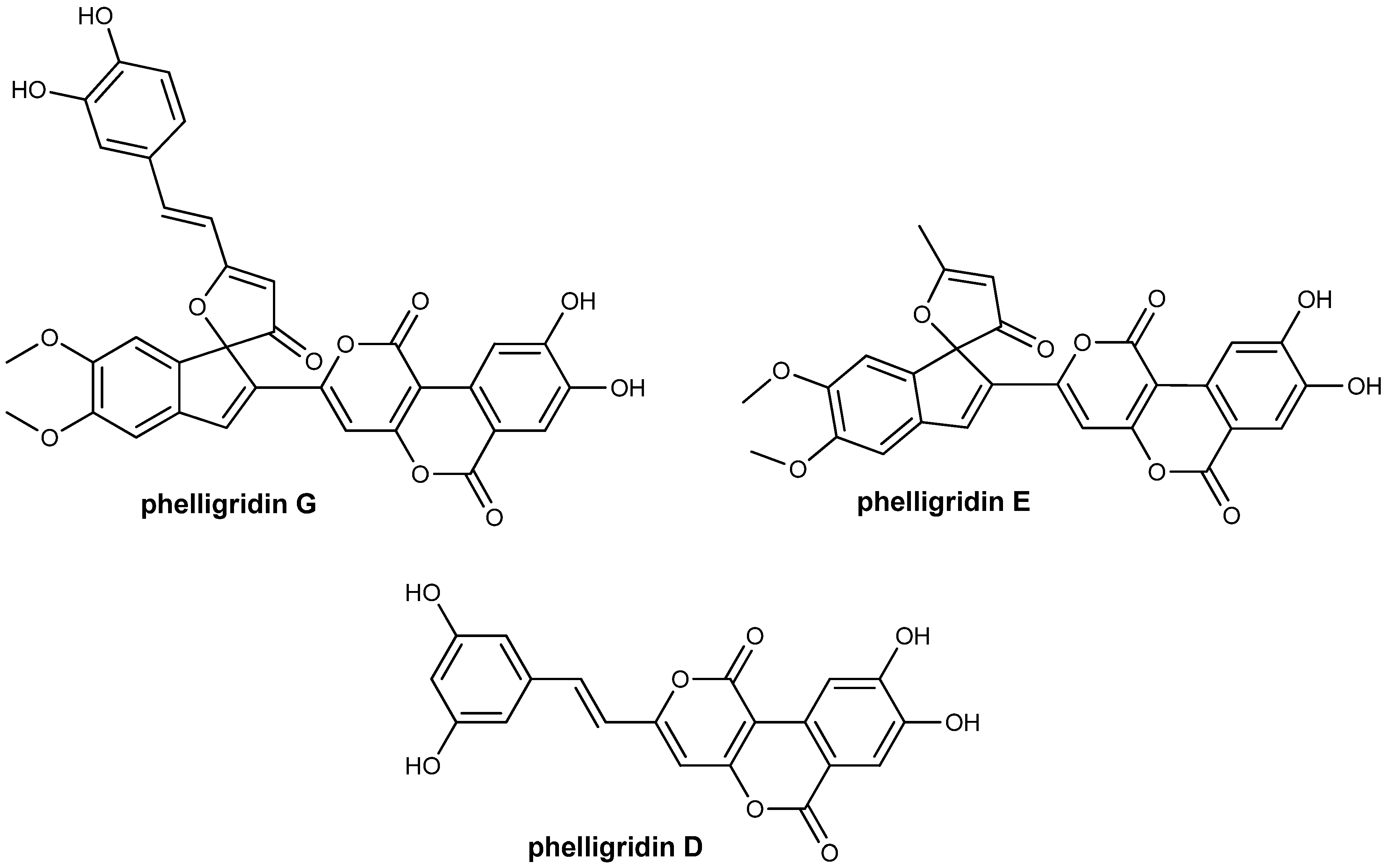
General
All reactions were carried out under an inert argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Commercial grade reagents and solvents were purchased and used without further purification except as indicated below. Hexanes, tetrahydrofuran (THF), diethyl ether (Et2O), and dichloromethane (CH2Cl2) were used directly as obtained from a Baker Cycle-tainer™ system. Yields refer to chromatographically and spectroscopically (1H-NMR) homogenous materials, unless otherwise stated. Reactions were monitored by thin layer chromatography (TLC) carried out on Whatman silica gel 60 Å precoated plates using UV light as the visualizing agent and an acidic mixture of DNP or basic aqueous potassium permanganate (KMnO4) and heat as developing agents. Flash chromatography was performed using Baker silica gel (60 Å particle size). NMR spectra were recorded on Bruker-500 and 400 instruments and calibrated using the residual undeuterated solvent signal as an internal reference (CHCl3 at δ 7.26 ppm for 1H-NMR, δ 77.1 ppm for 13C-NMR). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad. IR Spectra were recorded on Shimadzu FT-IR 8400 spectrometer. Melting points (m.p.) were uncorrected and measured with a Mel-Temp digital melting point apparatus. High resolution mass spectra (HRMS) were obtained from the University of Connecticut Mass Spectral Facility by using Direct Analysis in Real Time using an AccuTOF (JEOL) mass spectrometer.
