Supramolecular Ring Structures of 7-Methylguanine: A Computational Study of Its Self-assembly and Anion Binding


Abstract
:
1. Introduction

2. Theoretical Methods
2.1. Computational Details
2.2. Bonding Analyses
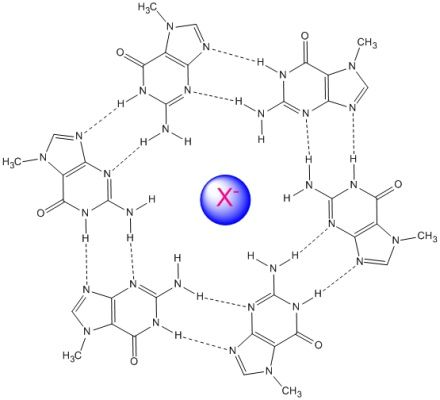
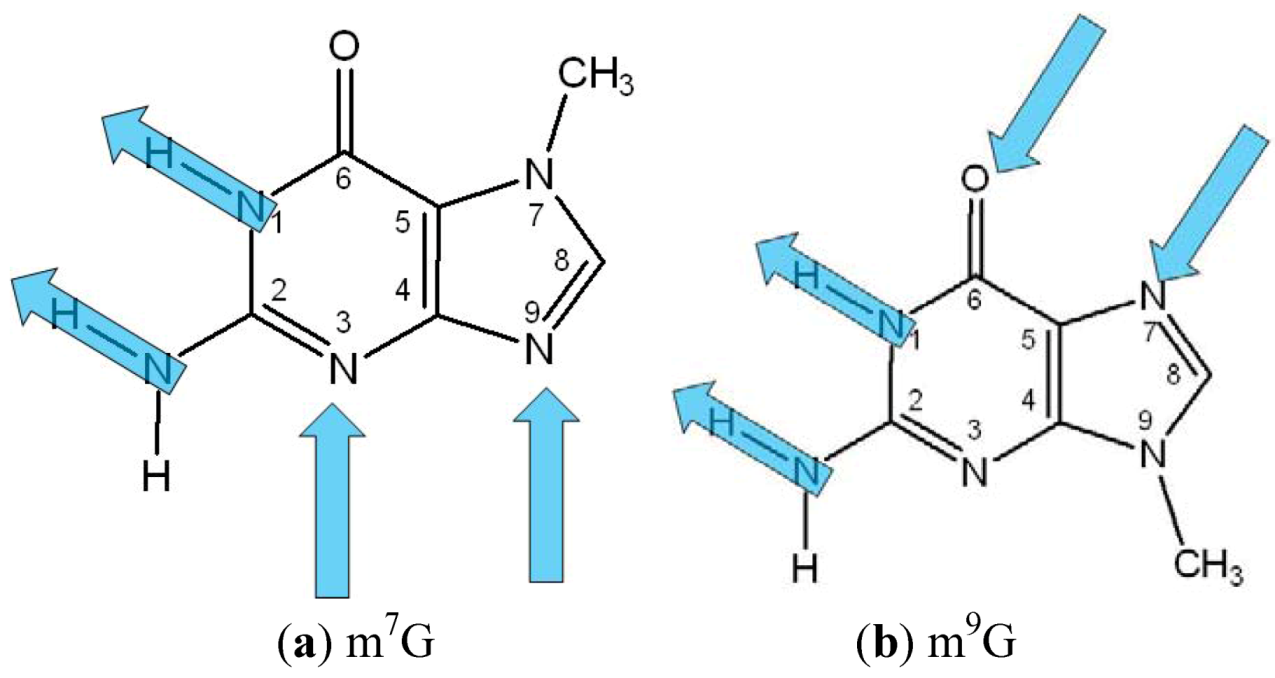
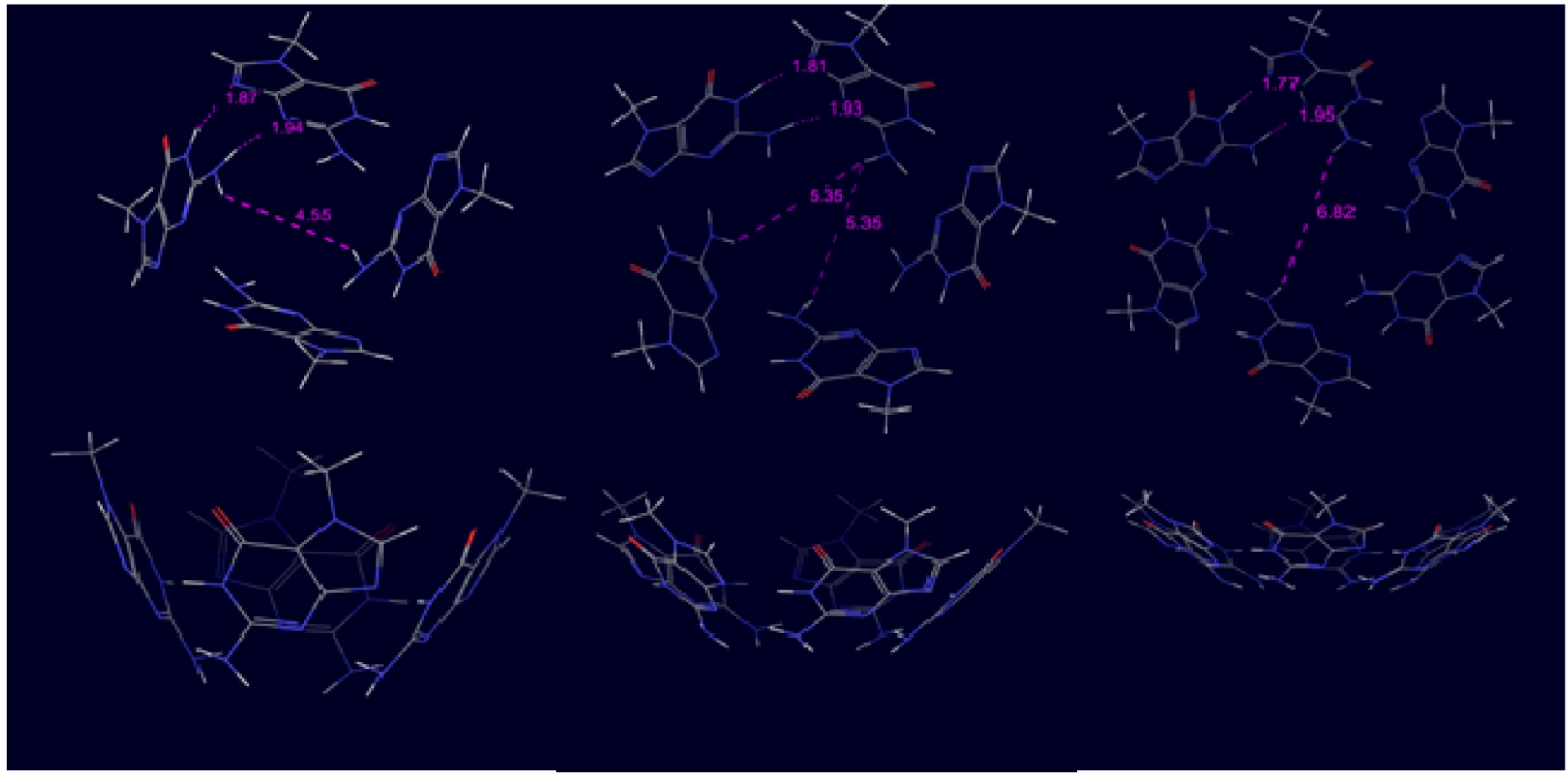
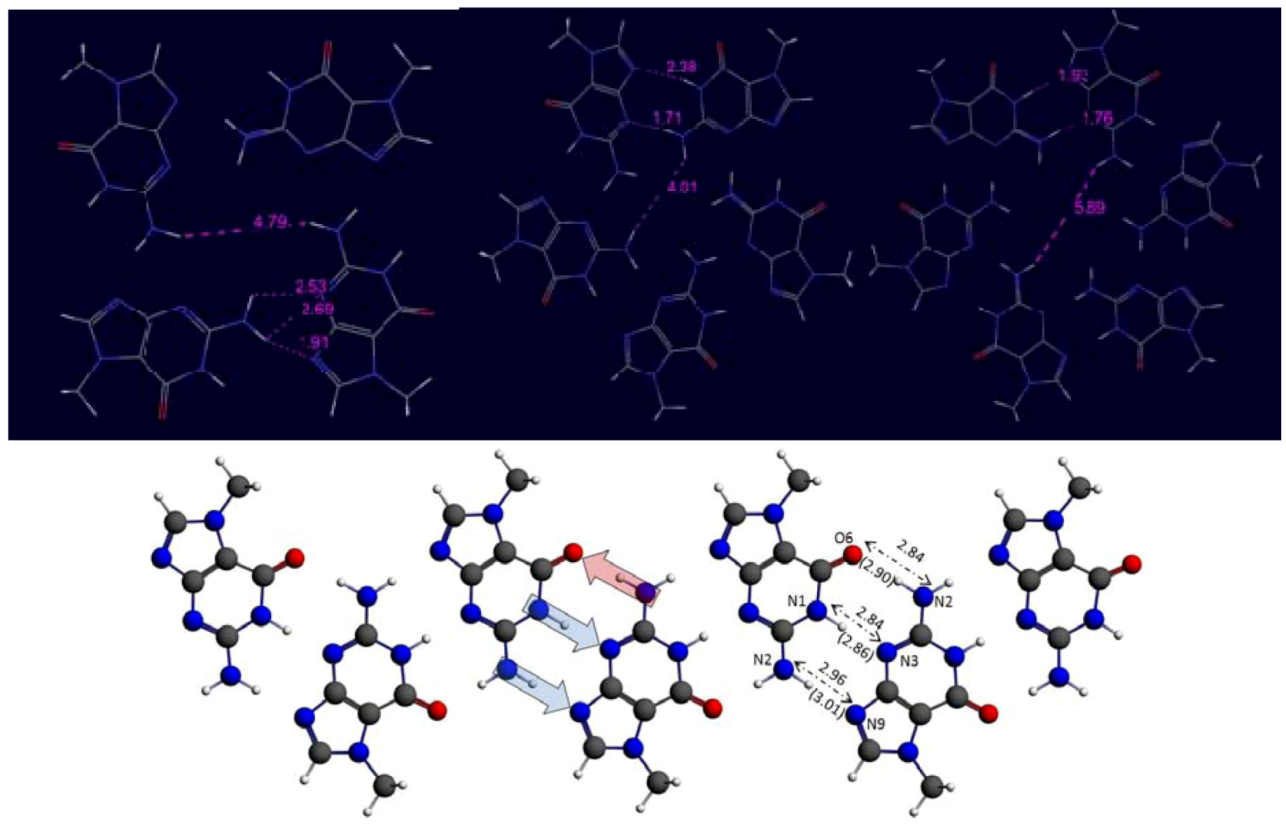
3. Results and Discussion
3.1. Systems without Ions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tetramer | Pentamer | Hexamer | Heptamer | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Energy (kcal/mol) | H-bonds | Energy (kcal/mol) | H-bonds | Energy (kcal/mol) | H-bonds | Energy (kcal/mol) | H-bonds | |||||
| Bonding | Average H-bond | Bonding | Average H-bond | Bonding | Average H-bond | Bonding | Average H-bond | |||||
| Ring without Cs sym. | −71.9 | −9.0 | 8 | −96.7 | −9.7 | 10 | −120.4 | −10.0 | 12 | - | ||
| Ring with Cs sym. | −53.3 | −6.7 | −76.6 | −7.7 | −114.0 | −9.5 | ||||||
| Ribbon with Cs sym. | −78.1 | −8.7 | 9 | −102.1 | −8.5 | 12 | −130.7 | −8.7 | 15 | −156.3 | −8.7 | 18 |

3.2. Hexameric Ring with Selected Anions

| Hexameric ring with anions | Cl− | Br− | NO3− | |||
|---|---|---|---|---|---|---|
| Bonding energy (in kcal/mol) | Ion bonding energy | Bonding energy (in kcal/mol) | Ion bonding energy | Bonding energy (in kcal/mol) | Ion bonding energy | |
| without Cs sym. | −170.8 | −50.3 | −168.3 | −47.6 | −177.0 | −56.3 |
| with Cs sym. | −170.3 | −50.1 | −167.7 | −47.6 | −174.3 | −54.1 |
4. Conclusions
Acknowledgments
References
- Steed, J.W.; Gale, P.A. Supramolecular Chemistry. From Molecules to Nanomaterials; John Wiley and Sons: Chichester, UK, 2012; Volumes 1–8. [Google Scholar]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; John Wiley and Sons: Chichester, UK, 2000; pp. 197–249. [Google Scholar]
