Conformational Analysis of Geometric Isomers of Pitavastatin Together with Their Lactonized Analogues

Abstract
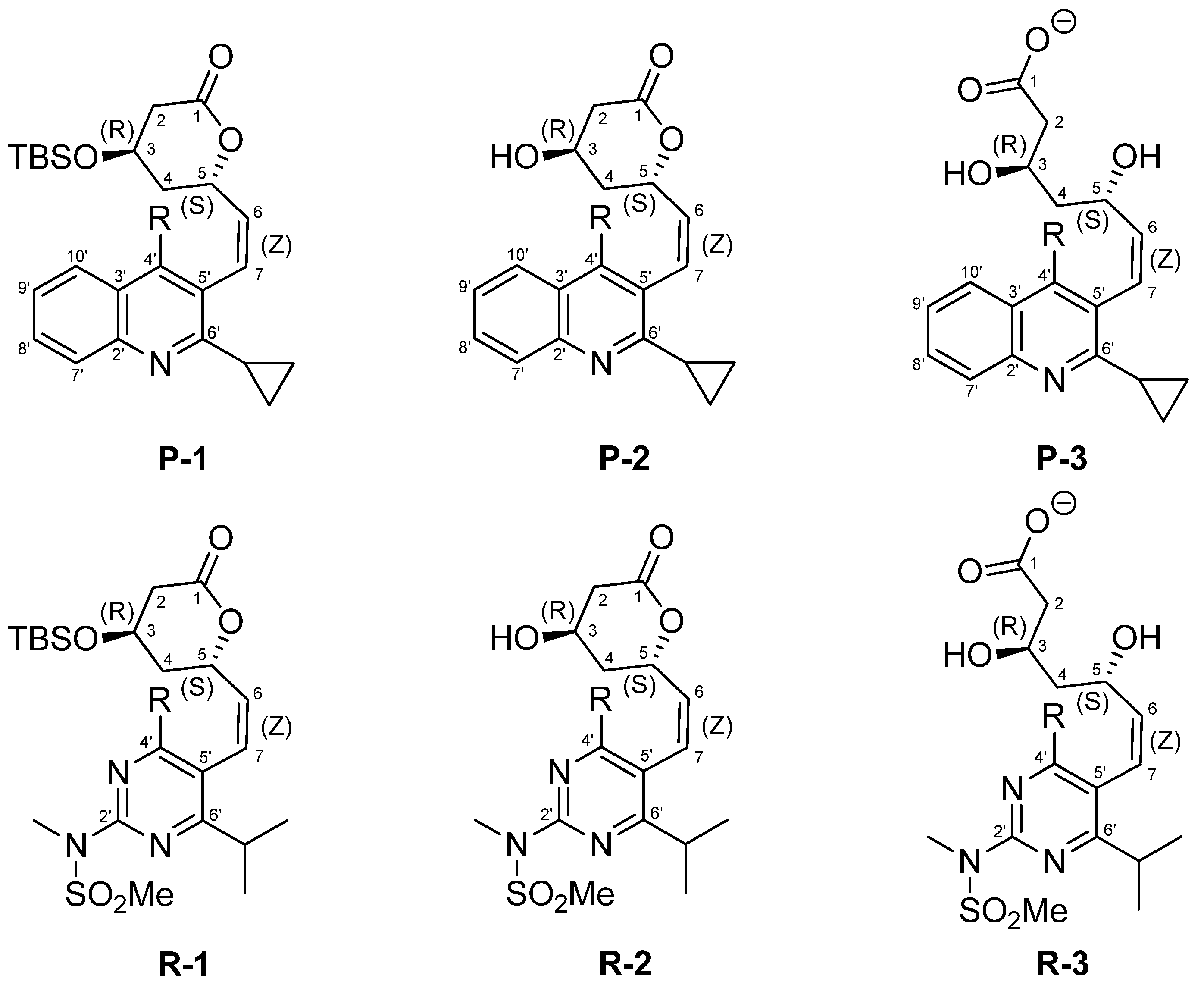
:
1. Introduction

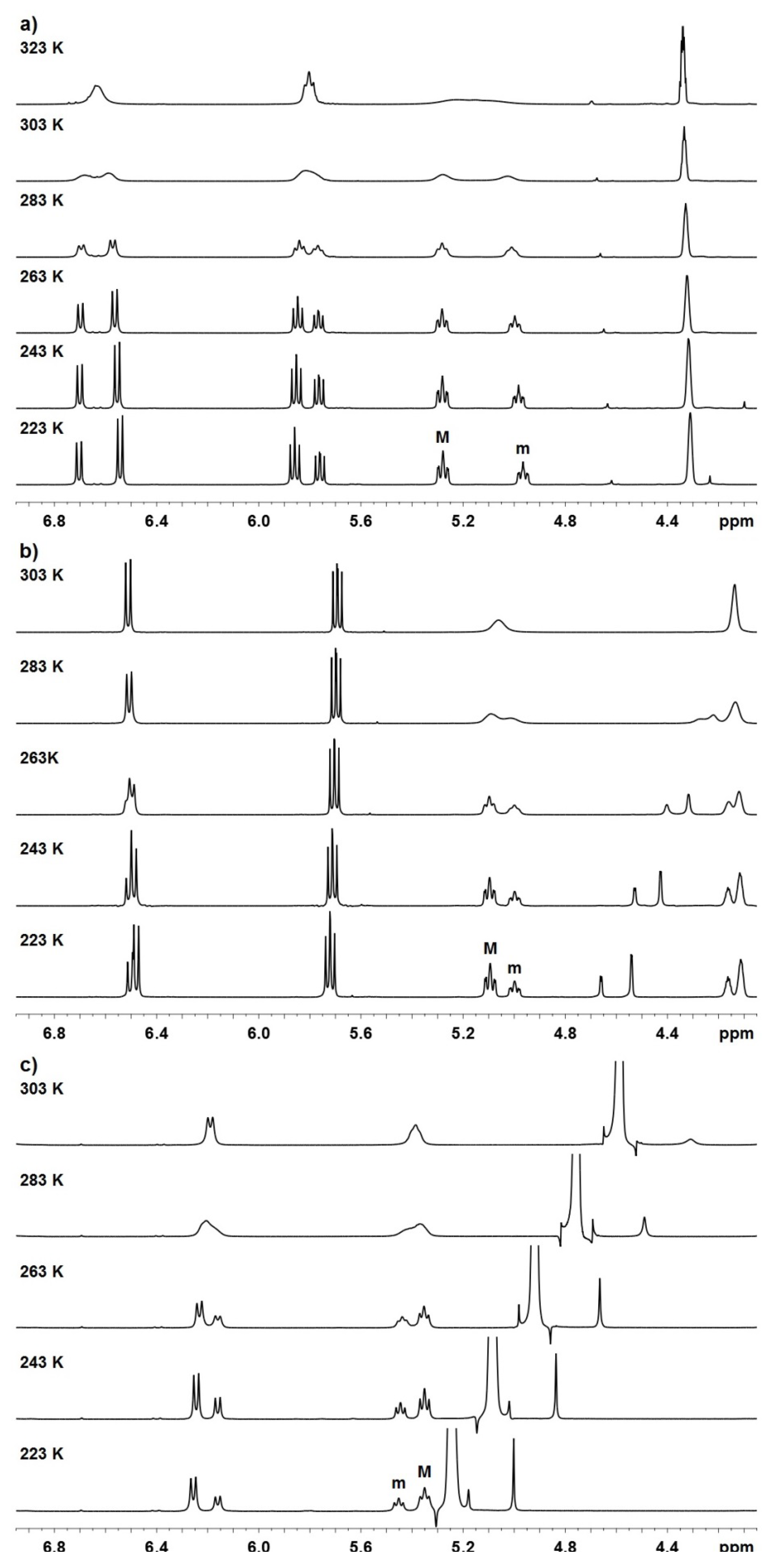
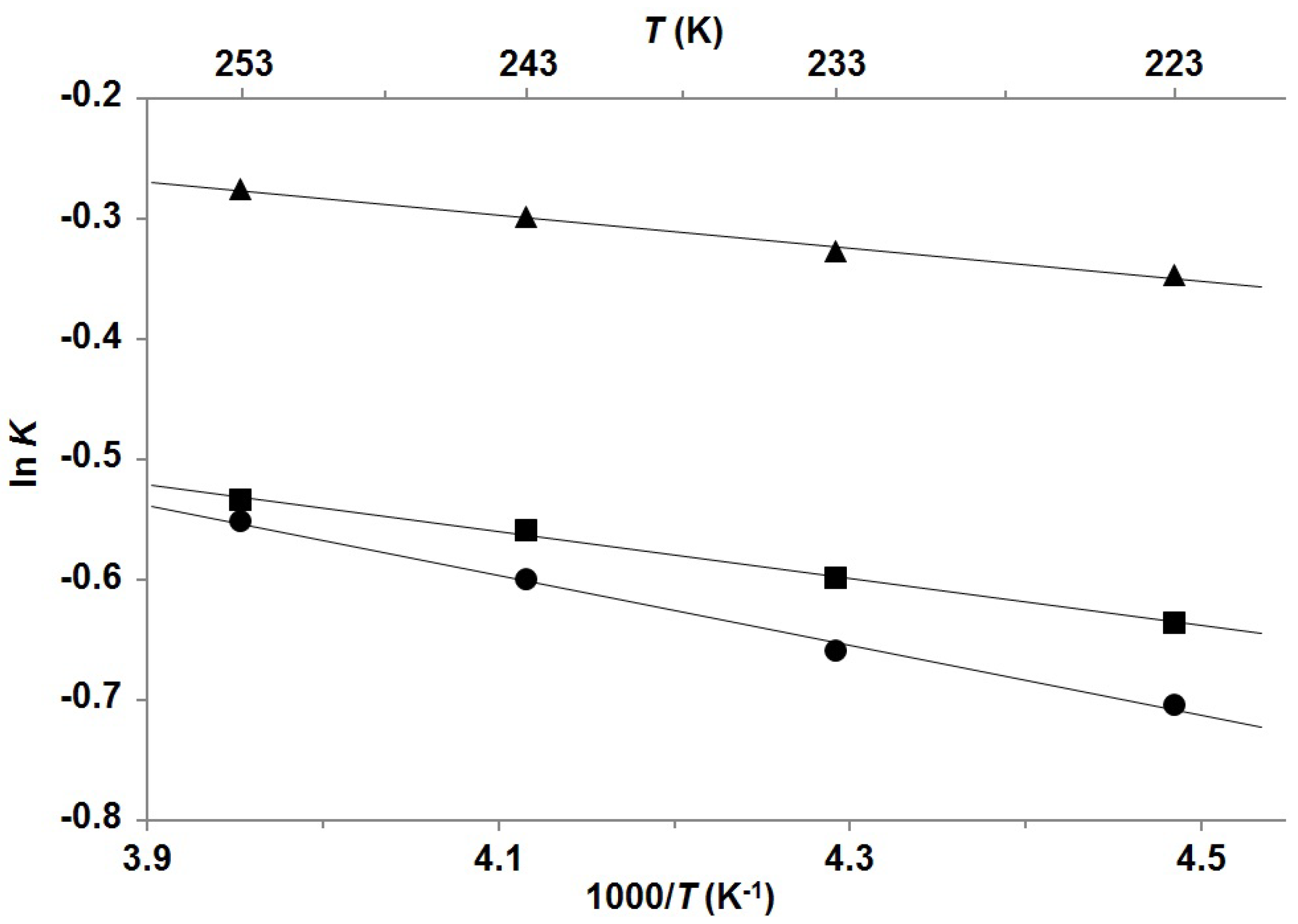
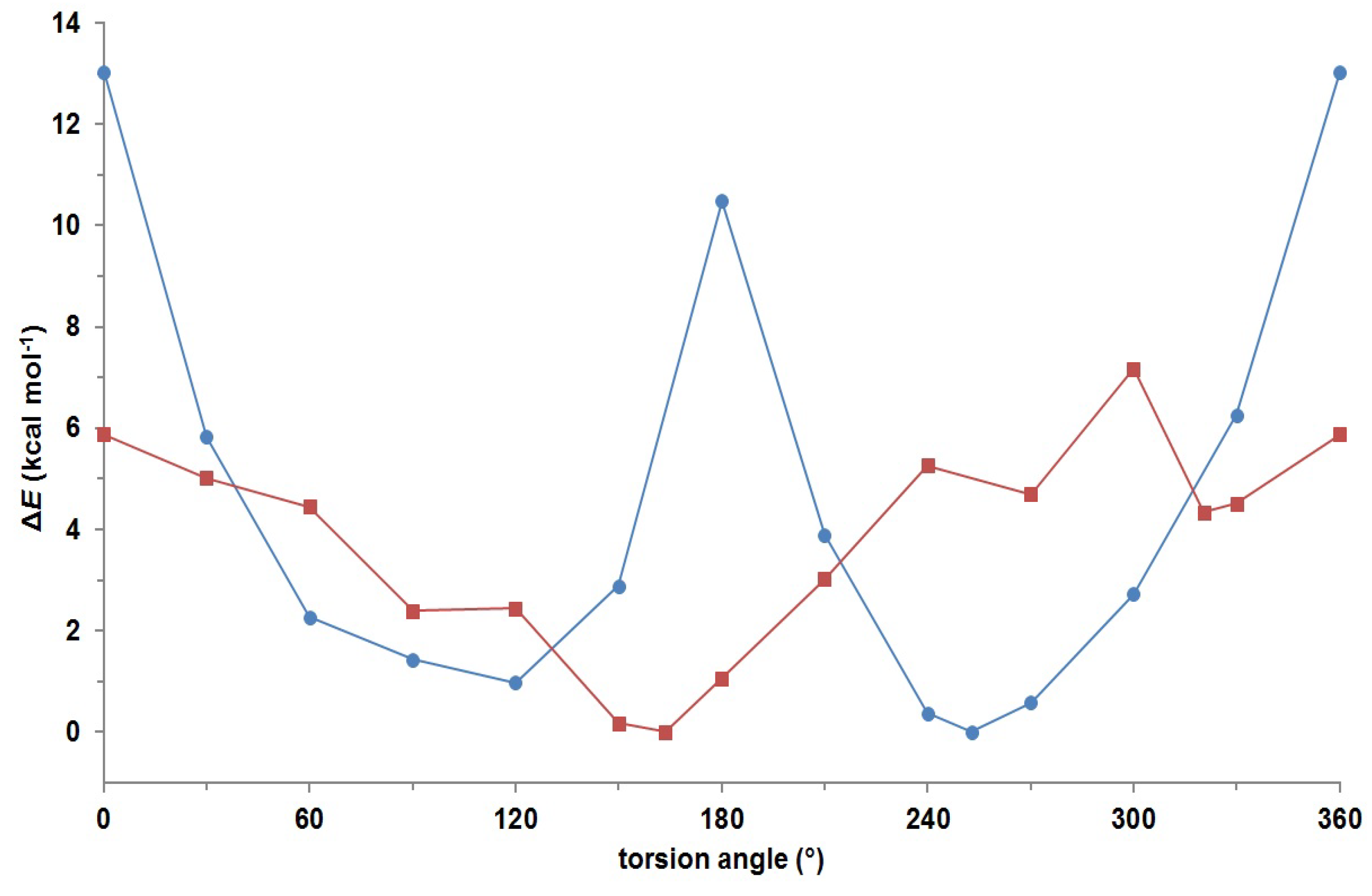
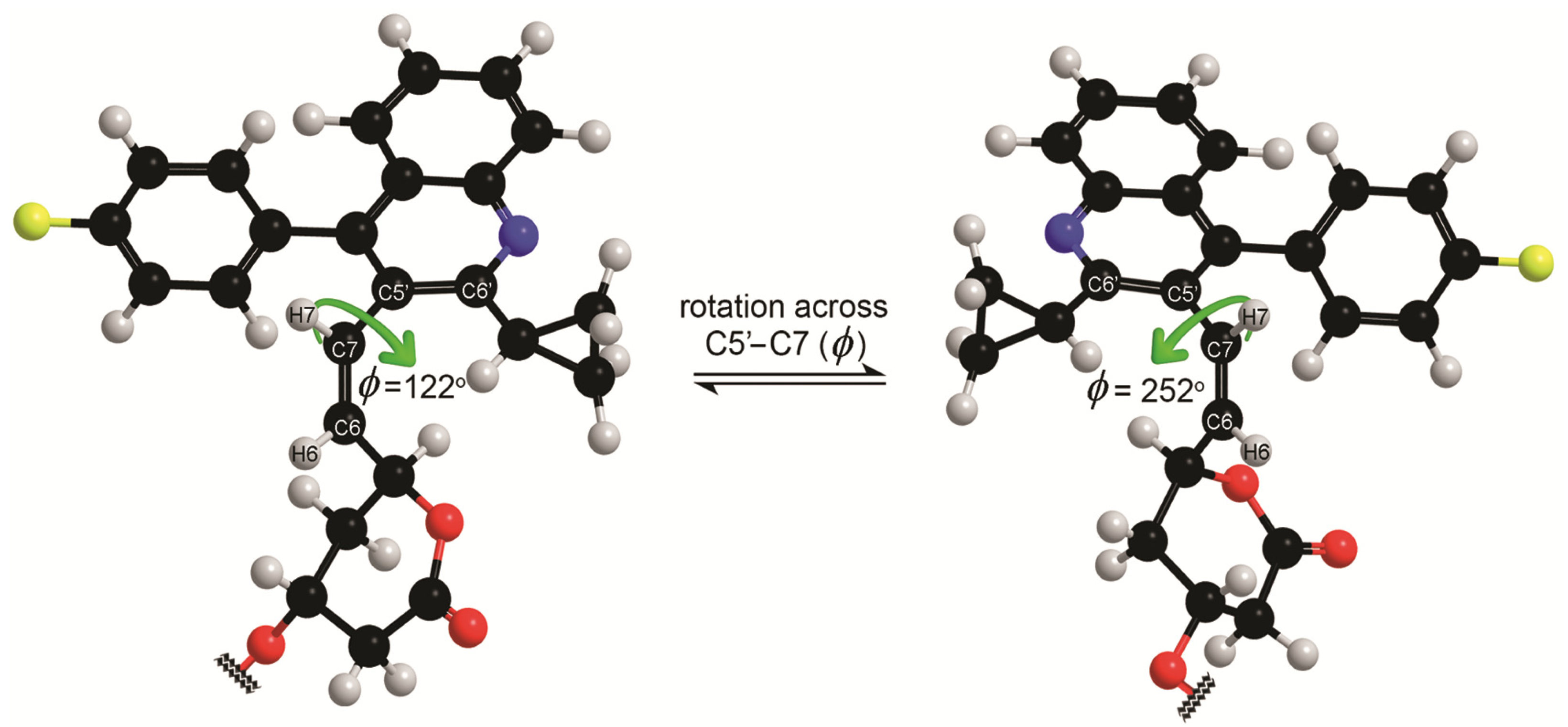
2. Results and Discussion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mole fractions of minor conformers | Thermodynamic parameters a | |||||
|---|---|---|---|---|---|---|---|
| 223 K | 233 K | 243 K | 253 K | Δ H° | – TΔS° | Δ G° | |
| P-1 b | 0.414 | 0.419 | 0.426 | 0.432 | 0.27 | −0.16 | 0.12 |
| P-2 b | 0.346 | 0.355 | 0.364 | 0.370 | 0.39 | −0.14 | 0.25 |
| P-3 c | 0.331 | 0.341 | 0.355 | 0.366 | 0.58 | −0.35 | 0.23 |
| Compound | Tcoal. (K) | Δν (Hz) | kE (s−1) | ΔG‡ (kcal mol-1) |
|---|---|---|---|---|
| P-1 a | 323 | 96.4 | 214 | 15.6 |