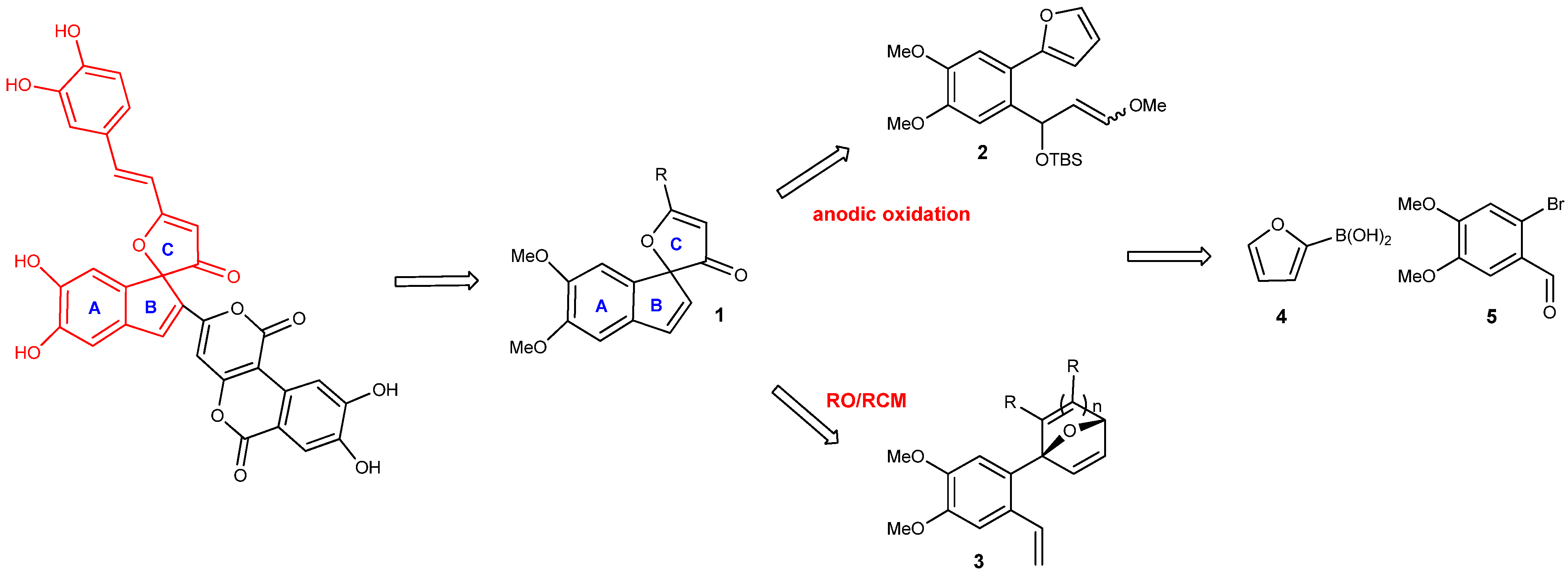
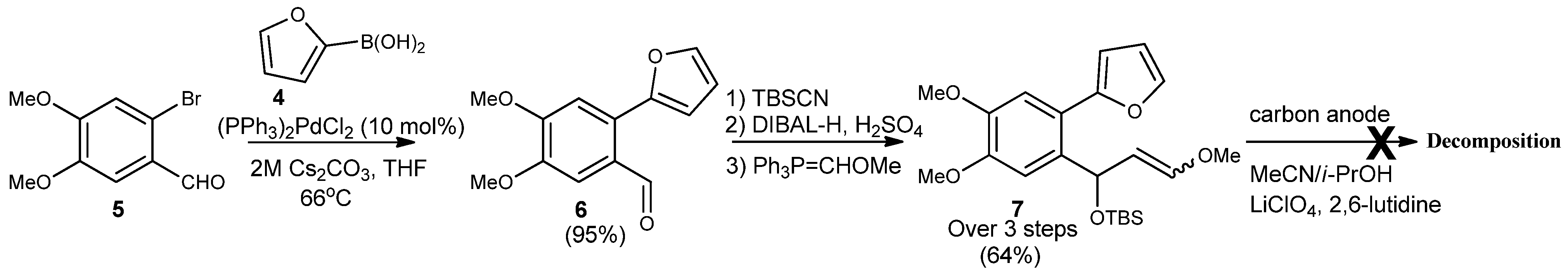
2-Furan-2-yl-4,5-dimethoxybenzaldehyde (6). Commercially available 3,4-dimethoxybromo-benzaldehyde 5 (0.5 g, 2.04 mmol) was added to an argon purged 25 mL flask. Anhydrous THF (10 mL) was added, followed by 2-furanboronic acid (2 equiv.), and PdCl2(PPh3)2 (0.1 mol equiv.). Once all the reagents were in solution, 2M aqueous solution of Cs2CO3 (3 mol equiv.) was added to the flask, which was immediately placed in a preheated oil bath and refluxed with vigorous stirring for 12–24 h. Once the reaction was deemed complete, it was cooled to room temperature, quenched with H2O, and diluted with Et2O. The aqueous layer was extracted with Et2O (3 × 5 mL) and the combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a crude yellow solid. The crude product was purified by column chromatography on silica gel (6:1 hexane/ethyl acetate) to yield 6 (95%) as a solid. Rf = 0.38; m.p. = 102 °C; IR (KBr, cm−1): υ: 730, 909, 1280, 2255, 3012 1H-NMR (400 MHz, CDCl3): δ = 3.96–4.04 (m, 6H), 6.58 (s, 2H), 7.13 (s, 1H), 7.54 (s, 1H), 7.62 (s, 1H), 10.27 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 190.58, 154.46, 153.48, 150.43, 149.04, 143.47, 128.85, 126.67, 115.40, 111.76, 110.38, 108.83, 77.44, 77.12, 76.80, 65.76, 56.00; HRMS (DART) calcd. for C13H13O4 [M+H]+: 233.0814; found 233.0823.
