Anti-Infectious Agents against MRSA

Abstract
:1. Introduction

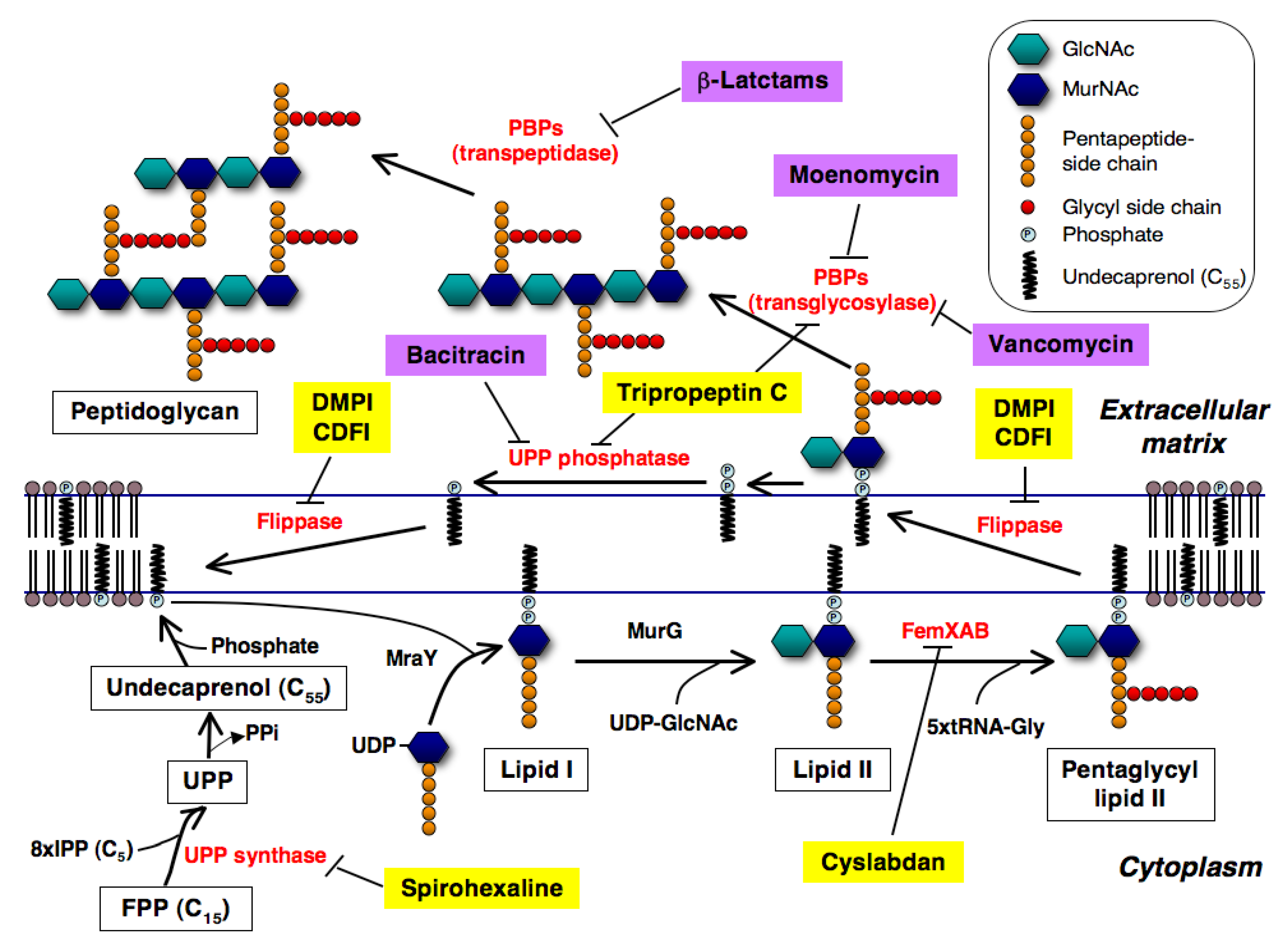
2. New Inhibitors of Bacterial Cell Wall Peptidoglycan
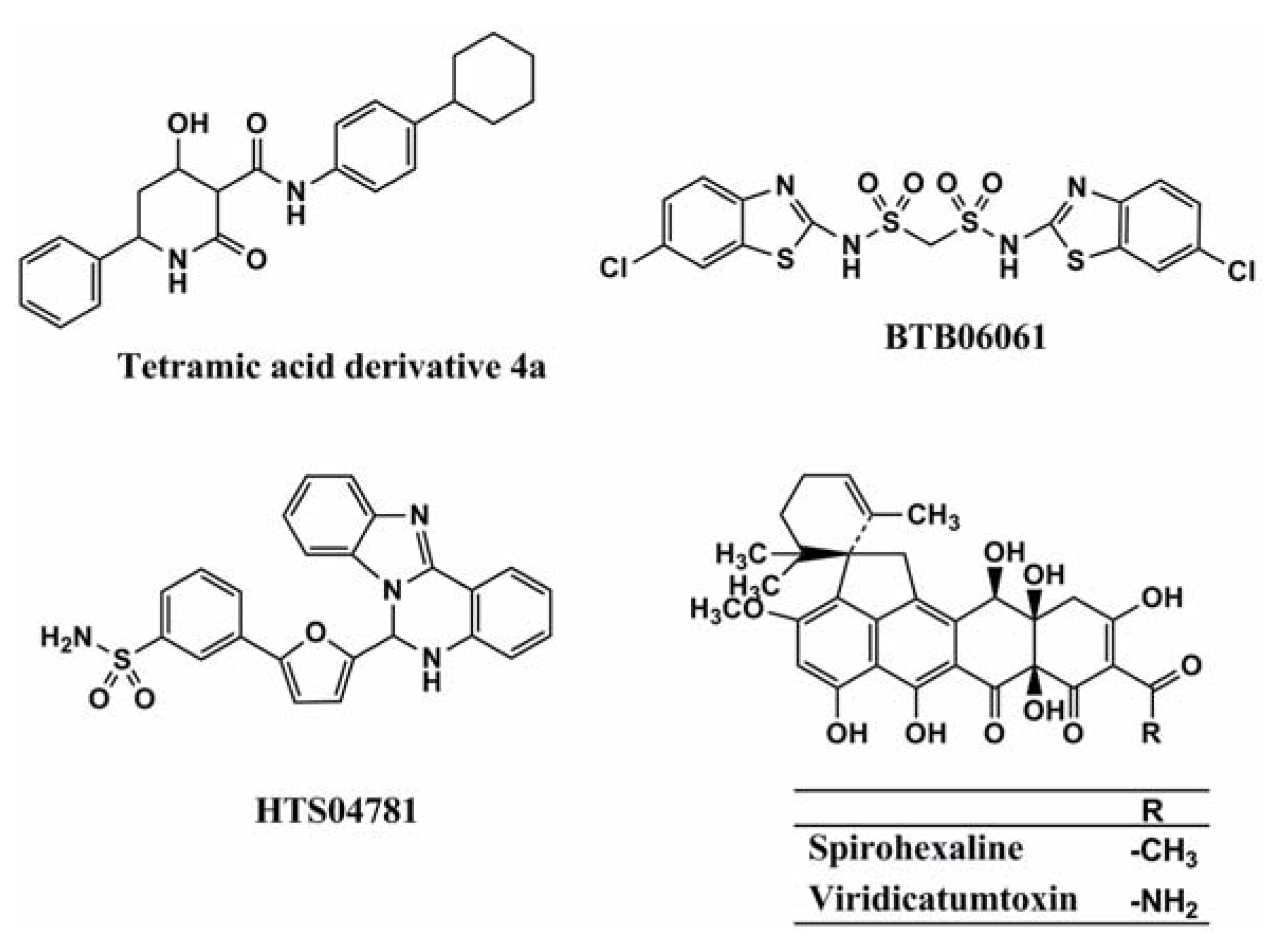
2.1. Spirohexaline
2.1.1. UPP Synthase as a Potential Target
2.1.2. Screening of UPP Synthase Inhibitors

2.1.3. Mechanism of Action of Spirohexaline

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure classification | Source | MIC against MRSA | Inhibitory activity against UPP synthase ** | Ref. |
|---|---|---|---|---|---|
| BTB06061 | Bisphosphonate | Synthetic origin | N.R. * | 71 M | [14] |
| Tetramic acid derivative 4a | Tetramic acid | Synthetic origin | N.R. | 0.2 M | [15] |
| Spirohexaline | Hexacyclic ring | Fungus (Penicillium brasilianum FKI-3368) | 6.25 g/mL | 9.0 M | [16] |
| Viridicatumtoxin | Hexacyclic ring | Fungus (Penicillium brasilianum FKI-3368) | 0.78 g/mL | 4.0 M | [17] |
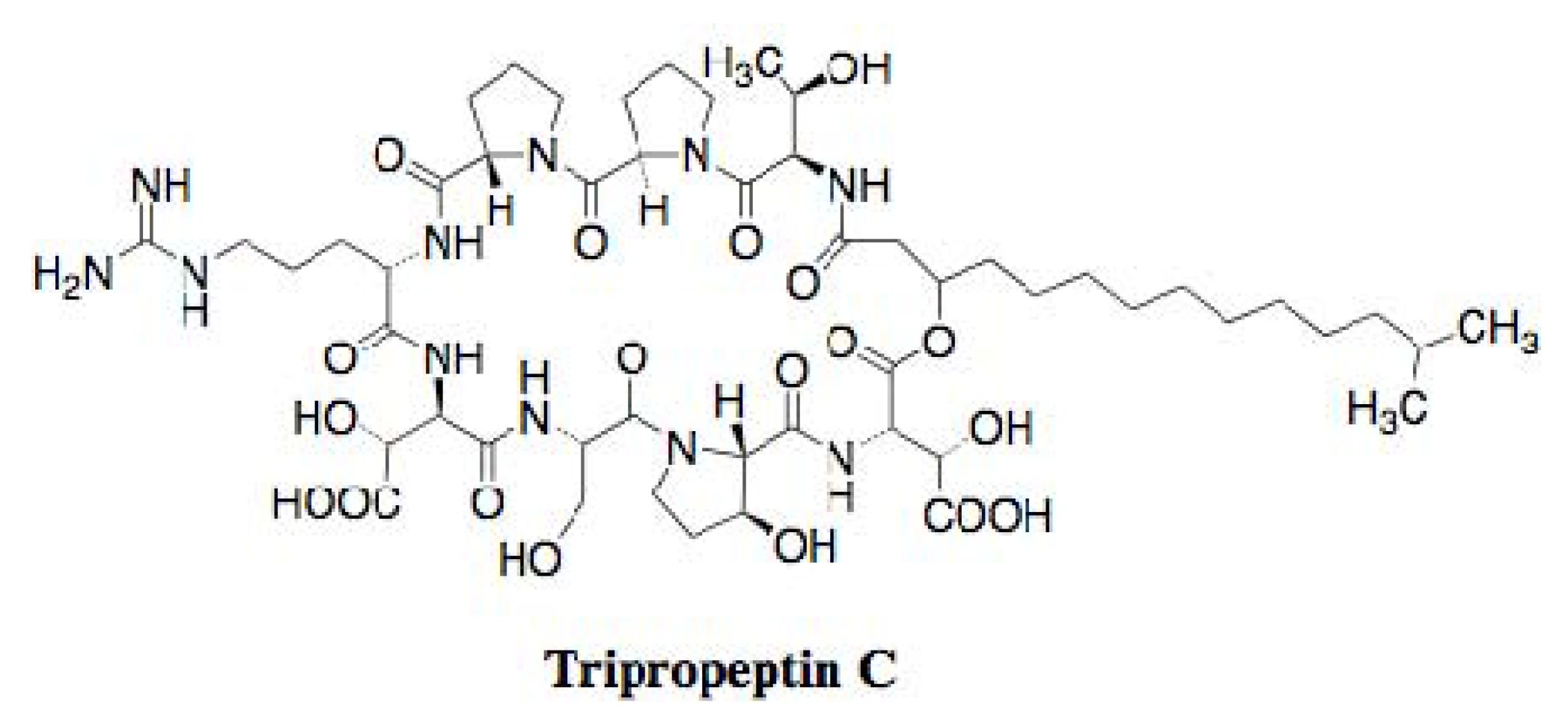
2.2. Tripropeptin C
2.2.1. Discovery

2.2.2. In Vitro and in Vivo Antimicrobial Activities
2.2.3. Mechanism of Action
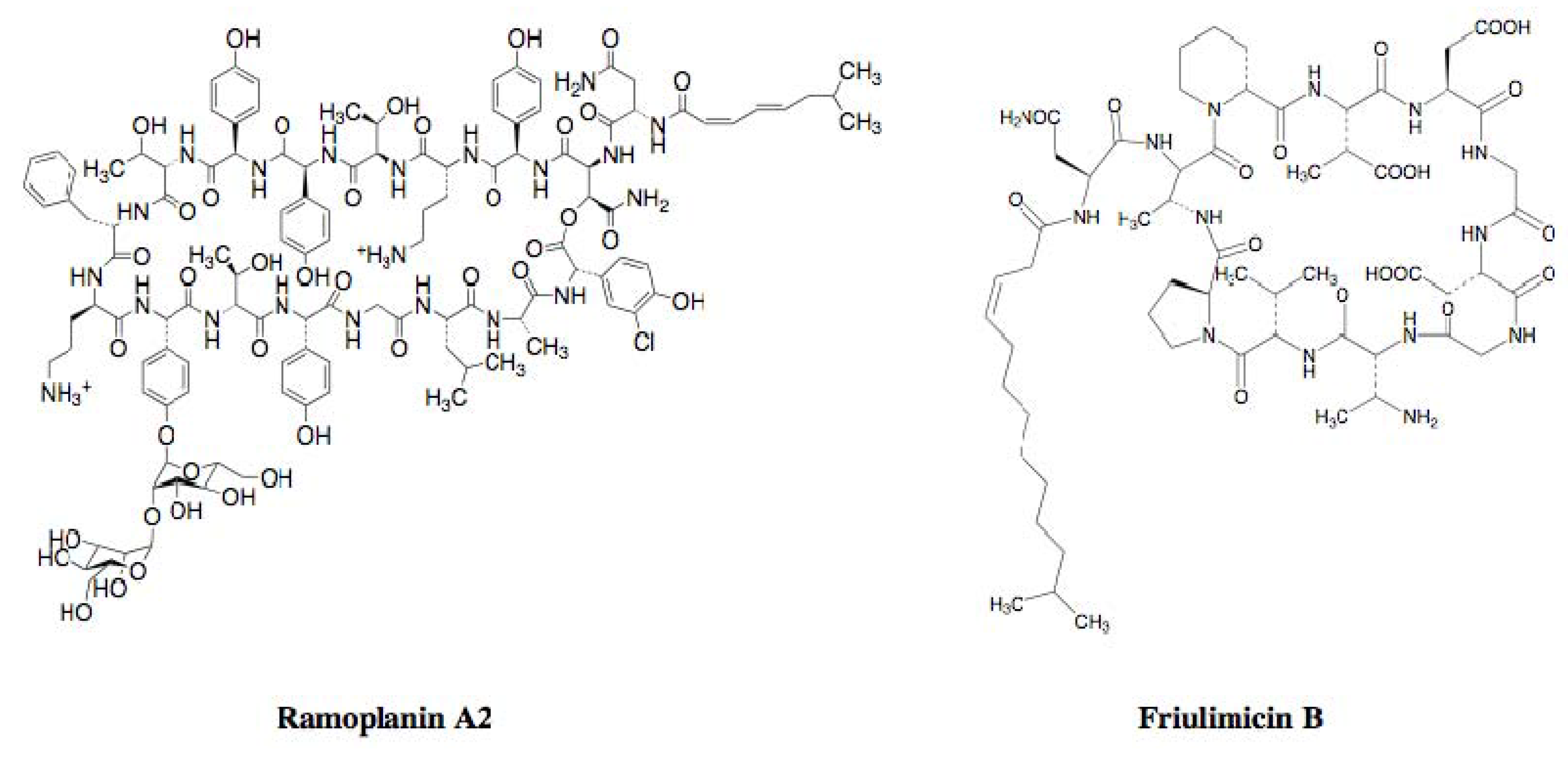
2.2.4. Cyclic Peptide Antibiotics Binding to Peptidoglycan Biosynthetic Molecules

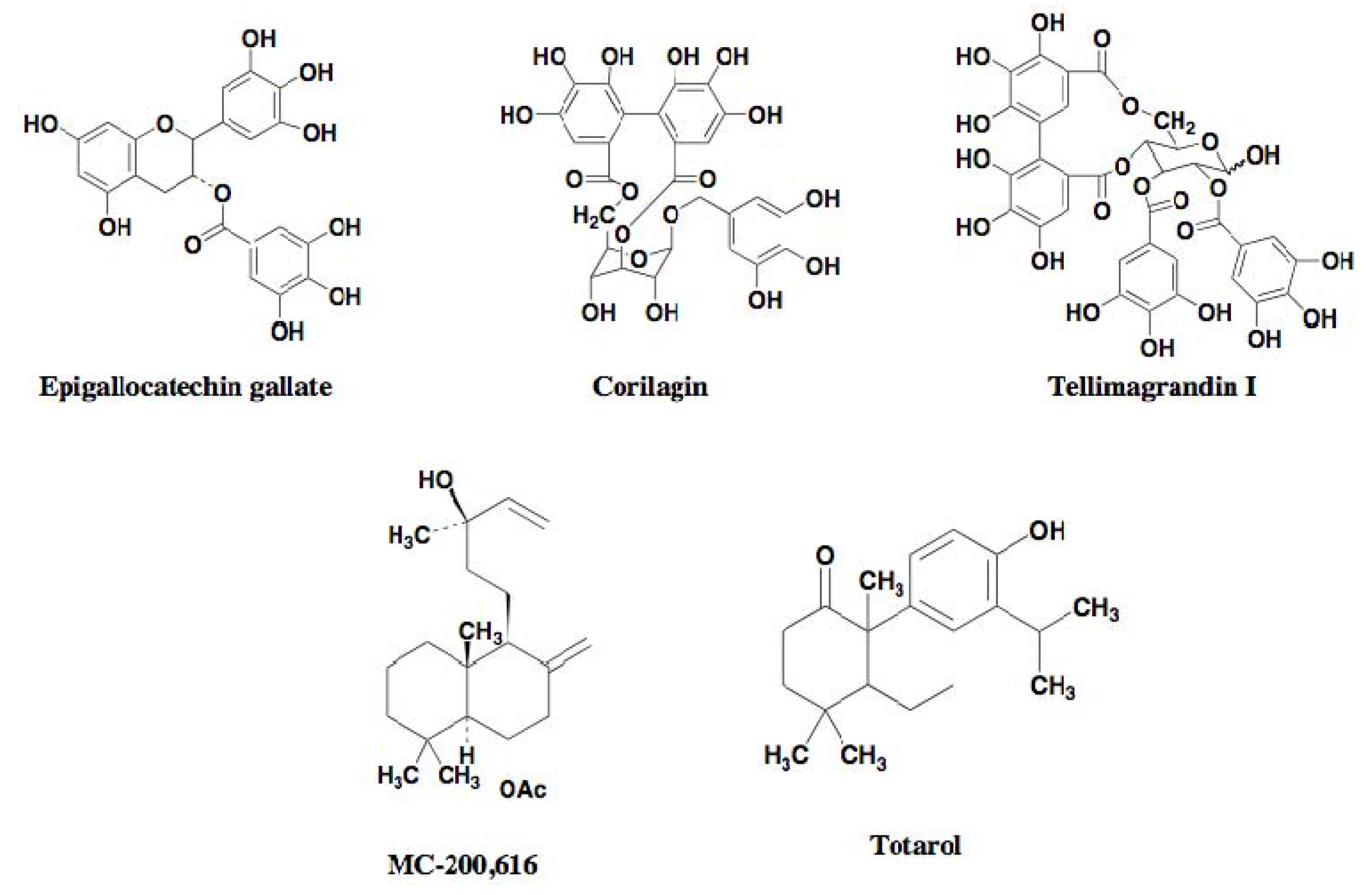
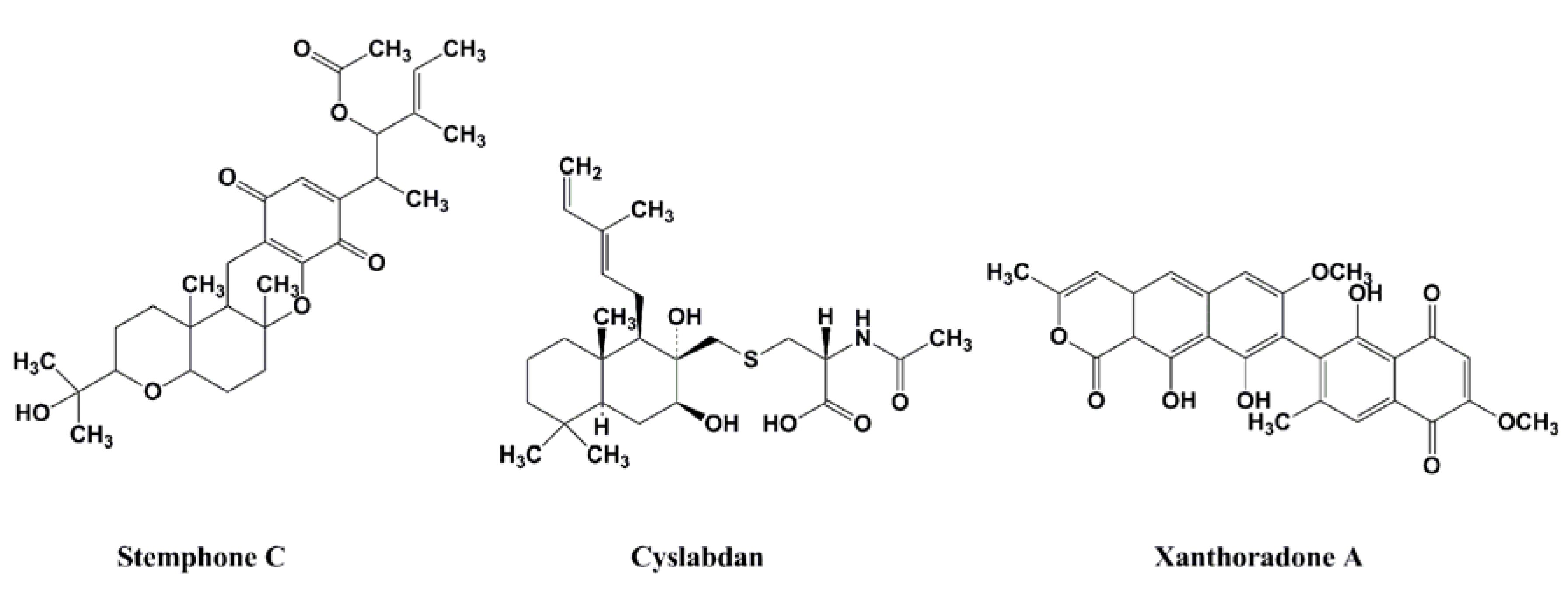
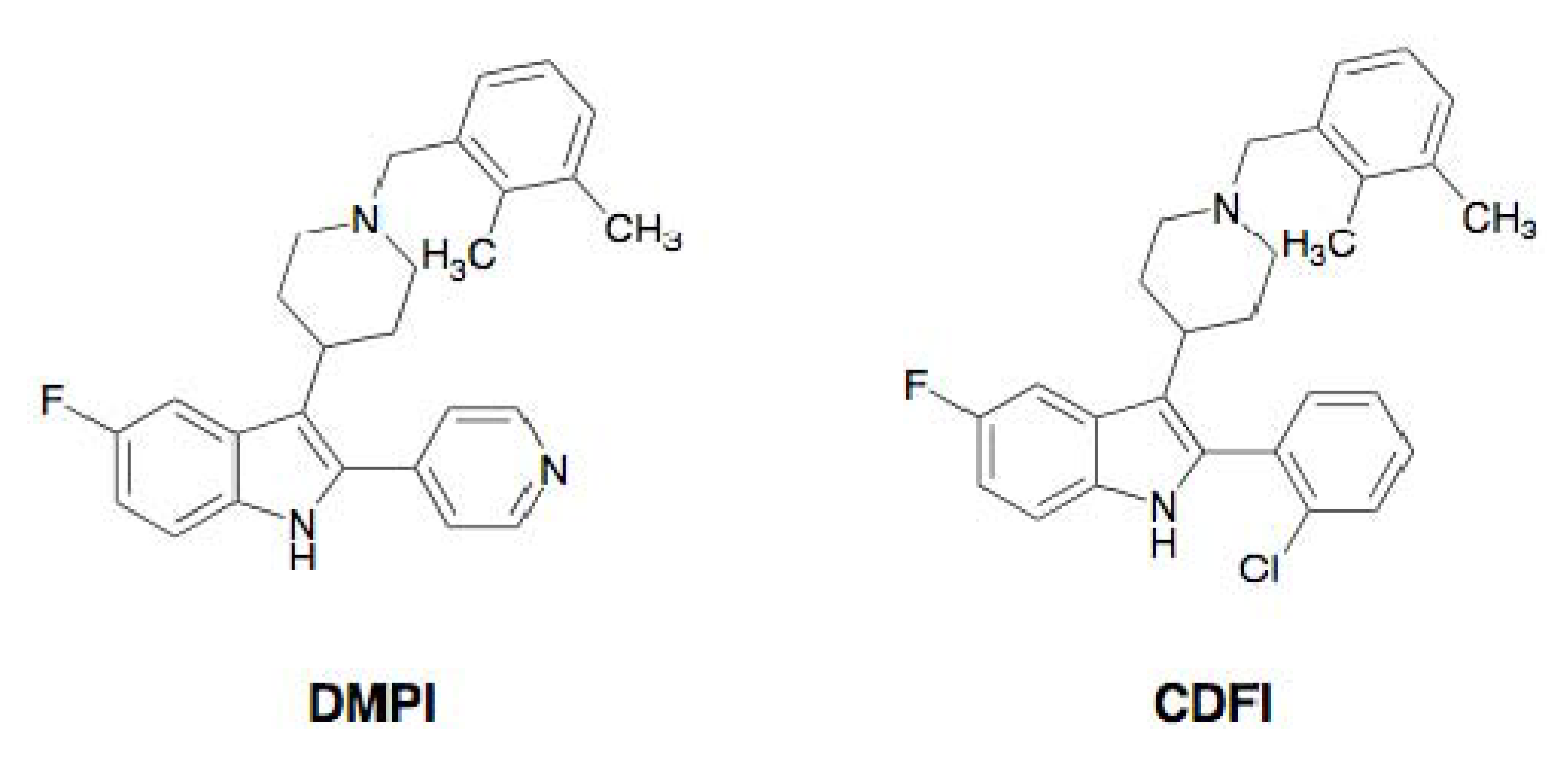
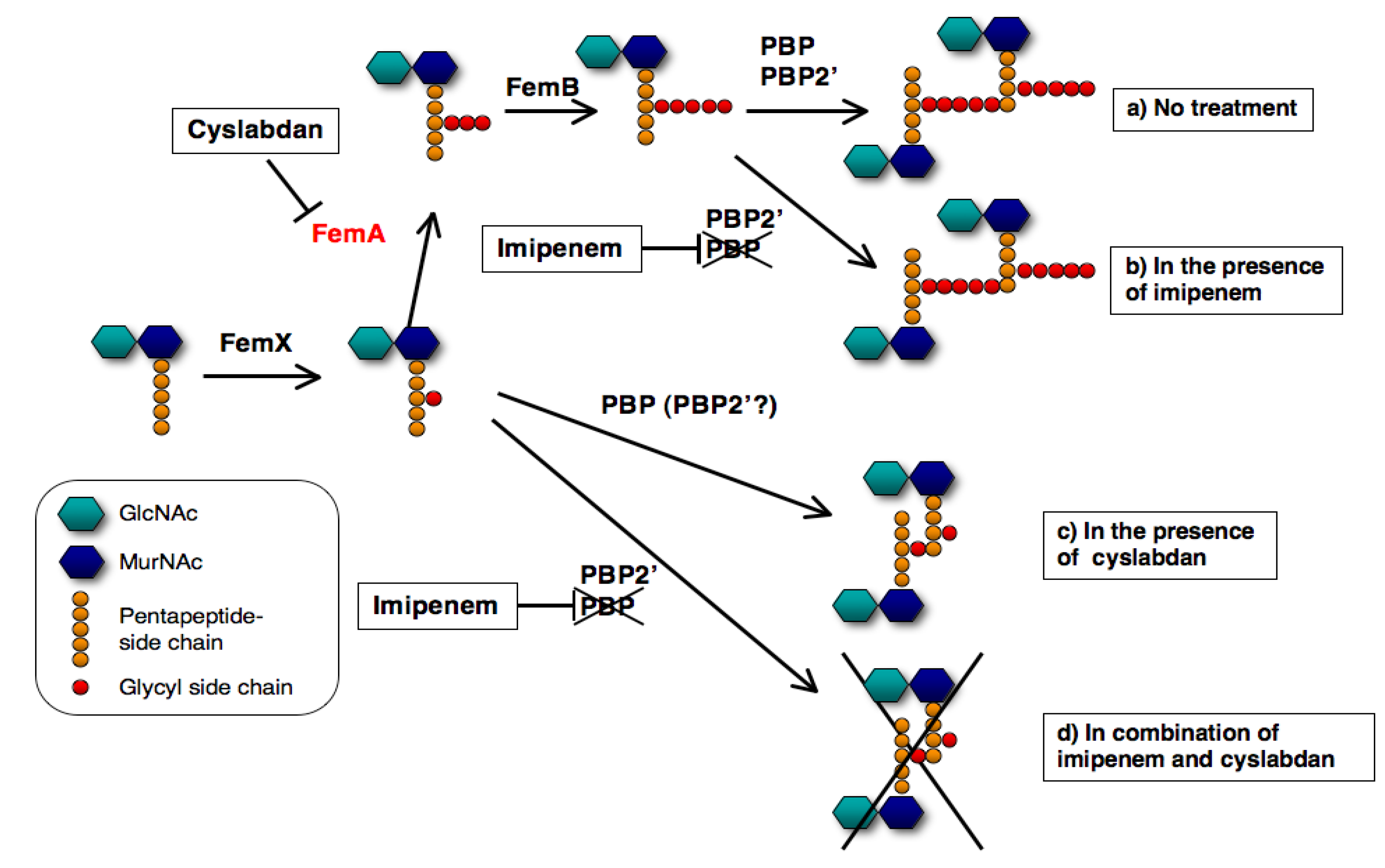
2.3. DMPI, CDFI, and Cyslabdan
2.3.1. Screening for β-Lactam Potentiators against MRSA
2.3.2. Potentiators of β-Lactam Activity against MRSA



2.3.3. Activity of β-Lactam Potentiators against MRSA
| Compound | Structure classification | Source | Potentiating activity of β-lactam * | Ref. | |||
|---|---|---|---|---|---|---|---|
| β-Lactam | MIC (µg/mL) | Potentiation ratio (fold) ** | |||||
| None | +Compound | ||||||
| MC-200,616 | Diterpene | Synthetic origin | Imipenem | 32 | 0.03 | 1067 | [25] |
| Totarol | Diterpene | Plant (totara tree) | Methicillin | 1024 | 4 | 256 | [26] |
| Epigallocatechin gallate | Polyphenol | Plant (tea) | Imipenem | 128 | 0.5 | 256 | [27] |