- Caltagirone, C.; Hiscock, J.R.; Hursthouse, M.B.; Light, M.E.; Gale, P.A. 1,3-Diindolylureas and 1,3-diindolylthioureas: Anion complexation studies in solution and the solid state. Chem. Eur. J. 2008, 14, 10236–10243. [Google Scholar] [CrossRef]
- Davis, J.T.; Gale, P.A.; Okunola, O.A.; Prados, P.; Iglesias-Sánchez, J.C.; Torroba, T.; Quesada, R. Using small molecules to facilitate exchange of bicarbonate and chloride anions across liposomal membranes. Nat. Chem. 2009, 1, 138–144. [Google Scholar] [CrossRef]
- Fisher, M.G.; Gale, P.A.; Hiscock, J.R.; Hursthouse, M.B.; Light, M.E.; Schmidtchen, F.P.; Tong, C.C. 1,2,3-triazole-strapped calix[4]pyrrole: A new membrane transporter for chloride. Chem. Commun. 2009, 3017–3019. [Google Scholar]
- Busschaert, N.; Wenzel, M.; Light, M.E.; Iglesias-Hernández, P.; Pérez-Tomás, R.; Gale, P.A. Structure–activity relationships in tripodal transmembrane anion transporters: The effect of fluorination. J. Am. Chem. Soc. 2011, 133, 14136–14148. [Google Scholar]
- Busschaert, N.; Kirby, I.L.; Young, S.; Coles, S.J.; Horton, P.N.; Light, M.E.; Gale, P.A. Squaramides as potent transmembrane anion transporters. Angew. Chem. Int. Ed. 2012, 51, 4426–4430. [Google Scholar]
- Sowerby, S.J.; Petersen, G.B. Scanning tunnelling microscopy and molecular modelling of xanthine monolayers self-assembled at the solid-liquid interface: Relevance to the origin of life. Origins Life Evol. B 1999, 29, 597–614. [Google Scholar] [CrossRef]
- Otero, R.; Schöck, M.; Molina, L.M.; Lægsgaard, E.; Stensgaard, I.; Hammer, B.; Besenbacher, F. Guanine quartet networks stabilized by cooperative hydrogen bonds. Angew. Chem. Int. Ed. 2005, 44, 2270–2275. [Google Scholar] [CrossRef]