(tert-Butyldimethylsilanyloxy)-2-furan-2’-yl-‘4,5’-dimethoxyphenyl)acetonitrile. To a flame dried flask was added t-butyldimethylsilyl cyanide (0.141 g, 1.0 mmol), potassium cyanide (0.003 g, 0.05 mmol), and 18-crown-6 (0.053 g, 0.4 mmol) under an argon atmosphere. A solution of aldehyde 6 (0.116 g, 0.500 mmol) in dry methylene chloride (5 mL) was added dropwise over 30 min to the homogenous mixture. The reaction was stirred vigorously for 24 h and monitored by TLC. Upon reaction completion the solvent is removed in vacuo and the crude oily residue was purified by flash chromatography using 15% EtOAc in hexanes to obtain a pure yellow oil (0.177g, 95% yield). Rf = 0.67; IR (KBr, cm−1) υ: 654, 735, 908, 2254, 2962, 3012 1H-NMR (500 MHz, CDCl3): δ = 0.02–0.12 (d, J = 50 Hz, 6H), 0.89 (s, 9H), 3.93–3.95 (d, J = 10 Hz, 6H), 5.89 (s, 1H), 6.53 (s, 2H), 7.02 (s, 1H), 7.30 (s,1H), 7.55 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 151.8, 149.5, 147.2, 126.6, 121.8, 119.7, 111.8, 110.9, 110.1, 108.9, 61.2, 56.2, 25.7, 18.3, −5.1; HRMS (DART) calcd. for C20H28 NO4Si [M+H]+: 374.1788; found 374.1776.