| Corilagin | Polyphenol | Plant (Arctostaphylos uva-urs) | Imipenem | 64 | 0.03 | 2133 | [28] |
| Tellimagrandin I | Polyphenol | Plant (Rosa canina L. | Oxacillin | 512 | 1 | 512 | [29] |
| Stemphone C | Tetracyclic quinone | Fungus (Aspergillus sp. FKI-2136) | Imipenem | 16 | 0.03 | 533 | [30,31] |
| Cyslabdan | Diterpene | Actinomycete (Streptomyces sp. K04-0144) | Imipenem | 16 | 0.015 | 1067 | [32,33,34] |
| Xanthoradone A | An aromatic ring-containing heterodimer | Fungus (P.radicum FKI-3765-2) | Imipenem | 16 | 0.03 | 533 | [35,36] |
| DMPI | Indole | Synthetic origin | Imipenem | 32 | 2 | 16 | [37] |
| CDFI | Indole | Synthetic origin | Imipenem | 32 | 2 | 16 | [37] |
2.3.4. Mechanism of Action of DMPI and CDFI
2.3.5. Mechanism of Action of Cyslabdan


3. Inhibitors of Wall Teichoic Acid
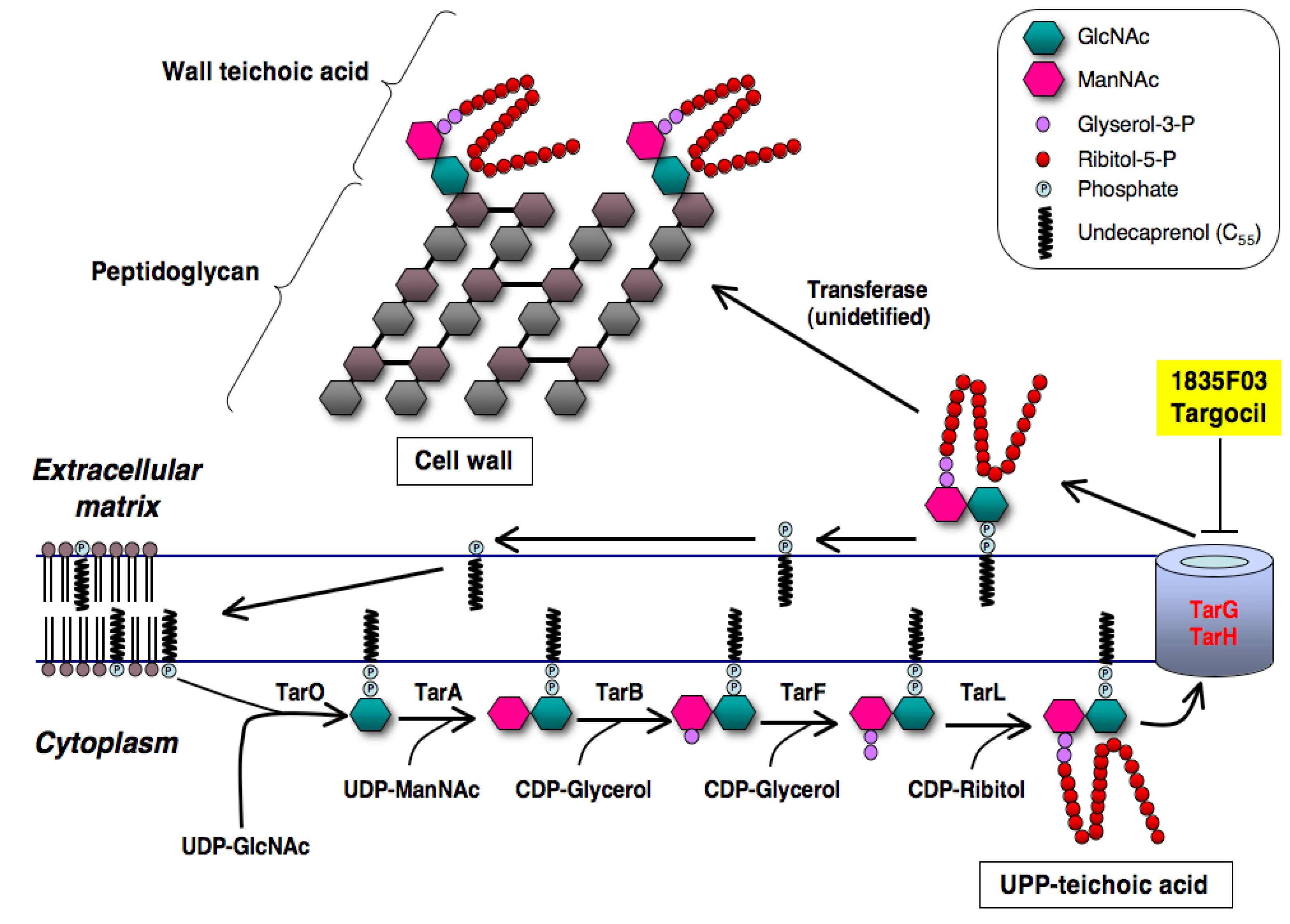
3.1. Wall Teichoic Acid as a Target of Anti-Infectious Agents against S. aureus

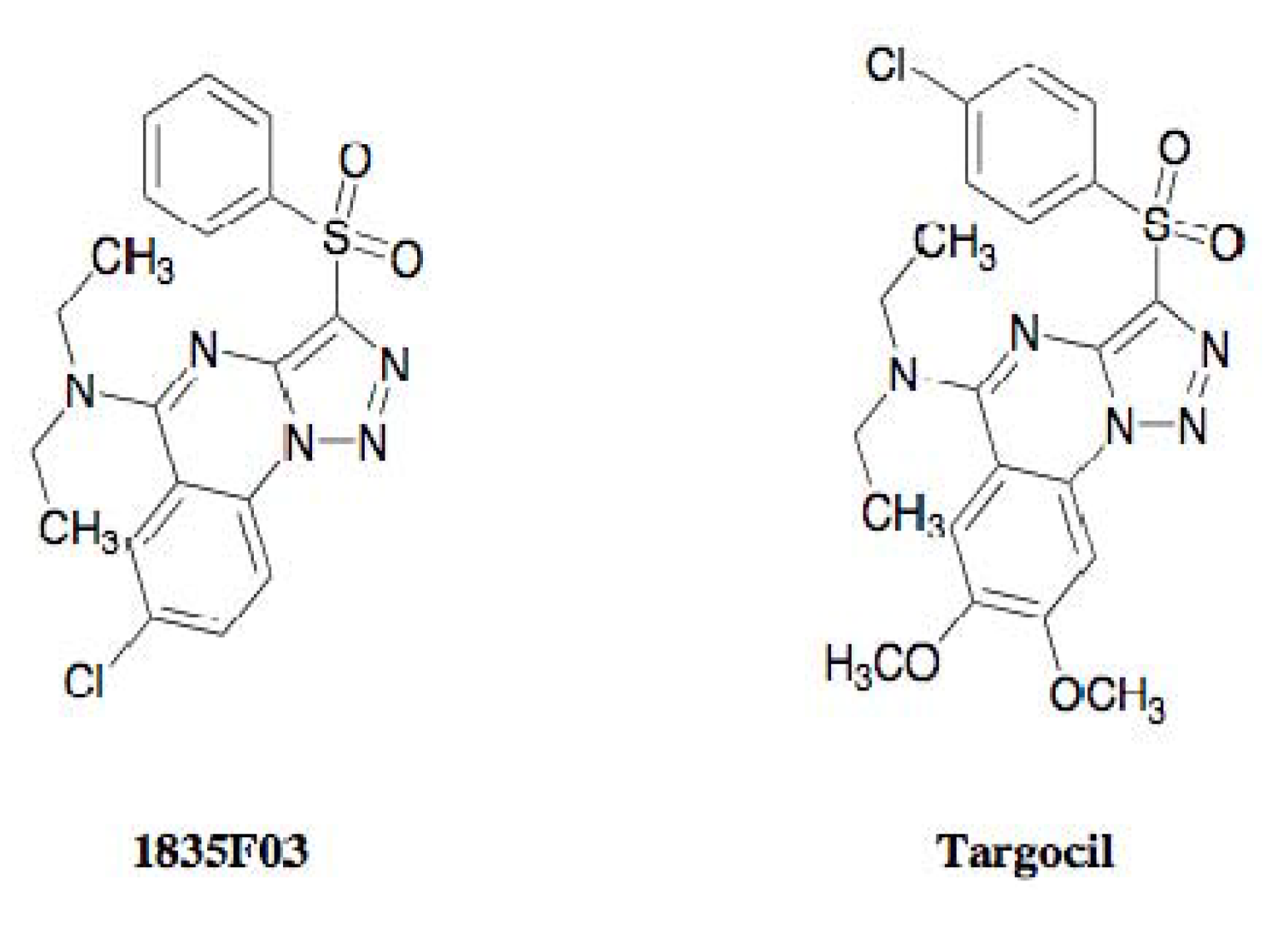
3.2. Discovery of 1835F03

3.3. Molecular Target of 1835F03
3.4. Development of Targocil as an Improved WTA Inhibitor
4. Inhibitors of a Virulence Factor
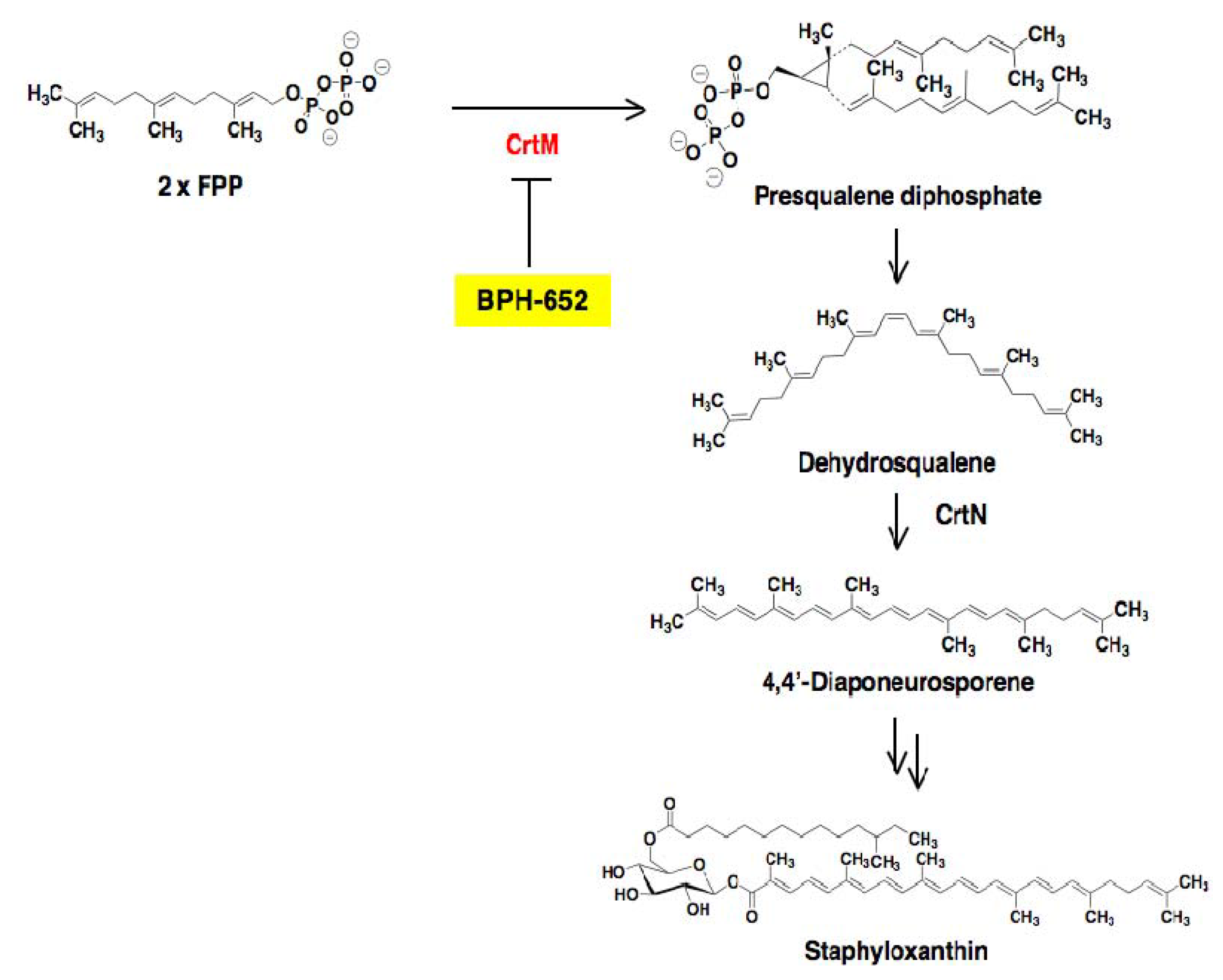
4.1. The Virulence Factor Staphyloxanthin as a Target of Anti-Infectious Agents against S. aureus

4.2. Screening for Inhibitors of Staphyloxanthin Biosynthesis in S. aureus
| Compound | Structure classification | Source | Ref. |
|---|---|---|---|
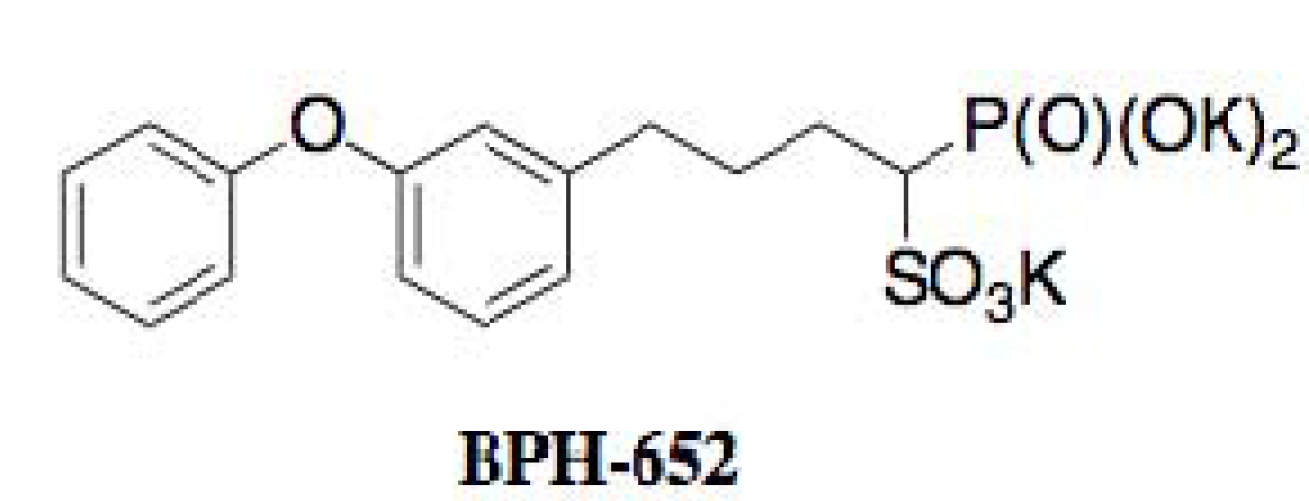
| BPH-652 | Biphenyl ether | Synthetic origin | [50] |
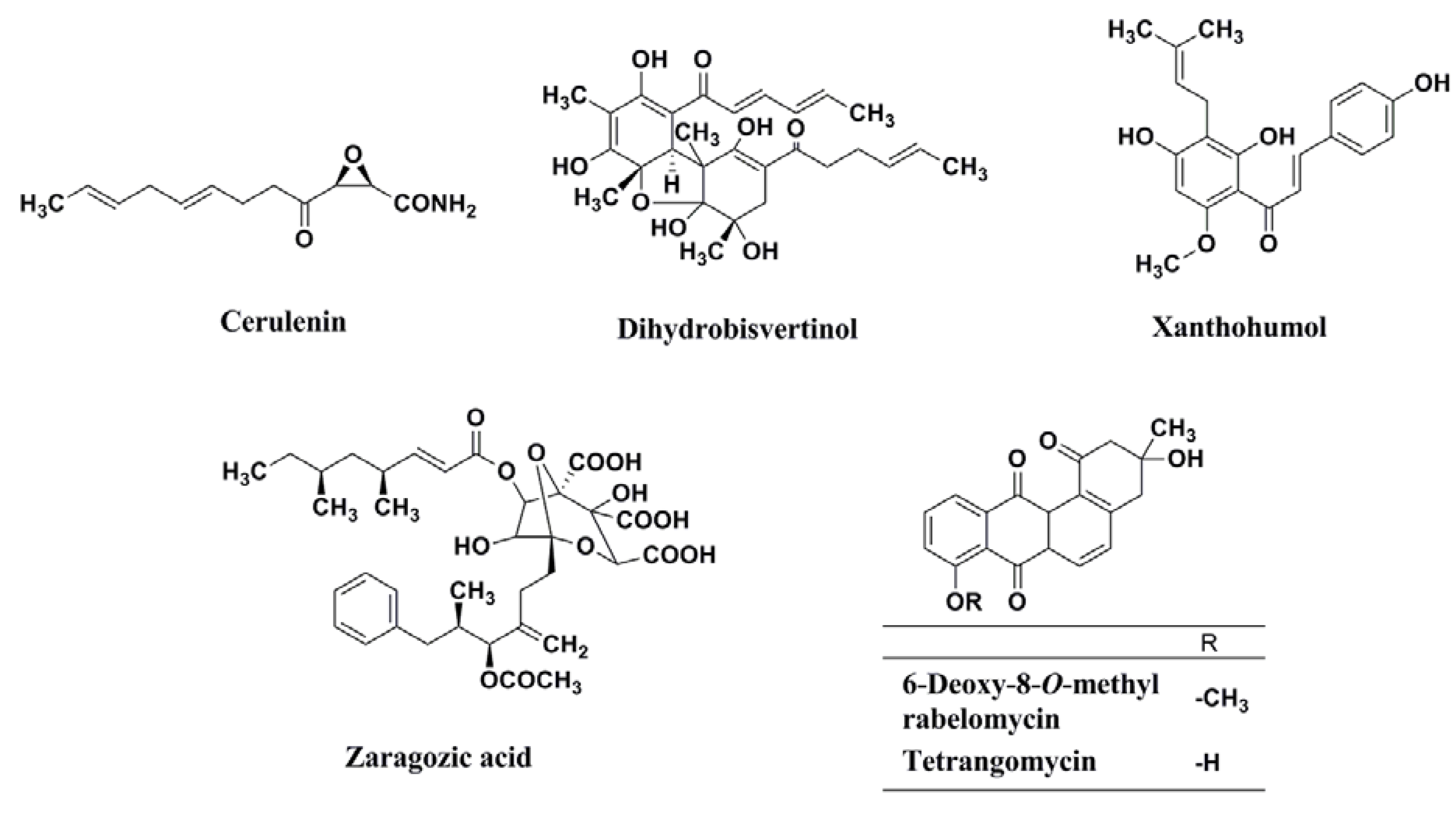
| Cerulenin | Epoxy fatty acid | Fungus (Cephalosporium caerulens KF-140) | [51] |
| Dihydrobisvertinol | Dibenzofuran | Fungus (Verticillium intertextum) | [51] |
| Xanthohumol | Chalcone | Hops plant | [51] |
| Zaragozic acid | Bicyclo ring | Fungus (Phoma sp.) | [51] |
| 6-Deoxy-8-O-methylrabelomycin | Anthraquinone | Actinomycete (Streptomyces badius 4-6) | [51] |
| Tetrangomycin | Anthraquinone | Actinomycete (Streptomyces badius 4-6) | [51] |