- Yu, M.; Wang, J.; Mura, M.; Meng, Q.-Q.; Xu, W.; Gersen, H.; Lægsgaard, E.; Stensgaard, I.; Kelly, R.E.A.; Kjems, J.; et al. Homochiral xanthine quintet networks self-assembled on Au(111) surfaces. ACS Nano 2011, 5, 6651–6660. [Google Scholar]
- Ciesielski, A.; Lena, S.; Masiero, S.; Spada, G.P.; Samori, P. Dynamers at the solid–liquid interface: controlling the reversible assembly/reassembly process between two highly ordered supramolecular guanine motifs. Angew. Chem. Int. Ed. 2010, 49, 1963–1966. [Google Scholar] [CrossRef]
- Davis, J.T.; Spada, G.P. Supramolecular architectures generated by self-assembly of guanosine derivatives. Chem. Soc. Rev. 2007, 36, 296–313. [Google Scholar] [CrossRef]
- Ferenc, G.; Pádár, P.; Szolomájer, J.; Kovács, L. N-Alkylated guanine derivatives. Curr. Org. Chem. 2009, 13, 1085–1135. [Google Scholar]
- Hudson, R.H.E.; Goncharenko, M.; Wallman, A.P.; Wojciechowski, F. PNA-directed triple-helix formation by N-7-xanthine. Synlett 2005, 1442–1446. [Google Scholar]
- Ferenc, G.; Pádár, P.; Szolomájer, J.; Howarth, N.M.; Kovács, L. Transition metal ion complexes of N-alkylguanines. Curr. Org. Chem. 2011, 15, 2871–2892. [Google Scholar] [CrossRef]
- Bald, I.; Wang, Y.-G.; Dong, M.; Rosen, C.B.; Ravnsbaek, J.B.; Zhuang, G.-L.; Gothelf, K.V.; Wang, J.-G.; Besenbacher, F. Control of self-assembled 2D nanostructures by methylation of guanine. Small 2011, 7, 939–949. [Google Scholar] [CrossRef]
- Guille, K.; Clegg, W. Anhydrous guanine: A synchrotron study. Acta Cryst. C 2006, 62, o515–o517. [Google Scholar] [CrossRef]
- Kozma, Á.; Ibáñez, S.; Silaghi-Dumitrescu, R.; Sanz Miguel, P.J.; Gupta, D.; Lippert, B. 7-Methylguanine: Protonation, formation of linkage isomers with trans-(NH3)2PtII, and base pairing properties. Dalton Trans. 2012, 41, 6094–6103. [Google Scholar] [CrossRef]