(tert-Butyldimethylsilanyloxy)-2-furan-2’-yl-‘4,5’-dimethoxyphenyl)acetaldehyde. Silyl cyanide (0.455 g, 1.22 mmol) was dissolved in dry toluene (4 mL) and cooled to −78 °C. DIBAL (0.868 g, 6.1 mmol) was added dropwise to the flask and the mixture stirred 2 h at this temp. Once the imine is formed, the reaction was quenched with methanol (2.1 mL) and warmed up to 0 °C. After 2 h, 1N H2SO4 (2 mL) was added dropwise to the mixture while stirring for 1 h at this temperature. Diethyl ether was added to dilute the reaction and the aqueous layer was separated. The aqueous layer was extracted with ether (3 × 3 mL) and the ethereal layer collected, washed with brine, dried over Na2SO4, and the solvent removed in vacuo. The crude oil was purified by flash chromatography using a 10% EtOAc in hexanes solution to obtain a pure oil (0.200 g, 44% yield). Rf = 0.49; IR (KBr, cm−1) υ: 732, 913, 1735, 2934, 2959, 3012 1H-NMR (500 MHz, CDCl3): δ= −0.09−0.02 (d, J = −35 Hz, 6H), 0.87 (s, 9H), 3.91–3.92 (d, J = 5 Hz, 6H), 5.50 (s, 1H), 6.49–6.61 (dd, J = 10, 5 Hz, 2H), 7.07–7.09 (d, J = 10 Hz, 2H), 7.51 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 198.9, 152.3, 149.3, 148.9, 142.4, 127.2, 122.8, 111.5, 111.2, 110.5, 108.9, 76.2, 56.0, 25.7, 25.7, 18.3, −4.9; HRMS (DART) calcd. for C20H29O5Si [M+H]+: 377.1784; found 377.1760.
tert-Butyl-[1-(2-furan-2’yl-4’,5’-dimethoxyphenyl)-3-methoxyallyloxy]dimethylsilane (7). To a stirred solution of dry THF (3 mL) was added methoxymethyltriphenylphosphonium bromide (0.36 g, 0.105 mmol) under argon. The homogenous mixture was cooled to 0 °C followed by addition of NaOt-Bu base (2 eq.) in one portion. The red-orange suspension was stirred about 30 min as the color persisted under the above temp. A 0.20 mM solution of 2-furyl benzaldehyde (0.802 g, 3.45 mmol) in THF was slowly added, and the reaction was monitored by TLC at 0 °C. The reaction was quenched with H2O (2 mL) and diluted with ether (4 mL). The aqueous layer was extracted with Et2O, and the combined extracts were washed with H2O and brine, then dried over Na2SO4. Removal of all solvents was done in vacuo to yield crude an oily residue as E/Z isomers of 7 (0.020 g, 64%). Rf = 0.33; IR (KBr, cm−1): υ: 812, 1278, 1687, 2857, 3233; 1H-NMR (500 MHz, CDCl3): δ = −0.11−0.05 (d, J = −30 Hz, 6H), 0.86 (s, 9H), 3.48 (s,1H), 3.91–3.92 (d, J = 5 Hz, 6H), 4.90–4.94 (m, J = 20 Hz, 1H), 5.59–5.60 (d, J = 5 Hz, 1H), 6.29–6.32 (d, J = 15 Hz, 1H), 6.37–6.48 (m, J = 55 Hz, 2H), 6.97 (s, 1H), 7.22 (s, 1H), 7.49 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 153.0, 149.2, 148.8, 147.7, 141.8, 135.7, 120.7, 111.4, 110.9, 109.9, 108.1, 107.3, 69.1, 56.1, 56.0, 25.9, 18.4, −4.7, −4.7; HRMS (DART) calcd. for C22H33O5Si [M+H]+: 405.2097; not detected.