4.3. Biological Activity of BPH-652

5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bugg, T.D.; Braddick, D.; Dowson, C.G.; Roper, D.I. Bacterial cell wall assembly: Still an attractive antibacterial target. Trends. Biotechnol. 2011, 29, 167–173. [Google Scholar] [CrossRef]
- Giesbrecht, P.; Kersten, T.; Maidhof, H.; Wecke, J. Staphylococcal cell wall: Morphogenesis and fatal variations in the presence of penicillin. Microbiol. Mol. Biol. Rev. 2007, 62, 1371–1414. [Google Scholar]
- Tomasz, A. Penicillin-binding proteins and the antibacterial effectiveness of beta-lactam antibiotics. Rev. Infect. Dis. 1986, 8, S260–S278. [Google Scholar] [CrossRef]
- Hammes, W.P.; Neuhaus, F.C. On the mechanism of action of vancomycin: Inhibition of peptidoglycan synthesis in Gaffkya homari. Antimicrob. Agents Chemother. 1974, 6, 722–728. [Google Scholar] [CrossRef]
- Ostash, B.; Walker, S. Moenomycin family antibiotics: Chemical synthesis, biosynthesis, and biological activity. Nat. Prod. Rep. 2010, 27, 1595–1617. [Google Scholar]
- Stone, K.J.; Strominger, J.L. Mechanism of action of bacitracin: Complexation with metal ion and C55-isoprenyl pyrophosphate. Proc. Natl. Acad. Sci.USA 1971, 68, 3223–3227. [Google Scholar] [CrossRef]
- Tomasz, A. Multiple-antibiotic resistant pathogenic bacteria. N. Engl. J. Med. 1994, 330, 1247–1251. [Google Scholar] [CrossRef]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 2007, 298, 1763–1771. [Google Scholar]
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Staphylococcus aureus with reduced susceptibility to vancomycin-United States, 1997. MMWR Morb. Mortal. Wkly. Rep. 1997, 46, 765–766.
- Bachmann, B.O.; Li, R.; Townsend, C.A. Lactam synthetase: A new biosynthetic enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9082–9086. [Google Scholar] [CrossRef]
- Shimizu, N.; Koyama, T.; Ogura, K. Molecular cloning, expression, and purification of undecaprenyl diphosphate synthase. No sequence similarity between E- and Z-prenyl diphosphate synthases. J. Biol. Chem. 1998, 273, 19476–19481. [Google Scholar]