| P-2 a | 303 | 14.4 | 32 | 15.9 |
| P-3 b | 303 | 57.3 | 127 | 15.0 |


3. Experimental
3.1. General
3.2. NMR Experiments
3.3. Ab Initio Calculations
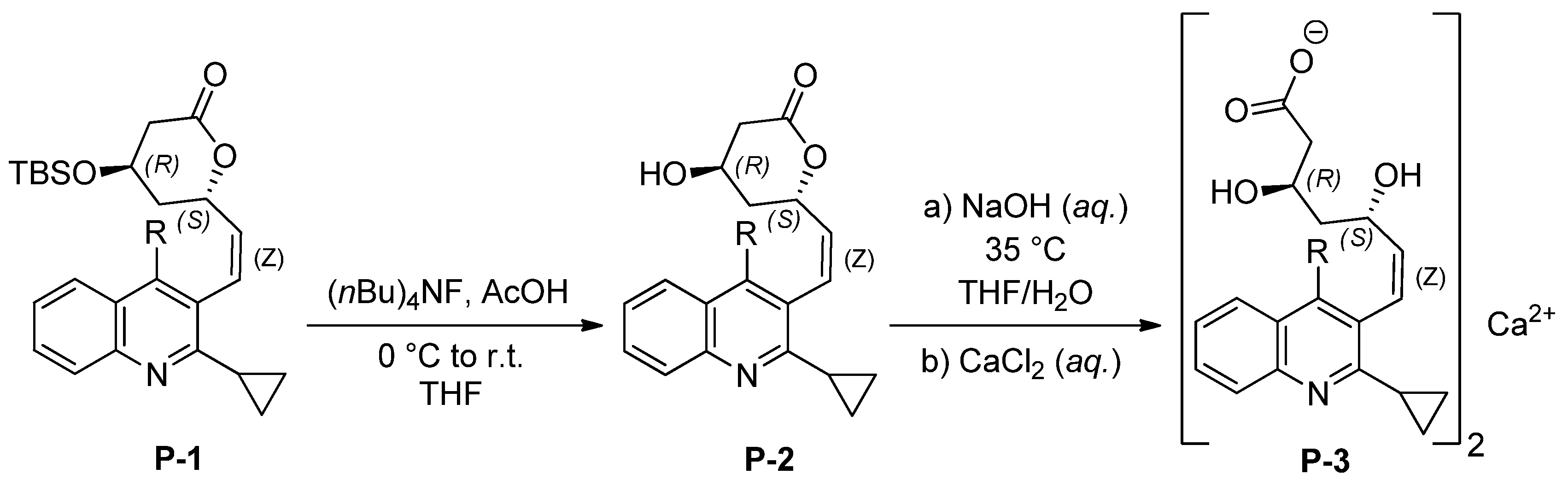
3.4. Synthetic Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Časar, Z. Historic overview and recent advances in the synthesis of super-statins. Curr. Org. Chem. 2010, 14, 816–845. [Google Scholar] [CrossRef]
- Singh, N.; Tamariz, J.; Chamorro, G.; Medina-Franco, J.L. Inhibitors of HMG-CoA reductase: Current and future prospects. Mini Rev. Med. Chem. 2009, 9, 1272–1283. [Google Scholar] [CrossRef]
- Fuenfschilling, P.C.; Pascale, H.; Mutz, J.-P. An improved manufacturing process for fluvastatin. Org. Process Res. Dev. 2007, 11, 13–18. [Google Scholar] [CrossRef]
- Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; et al. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran 2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J. Med. Chem. 1991, 34, 357–366. [Google Scholar] [CrossRef]