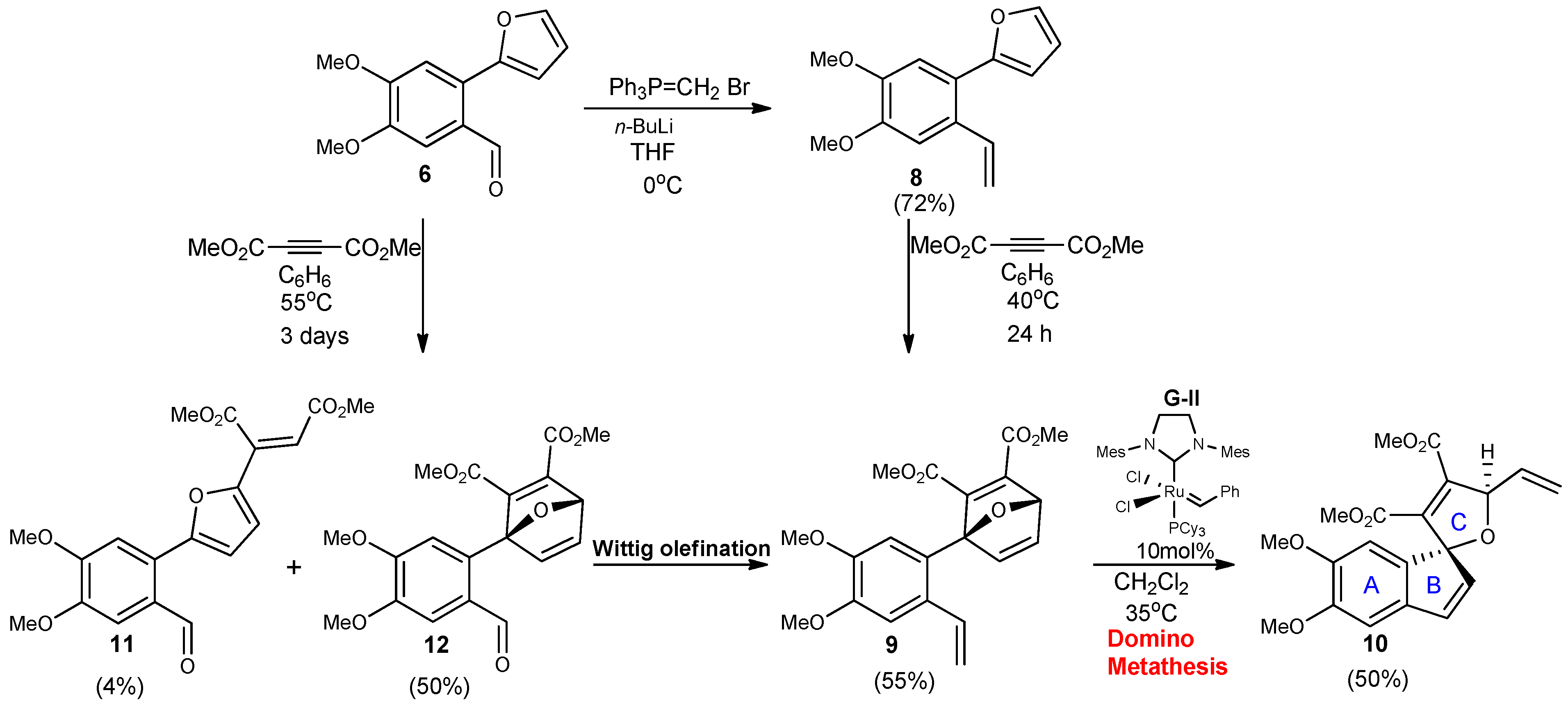
2-(4,4-Dimethoxy-2-vinylphenyl)furan (8). To a stirred solution of dry THF (36 mL) was added methyl triphenylphosphonium bromide (1.52 g, 4.32 mmol) under argon. The homogenous mixture was cooled to 0 °C followed by dropwise addition of a 2.5 M solution n-BuLi in hexanes (1.25 eq.). The red-orange suspension was stirred about 30 min as the color persisted under the above temp. A 0.53 mM solution of 2-furylbenzaldehyde (6, 0.802 g, 3.45 mmol) in THF was slowly added, and the reaction was complete at 0 °C after 10–15 mins. The reaction was quenched with sat. aq. NH4Cl (10 mL) and diluted with EtOAc (20 mL). The aqueous layer was extracted with EtOAc, and the combined extracts were washed with H2O and brine and then dried over Na2SO4. Removal of all solvents was done in vacuo to yield a crude solid material, which was purified by silica gel column chromatography (hexane/EtOAc, 10:1) to yield olefin 8 as a yellow oil (0.568 g, 72%). Rf = 0.43; IR (KBr, cm−1): υ: 730, 909, 1266, 2254, 3007 1H-NMR (500 MHz, CDCl3): δ = 3.93–3.95 (d, 6H, J = 3.3 Hz), 5.24–5.26 (dd, J = 5, 5 Hz, 1H), 5.59–5.62 (d, J = 15 Hz, 1H), 6.42–6.49 (d, J = 35 Hz, 2H), 7.99–7.05 (m, J = 30 Hz, 1H), 7.03 (s, 1H), 7.12 (s, 1H), 7.48 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 152.64, 148.91, 141.91, 135.98, 128.73, 122.63, 114.14, 111.56, 110.39, 109.39, 56.16, 56.11; HRMS (DART) calcd. for C14H15O3 [M+H]+: 231.1021; found 231.1000.