- Li, H.; Huang, J.; Jiang, X.; Seefeld, M.; McQueney, M.; Macarron, R. The effect of triton concentration on the activity of undecaprenyl pyrophosphate synthase inhibitors. J. Biomol. Screen. 2003, 8, 712–715. [Google Scholar] [CrossRef]
- Peukert, S.; Sun, Y.; Zhang, R.; Hurley, B.; Sabio, M.; Shen, X.; Gray, C.; Dzink-Fox, J.; Tao, J.; Cebula, R.; Wattanasin, S. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): Tetramic, tetronic acids and dihydropyridin-2-ones. Bioorg. Med. Chem. Lett. 2008, 18, 1840–1844. [Google Scholar]
- Kuo, C.J.; Guo, R.T.; Lu, I.L.; Liu, H.G.; Wu, S.Y.; Ko, T.P.; Wang, A.H.; Liang, P.H. Structure-based inhibitors exhibit differential activities against Helicobacter pylori and Escherichia coli undecaprenyl pyrophosphate synthases. J. Biomed. Biotechnol. 2008, 2008, 1–6. [Google Scholar]
- Durrant, J.D.; Cao, R.; Gorfe, A.A.; Zhu, W.; Li, J.; Sankovsky, A.; Oldfield, E.; McCammon, J.A. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem. Biol. Drug Des. 2011, 78, 323–332. [Google Scholar] [CrossRef]
- Inokoshi, J.; Nakamura, Y.; Hongbin, Z.; Uchida, R.; Nonaka, K.; Masuma, R.; Tomoda, H. Spirohexalines, New Inhibitors of Bacterial Undecaprenyl Pyrophosphate Synthase, produced by Penicillium brasilianum FKI-3368. J. Antibiot. 2012. [Google Scholar] [CrossRef]
- Hashizume, H.; Igarashi, M.; Hattori, S.; Hori, M.; Hamada, M.; Takeuchi, T. Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. I. Taxonomy, isolation and biological activities. J. Antibiot. 2001, 54, 1054–1059. [Google Scholar]
- Hashizume, H.; Hirosawa, S.; Sawa, R.; Muraoka, Y.; Ikeda, D.; Naganawa, H.; Igarashi, M. Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. J. Antibiot. 2004, 57, 52–58. [Google Scholar] [CrossRef]
- Hashizume, H.; Sawa, R.; Harada, S.; Igarashi, M.; Adachi, H.; Nishimura, Y.; Nomoto, A. Tripropeptin C blocks the lipid cycle of cell wall biosynthesis by complex formation with undecaprenyl pyrophosphate. Antimicrob. Agents Chemother. 2011, 55, 3821–3828. [Google Scholar] [CrossRef]
