {4-[(1-{2-[6-[14C]cyano-1-(cyclohexylmethyl)-1H-indol-3-yl]-3,3,3-trifluoro-2-hydroxy-propyl}piperidin-4-yl)oxy]-3-methoxyphenyl}acetate (−)-12. A mixture of (−)-11 (30 mg, 43.1 μmol), [14C] Zn(CN)2 (5.1 mg, 42.0 μmol, 4.83 mCi, 115 mCi/mmol) and Zn(CN)2 (5.2 mg, 44.3 μmol) in dry DMF (1.0 mL) was stirred at room temperature for 30 min under argon. Pd(t-Bu3P)2 (4.4 mg, 8.61 μmol) was added to the mixture which was stirred at 150 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with saturated NaHCO3 solution and EtOAc. The mixture was filtered through Celite. The filtrate was extracted with EtOAc (×2). The organic layer was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (EtOAc/n-hexane = 1/3) to afford (−)-12 (21.7 mg, 78%) as colorless oil. The data of unlabeled (−)-12; Anal. Calcd for C35H42BrF3N3O5: C, 65.51; H, 6.60; N, 6.55. Found: 65.43; H, 6.60; N, 6.47; [α]D25 −2.42 (c 2.06, CHCl3); 1H-NMR (DMSO-d6) δ 8.16 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.72 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H), 6.89–6.79 (m, 2H), 6.69 (dd, J = 8.4, 1.6 Hz, 1H), 6.29 (s, 1H, OH), 4.19–3.99 (m, 5H), 3.68 (s, 3H), 3.53 (s, 2H), 3.15 (d, J = 13.8 Hz, 1H), 2.94 (d, J = 13.8 Hz, 1H), 2.77–2.60 (m, 2H), 2.37–2.21 (m, 2H), 1.82–1.52 (m, 6H), 1.50–1.35 (m, 4H), 1.19–1.02 (m, 6H), 1.01–0.87 (m, 2H); 13C-NMR (DMSO-d6) δ 171.2, 150.0, 145.1, 135.4, 132.6, 129.1, 127.7, 126.1 (q, 1JC-F = 287.6 Hz), 122.5, 121.3, 120.5, 116.6, 115.6, 113.8, 111.9, 102.5, 74.5 (q, 2JC-F = 26.8 Hz), 73.4, 60.1, 59.9, 55.5, 51.8, 51.6, 39.8, 38.1, 30.7, 30.6, 29.9, 29.8, 25.9, 25.1, 14.; IR (ATR) ν 3680, 2220, 1731, 1615, 1589, 1541, 1509, 1471, 1451, 1420, 1388, 1367, 1332, 1264, 1226, 1139, 1097, 1033 cm−1; MS (ESI) m/z 642 (M+H)+.
(−)-{4-[(1-{2-[6-[14C]cyano-1-(cyclohexylmethyl)-1H-indol-3-yl]-3,3,3-trifluoro-2-hydroxypropyl}-piperidin-4-yl)oxy]-3-methoxyphenyl}acetic acid (−)-1. To a mixture of (−)-12 (21.7 mg, 33.7 μmol) in THF (0.34 mL) and MeOH (0.34 mL) was added 1 M NaOH (0.34 mL) at room temperature. The mixture was stirred at room temperature for 2 h. The reaction mixture was neutralized with saturated NH4Cl solution. The mixture was extracted with EtOAc. The organic layer was washed with brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (MeOH/CHCl3 = 1/20) to afford (−)-1 (11.0 mg, 53%, 57.0 mCi/mmol, >99% ee) as an amorphous solid. HPLC (retention time = 24.3 min) and radio-TLC (plate developed in 10% MeOH/CHCl3) showed the product to be more than 99.5% pure. Chemical and radiochemical purity was measured by HPLC at 40 °C using a Shimadzu LC-10AD pump equipped with a Shimadzu SPD-M20A UV detector (λ = 220 nm) and a PerkinElmer 150TR radioactivity detector with a Sumika Chemical Analysis Service Sumipax ODS A-212 column (6 mm × 150 mm, 5 μm), eluted at 1.0 mL/min with a 45 min gradient (from 20% B to 95% B), where solvent A is water (0.1% TFA solution) and solvent B is acetonitrile (0.1% TFA solution). Enantiomeric excess was measured by HPLC [Chiralcel OD-RH (4.6 mm × 150 mm, 5 μm), water (1% TFA solution)/acetonitrile (1% TFA solution) = 66/34, flow rate 1.0 mL/min, λ = 220 nm, retention times: (+)-isomer 30.8 min, (−)-isomer 33.3 min]. Co-injection of the radiolabeled product with an unlabeled standard of product (−)-1 produced a single peak.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