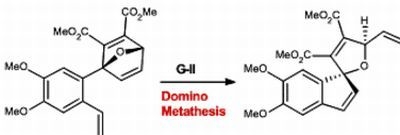
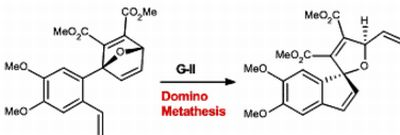
1-(4’,5’-Dimethoxy-2’vinylphenyl)-7-oxabicyclo[2.2.1]hepta-2,5diene-2,3-dicarboxylic acid dimethyl ester (9). Dimethoxybenzylfuran 8 (343 mg, 1.49 mmol) was added to a flame dried vial in benzene (0.37mL) to make a 3 mM solution, followed by addition of DMAD (233 mg,1.64 mmol) at room temperature. The reaction stirred vigorously until everything was in solution, then placed in a preheated oil bath at 40 °C. The reaction was monitored by TLC using a 30% EtOAc in hexanes solution. Upon completion the solvents were removed in vacuo to afford a crude oil, which was purified by silica gel chromatography (SiO2, 7 g, 25% EtOAc in hexanes to yield cycloadduct 9 as an oil (0.264 g, 55%). Rf = 0.21; IR (KBr, cm−1): υ: 731, 908, 1268, 2345, 3016; 1H-NMR (500 MHz, CDCl3): δ = 3.57 (s, 3H), 3.74 (s, 3H), 3.84–3.86 (d, J = 10 Hz, 6H), 5.14–5.16 (d, J = 10 Hz, 1H), 5.50–5.54 (d, J = 20 Hz, 1H), 5.75 (s, 1H), 6.92–7.02 (dd, J = 15, 15 Hz, 1H), 7.01–7.03 (d, J = 10 Hz, 2H), 7.27–7.28 (dd, J = 5 Hz, 1H), 7.47–7.48 (d, J = 5 Hz, 1H). 13C-NMR (126 MHz, CDCl3) δ = 164.9, 162.7, 158.8, 149.4, 149.2, 148.6, 145.2, 144.7, 134.7, 130.0, 123.6, 114.7, 110.9, 109.4, 97.9, 83.5, 55.9, 52.4; HRMS (DART) calcd. for C20H21O7 [M+H]+: 373.1287; found 373.1280.
4’5’Dimethoxyspiro[5-oxafuryl-2,3 di(methoxycarbonyl)-4-vinyl]-1,1’[1H]indene (10). To a solution of oxabicyclic ether 9 (177 mg, 0.475 mmol) in dry dichloromethane (47.5 mL) was added a solution of Grubbs 2nd generation catalyst G-II (40 mg, 0.0475 mmol) in 1 mL CH2Cl2 at room temperature. The reaction was stirred vigorously at room temperature then placed into a preheated oil bath at 35 °C. The reaction was monitored by 1H-NMR. After 24 hrs of heating, the reaction was removed from heat and concentrated to a crude brown oil. Purification of the residue by flash chromatography using 25% EtOAc in hexanes afforded pure spirocyclic compound 10 (0.048g, 50%). Rf = 0.21; IR (KBr, cm−1): υ: 732, 907, 1255, 2253, 2955; 1H-NMR (500 MHz, CDCl3): δ = 3.56 (s, 3H), 3.81 (s, 3H), 3.87–3.89 (d, J = 10 Hz, 6H), 5.28–5.30 (d, J = 20 Hz, 1H), 5.48–5.51 (d, J = 15, 1H), 5.76–5.78 (s,1H) 5.99–6.04 (m, 1H), 6.16–6.17 (d, J = 5.5 Hz, 1H), 6.61–6.62 (d, J = 5.5 Hz, 1H), 6.76 (s, 1H), 6.89 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 163.1, 162.3, 150.3, 148.4, 142.9, 136.7, 136.6, 135.7, 133.7, 128.8, 127.0, 118.1, 107.8, 105.8, 99.38 86.4, 56.5, 56.6, 52.6; HRMS (DART) calcd. for C20H21O7 [M+H]+: 373.1287; found 373.1283.