- Fulco, P.; Wenzel, R.P. Ramoplanin: A topical lipoglycodepsipeptide antibacterial agent. Expert. Rev. Anti Infect. Ther. 2006, 4, 939–945. [Google Scholar] [CrossRef]
- Walker, S.; Chen, L.; Hu, Y.; Rew, Y.; Shin, D.; Boger, D.L. Chemistry and biology of ramoplanin: A lipoglycodepsipeptide with potent antibiotic activity. Chem. Rev. 2005, 105, 449–476. [Google Scholar]
- Vértesy, L.; Ehlers, E.; Kogler, H.; Kurz, M.; Meiwes, J.; Seibert, G.; Vogel, M.; Hammann, P. Friulimicins: Novel lipopeptide antibiotics with peptidoglycan synthesis inhibiting activity from Actinoplanes friuliensis sp. nov. II. Isolation and structural characterization. J. Antibiot. 2000, 53, 816–827. [Google Scholar] [CrossRef]
- Schneider, T.; Gries, K.; Josten, M.; Wiedemann, I.; Pelzer, S.; Labischinski, H.; Sahl, H.G. The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 2009, 53, 1610–1618. [Google Scholar] [CrossRef]
- Chamberland, S.; Blais, J.; Boggs, A.F.; Bao, Y.; Malouin, F.; Hecker, S.J.; Lee, V.J. MC-207,252 Abolishes PBP2a mediated β-lactam resistance in Staphylococci. 35th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco, CA, USA; 14–20 September 1995; pp. F138–F144. [Google Scholar]
- Nicolson, K.; Evans, G.; O’Toole, P.W. Potentiation of methicillin activity against methicillin-resistant Staphylococcus aureus by diterpenes. FEMSMicrobiol. Lett. 1999, 179, 233–239. [Google Scholar] [CrossRef]
- Zhao, W.H.; Hu, Z.Q.; Okubo, S.; Hara, Y.; Shimamura, T. Mechanism of synergy between epigallocatechin gallate and beta-lactams against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1737–1742. [Google Scholar] [CrossRef]
- Shimizu, M.; Shiota, S.; Mizushima, T.; Ito, H.; Hatano, T.; Yoshida, T.; Tsuchiya, T. Marked potentiation of activity of beta-lactams against methicillin-resistant Staphylococcus aureus by corilagin. Antimicrob. Agents Chemother. 2001, 45, 3198–3201. [Google Scholar] [CrossRef]