- Shi, X.D.; Mullaugh, K.M.; Fettinger, F.C.; Jiang, Y.; Hofstadler, S.A.; Davis, J.T. Lipophilic G-quadruplexes are self-assembled ion pair receptors, and the bound anion modulates the kinetic stability of these complexes. J. Am. Chem. Soc. 2003, 125, 10830–10841. [Google Scholar]
- Fonseca Guerra, C.; van der Wijst, T.; Poater, J.; Swart, M.; Bickelhaupt, F.M. Adenine versus guanine quartets in aqueous solution: Dispersion-corrected DFT study on the differences in pi-stacking and hydrogen-bonding behavior. Theor. Chem. Acc. 2010, 125, 245–252. [Google Scholar] [CrossRef]
- Meyer, M.; Sühnel, J. Self-association of isoguanine nucleobases and molecular recognition of alkaline ions: tetrad vs pentad structures. J. Phys. Chem. A 2003, 107, 1025–1031. [Google Scholar] [CrossRef]
- Gu, J.; Leszczynski, J. Isoguanine complexes: Quintet versus tetrad. J. Phys. Chem. B 2003, 107, 6609–6613. [Google Scholar]
- Pierce, S.E.; Wang, J.; Jayawickramarajah, J.; Hamilton, A.D.; Brodbelt, J.S. Examination of the effect of the annealing cation on higher order structures containing guanine or isoguanine repeats. Chem. Eur. J. 2009, 15, 11244–11255. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; et al. Computer code ADF2010.01. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 24 December 2012).
- Swart, M.; Bickelhaupt, F.M. Optimization of strong and weak coordinates. Int. J. Quantum Chem. 2006, 106, 2536–2544. [Google Scholar] [CrossRef]
- Swart, M.; Bickelhaupt, F.M. QUILD: Quantum-regions interconnected by local descriptions. J. Comput. Chem. 2008, 29, 724–734. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- van der Wijst, T.; Fonseca Guerra, C.; Swart, M.; Bickelhaupt, F.M.; Lippert, B. A ditopic ion-pair receptor based on stacked nucleobase quartets. Angew. Chem. Int. Ed. 2009, 48, 3285–3287. [Google Scholar]