1-(2’-Formyl-4’,5’dimethoxyphenyl)-7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylic acid dimethyl ester (12). Dimethoxyfurylbenzaldehyde 6 (343 mg,1.49 mmol) in benzene (0.37 mL) was added to a flame dried vial to make a 3 mM solution, followed by addition of DMAD (233 mg, 1.64 mmol) at room temperature. The reaction was stirred vigorously until everything was in solution, then added to a preheated oil bath at 50 °C. The reaction was monitored by TLC using a 30% EtOAc solution in hexanes as eluent. Upon completion the solvents were removed in vacuo to afford a crude oil, which was purified by silica gel chromatography (SiO2, 7 g, 25% EtOAc in hexanes) to yield cycloadduct 12 as an oil. Diastereomers were obtained, which could be separated via column chromatography (0.202 g, 50% of desired product 12). Rf = 0.085; Rf = 0.17; m.p. = 175 °C; IR (KBr, cm−1): υ: 730, 912, 1291, 1684, 3024; 1H NMR (500 MHz, CDCl3): δ = 3.63 (s, 3H), 3.81 (s, 3H), 3.97–3.98 (d, J = 5 Hz, 6H), 5.76 (s, 1H), 7.21 (s, 1H), 7.25 (s, 2H), 7.30–7.33 (dd, J = 5, 5 Hz, 1H), 7.45 (s, 1H), 10.03 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 190.9, 164.1, 162.7, 156.8, 153.5, 148.9, 148.3, 144.9, 143.3, 129.9, 126.9, 114.6, 110.3, 96.6, 82.6, 56.2, 56.1, 52.3, 52.2; HRMS (DART) calcd. for C19H18O8: [M+H]+: 375.1080; found 375.1069.
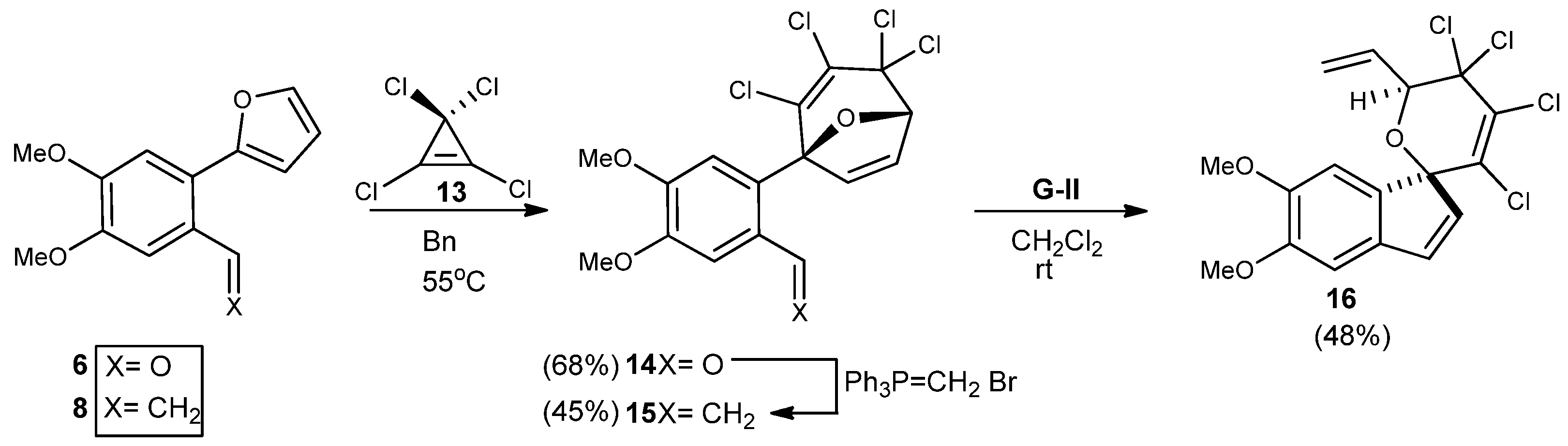
4’,5’-Dimethoxy-2-(2,3,4,4-tetrachloro-8-oxabicyclo[3.2.1]octa-2,6-dien-1-yl-benzaldehyde (14). Dimethoxyfurylbenzaldehyde 6 (0.237 mmol) was dissolved in benzene to make a 2.97 M solution at room temperature, followed by addition of tetrachlorocyclopropene (1 eq.). The homogeneous mixture was stirred vigorously, heated to 55 °C and stirred for 3–4 days. The reaction was monitored by TLC using a 25% EtOAc in hexanes solution. Upon completion the solvents were removed in vacuo to afford a crude solid, which was purified by flash chromatography (SiO2, 9 g, 25% EtOAc in hexanes) to yield a