- Shiota, S.; Shimizu, M.; Mizushima, T.; Ito, H.; Hatano, T.; Yoshida, T.; Tsuchiya, T. Restoration of effectiveness of beta-lactams on methicillin-resistant Staphylococcus aureus by tellimagrandin I from rose red. FEMS Microbiol. Lett. 2000, 185, 135–138. [Google Scholar]
- Koyama, N.; Nagahiro, T.; Yamaguchi, Y.; Masuma, R.; Tomoda, H.; Omura, S. Stemphones, novel potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus, produced by Aspergillus sp. FKI-2136. J. Antibiot. 2005, 58, 695–703. [Google Scholar]
- Yamazaki, H.; Koyama, N.; Omura, S.; Tomoda, H. Structure-activity relationships of stemphones, potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus. J. Antibiot. 2008, 61, 426–441. [Google Scholar] [CrossRef]
- Fukumoto, A.; Kim, Y.P.; Matsumoto, A.; Takahashi, Y.; Shiomi, K.; Tomoda, H.; Omura, S. Cyslabdan, a new potentiator of imipenem activity against methicillin-resistant Staphylococcus aureus, produced by Streptomyces sp. K04-0144. I. Taxonomy, fermentation, isolation and structural elucidation. J. Antibiot. 2008, 61, 1–6. [Google Scholar]
- Fukumoto, A.; Kim, Y.P.; Hanaki, H.; Shiomi, K.; Tomoda, H.; Omura, S. Cyslabdan, a new potentiator of imipenem activity against methicillin-resistant Staphylococcus aureus, produced by Streptomyces sp. K04-0144. II. Biological activities. J. Antibiot. 2008, 61, 7–10. [Google Scholar]
- Koyama, N.; Tokura, Y.; Takahashi, Y.; Tomoda, H. New cyslabdans B and C, potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus produced by Streptomyces sp. K04-0144. Acta. Pharm. Sin. B 2011, 1, 236–239. [Google Scholar]
- Yamazaki, H.; Nonaka, K.; Masuma, R.; Omura, S.; Tomoda, H. Xanthoradones, new potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus, produced by Penicillium radicum FKI-3765-2: I. Taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2009, 62, 431–434. [Google Scholar]
- Yamazaki, H.; Omura, S.; Tomoda, H. Xanthoradones, new potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus, produced by Penicillium radicum FKI-3765-2 II. Structure elucidation. J. Antibiot. 2009, 62, 435–437. [Google Scholar] [CrossRef]