- Fonseca Guerra, C.; Zijlstra, H.; Paragi, G.; Bickelhaupt, F.M. Telomere structure and stability: Covalency in hydrogen bonds, not resonance assistance, causes cooperativity in guanine quartets. Chem. Eur. J. 2011, 17, 12612–12622. [Google Scholar]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree-Fock-Slater method. Theoret. Chim. Acta 1977, 46, 1–10. [Google Scholar]
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3 and CNCH3 as σ donors and π acceptors. A theoretical study by Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar]
- Xantheas, S.S. Cooperativity and hydrogen bonding network in water clusters. Chem. Phys. 2000, 258, 225–231. [Google Scholar] [CrossRef]
- Sample Availability: Sample of 7-methylguanine is available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paragi, G.; Kupihár, Z.; Guerra, C.F.; Bickelhaupt, F.M.; Kovács, L. Supramolecular Ring Structures of 7-Methylguanine: A Computational Study of Its Self-assembly and Anion Binding. Molecules 2013, 18, 225-235. https://doi.org/10.3390/molecules18010225
Paragi G, Kupihár Z, Guerra CF, Bickelhaupt FM, Kovács L. Supramolecular Ring Structures of 7-Methylguanine: A Computational Study of Its Self-assembly and Anion Binding. Molecules. 2013; 18(1):225-235. https://doi.org/10.3390/molecules18010225
Chicago/Turabian StyleParagi, Gábor, Zoltán Kupihár, Célia Fonseca Guerra, F. Matthias Bickelhaupt, and Lajos Kovács. 2013. "Supramolecular Ring Structures of 7-Methylguanine: A Computational Study of Its Self-assembly and Anion Binding" Molecules 18, no. 1: 225-235. https://doi.org/10.3390/molecules18010225
APA StyleParagi, G., Kupihár, Z., Guerra, C. F., Bickelhaupt, F. M., & Kovács, L. (2013). Supramolecular Ring Structures of 7-Methylguanine: A Computational Study of Its Self-assembly and Anion Binding. Molecules, 18(1), 225-235. https://doi.org/10.3390/molecules18010225