- Watanabe, M.; Koike, H.; Ishiba, T.; Okada, T.; Sea, S.; Hirai, K. Synthesis and biological activity of methanesulfonamide pyrimidine- and N-methanesulfonyl pyrrole-substituted 3,5-dihydroxy-6-heptenoates, a novel series of HMG-CoA reductase inhibitors. Bioorg. Med. Chem. 1997, 5, 437–444. [Google Scholar] [CrossRef]
- Saito, Y. Pitavastatin: An overview. Atheroscler. Suppl. 2011, 12, 271–276. [Google Scholar] [CrossRef]
- Stokker, G.E.; Hoffman, W.F.; Alberts, A.W.; Cragoe, E.J.; Deana, A.A.; Gilfillan, J.L.; Huff, J.W.; Novello, F.C.; Prugh, J.D.; Smith, R.L.; et al. 3-hydroxy-3-methylglutaryl-conezyme-A reductase inhibitors.1. Structural modification of 5-substituted 3,5-dihydroxypentanoic acids and their lactone derivatives. J. Med. Chem. 1985, 28, 347–358. [Google Scholar] [CrossRef]
- Beck, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Bergmann, A.; Granzer, E.; Jendralla, H.; Vonkerekjarto, B.; Krause, R.; Paulus, E.; et al. Synthesis and biological activity of new HMG-CoA reductase inhibitors.1. Lactones of pyridine-substituted and pyrimidine-substituted 3,5-dihydroxy-6-heptenoic (-heptanoic) acids. J. Med. Chem. 1990, 33, 52–60. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Holdgate, G. A.; Ward, W. H. J.; McTaggart, F. Molecular mechanism for inhibition of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase by rosuvastatin. Biochem. Soc. Trans. 2003, 31, 528–531. [Google Scholar] [CrossRef]
- Da Costa, R.F.; Freire, V.N.; Bezerra, E.M.; Cavada, B.S.; Caetano, E.W.S.; de Lima Filho, J.L.; Albuquerque, E.L. Explaining statin inhibition effectiveness of HMG-CoA reductase by quantum biochemistry computations. Phys. Chem. Chem. Phys. 2012, 14, 1389–1398. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Cirla, A.; Mann, J. Combretastatins. From natural products to drug discovery. Nat. Prod. Rep. 2003, 20, 558–564. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef]
- Lv, W.; Liu, J.; Lu, D.; Flockhart, D.A.; Cushman, M. Synthesis of mixed (E,Z)-, (E)-, and (Z)-norendoxifen with dual aromatase inhibitory and estrogen receptor modulatory activities. J. Med. Chem. 2013, 56, 4611–4618. [Google Scholar] [CrossRef]
- Fabris, J.; Makuc, D.; Časar, Z.; Plavec, J. Conformational analysis of E/Z-isomeric pairs of rosuvastatin and its lactonized analogues. Tetrahedron 2013, 69, 6262–6268. [Google Scholar] [CrossRef]
- Fabris, J.; Gazić Smilović, I.; Časar, Z. The use of a lactonized statin side-chain precursor in a concise and efficient assembly of pitavastatin. Synthesis 2012, 44, 1700–1710. [Google Scholar] [CrossRef]
- Časar, Z. Straightforward and efficient synthesis of (4R,6S)-4-(tert-Butyldimethylsiloxy)-6-(hydroxymethyl)tetrahydropyran-2-one. Synlett 2008, 13, 2036–2040. [Google Scholar]
- Časar, Z.; Košmrlj, J. The first convenient entry to δ-formyl-δ-valerolactone precursor for the synthesis of statins via lactonized side chain. Synlett 2009, 07, 1144–1148. [Google Scholar]
- Časar, Z.; Steinbücher, M.; Košmrlj, J. Lactone pathway to statins utilizing the wittig reaction. the synthesis of rosuvastatin. J. Org. Chem. 2010, 75, 6681–6684. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C. The relationship between proton–proton NMR coupling constants and substituent electronegativities–I. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.2; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sample Availability: Samples are not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Makuc, D.; Fabris, J.; Časar, Z.; Plavec, J. Conformational Analysis of Geometric Isomers of Pitavastatin Together with Their Lactonized Analogues. Molecules 2013, 18, 13283-13296. https://doi.org/10.3390/molecules181113283
Makuc D, Fabris J, Časar Z, Plavec J. Conformational Analysis of Geometric Isomers of Pitavastatin Together with Their Lactonized Analogues. Molecules. 2013; 18(11):13283-13296. https://doi.org/10.3390/molecules181113283
Chicago/Turabian StyleMakuc, Damjan, Jan Fabris, Zdenko Časar, and Janez Plavec. 2013. "Conformational Analysis of Geometric Isomers of Pitavastatin Together with Their Lactonized Analogues" Molecules 18, no. 11: 13283-13296. https://doi.org/10.3390/molecules181113283
APA StyleMakuc, D., Fabris, J., Časar, Z., & Plavec, J. (2013). Conformational Analysis of Geometric Isomers of Pitavastatin Together with Their Lactonized Analogues. Molecules, 18(11), 13283-13296. https://doi.org/10.3390/molecules181113283