- Huber, J.; Donald, R.G.; Lee, S.H.; Jarantow, L.W.; Salvatore, M.J.; Meng, X.; Painter, R.; Onishi, R.H.; Occi, J.; Dorso, K.; et al. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem. Biol. 2009, 16, 837–848. [Google Scholar] [CrossRef]
- Shiota, S.; Shimizu, M.; Sugiyama, J.; Morita, Y.; Mizushima, T.; Tsuchiya, T. Mechanisms of action of corilagin and tellimagrandin I that remarkably potentiate the activity of β-lactams against methicillin-resistant Staphylococcus aureus. Microbiol. Immunol. 2004, 48, 67–73. [Google Scholar]
- Koyama, N.; Tokura, Y.; Münch, D.; Sahl, H.-G.; Schneider, T.; Shibagaki, Y.; Ikeda, H.; Tomoda, H. The nonantibiotic small molecule cyslabdan enhances the potency of β-lactams against MRSA by inhibiting pentaglycine interpeptide bridge synthesis. PLoS One 2012, 7, e48981. [Google Scholar]
- Berger-Bächi, B.; Barberis-Maino, L.; Strässle, A.; Kayser, F.H. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus. Molecular cloning and characterization. Mol. Gen. Genet. 1989, 219, 263–269. [Google Scholar]
- Maidhof, H.; Reinicke, B.; Blümel, P.; Berger-Bächi, B.; Labischinski, H. femA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. J. Bacteriol. 1991, 173, 3507–3513. [Google Scholar]
- Strandén, A.M.; Ehlert, K.; Labischinski, H.; Berger-Bächi, B. Cell wall monoglycine cross-bridges and methicillin hypersusceptibility in a femAB null mutant of methicillin-resistant Staphylococcus aureus. J. Bacteriol. 1997, 179, 9–16. [Google Scholar]
- Brown, S.; Meredith, T.; Swoboda, J.; Walker, S. Staphylococcus aureus and Bacillus subtilis W23 make polyribitol wall teichoic acids using different enzymatic pathways. Chem. Biol. 2010, 17, 1101–1110. [Google Scholar] [CrossRef]
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S. Wall teichoic acid function, biosynthesis, and inhibition. Chembiochem 2010, 11, 35–45. [Google Scholar]
- Swoboda, J.G.; Meredith, T.C.; Campbell, J.; Brown, S.; Suzuki, T.; Bollenbach, T.; Malhowski, A.J.; Kishony, R.; Gilmore, M.S.; Walker, S. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem. Biol. 2009, 4, 875–883. [Google Scholar] [CrossRef]
- Schirner, K.; Stone, L.K.; Walker, S. ABC transporters required for export of wall teichoic acids do not discriminate between different main chain polymers. ACS Chem. Biol. 2011, 6, 407–412. [Google Scholar] [CrossRef]
- Lee, K.; Campbell, J.; Swoboda, J.G.; Cuny, G.D.; Walker, S. Development of improved inhibitors of wall teichoic acid biosynthesis with potent activity against Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2010, 20, 1767–1770. [Google Scholar]
- Pelz, A.; Wieland, K.P.; Putzbach, K.; Hentschel, P.; Albert, K.; Götz, F. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem. 2005, 280, 32493–32498. [Google Scholar]
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [Google Scholar] [CrossRef]
- Liu, C.I.; Liu, G.Y.; Song, Y.; Yin, F.; Hensler, M.E.; Jeng, W.Y.; Nizet, V.; Wang, A.H.; Oldfield, E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [Google Scholar]
- Sakai, K.; Koyama, N.; Fukuda, T.; Mori, Y.; Onaka, H.; Tomoda, H. Search method for inhibitors of Staphyloxanthin production by methicillin-resistant Staphylococcus aureus. Biol. Pharm. Bull. 2012, 35, 48–53. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koyama, N.; Inokoshi, J.; Tomoda, H. Anti-Infectious Agents against MRSA. Molecules 2013, 18, 204-224. https://doi.org/10.3390/molecules18010204
Koyama N, Inokoshi J, Tomoda H. Anti-Infectious Agents against MRSA. Molecules. 2013; 18(1):204-224. https://doi.org/10.3390/molecules18010204
Chicago/Turabian StyleKoyama, Nobuhiro, Junji Inokoshi, and Hiroshi Tomoda. 2013. "Anti-Infectious Agents against MRSA" Molecules 18, no. 1: 204-224. https://doi.org/10.3390/molecules18010204
APA StyleKoyama, N., Inokoshi, J., & Tomoda, H. (2013). Anti-Infectious Agents against MRSA. Molecules, 18(1), 204-224. https://doi.org/10.3390/molecules18010204