yellow solid of 14 (0.42g, 68%). Rf = 0.67; m.p. = 175 °C; IR (KBr, cm−1): υ: 733, 907, 1277, 1516, 2938; 1H-NMR (500 MHz, CDCl3): δ = 3.98–4.07 (d, J = 43 Hz, 6H), 5.46 (s, 1H), 7.00 (s, 1H), 7.30 (s, 1H), 7.59 (s, 1H), 10.23 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 190.1, 153.0, 150.2, 144.7, 138.7, 129.4, 127.9, 122.5, 112.6, 110.3, 94.2, 82.6, 75.2, 64.1, 63.3, 56.3, 56.2; HRMS (DART) calcd. for C16H13Cl4O4 [M+H]+: 410.9540; found 410.9540.
2,3,4,4,Tetrachloro-1-(4’,5’dimethoxy-2’-vinylphenyl-8-oxabicyclo[3.2.1]octa-2,6-diene (15). Cyclo-adduct 14 (0.237 mmol) was olefinated as mentioned above to give crude compound 15 which was purified using flash chromatography on silica gel (hexane/EtOAc, 20:1) to yield a yellow solid (0.57 g, 45%). Rf = 0.71; IR (KBr, cm−1): υ: 731, 908, 2252, 2941, 3014; 1H-NMR (500 MHz, CDCl3): δ = 3.95–3.99 (d, J = 16 Hz, 6H), 5.18–5.21 (d, J = 12Hz, 1H), 5.39 (s, 1H), 5.54–5.57 (d, J = 12 Hz, 1H), 6.88–7.00 (dd, J = 4 Hz, dd, J = 8 Hz, 1H), 6.93 (s, 1H), 6.96 (s, 1H), 7.09 (s, 1H), 7.19 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 150.3, 148.4, 145.2, 137.3, 135.1, 132.7, 120.8, 115.0, 113.5, 110.0, 95.6, 82.4, 75.5, 64.7, 63.6, 56.2, 56.1; HRMS (DART) calcd. for C17H15Cl4O3 [M+H]+: 408.9747; found 408.9767.
4’5’Dimethoxyspiro[6-oxapyrano-2,3,4,4 tetrachloro-5-vinyl]-1,1’[1H]indene (16). Purification of the crude residue 16 by flash chromatography using 25% EtOAc in hexanes afforded pure spiro-fused pyran. Yield: 0.048 g (48%). Rf = 0.30; IR (KBr, cm−1): υ: 733, 912, 2324, 2861, 2943; 1H-NMR (500 MHz, CDCl3): δ = 3.90–3.91 (d, J = 5 Hz, 6H), 5.07–5.08 (d, J = 5 Hz, 1H), 5.45–5.47 (d, J = 10 Hz, 1H), 5.52 (s, 1H) 6.12–6.13 (d, J = 5 Hz, 1H), 6.22–6.28 (m, J = 30 Hz, 1H), 6.59–6.60 (d, J = 5 Hz, 1H), 6.81(s, 1H), 7.68 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ = 150.9, 148.5, 144.9, 137.8, 137.3, 134.3, 132.6, 131.3, 124.9, 121.2, 111.8, 106.5, 84.0, 70.20, 59.53, 56.6, 56.4; HRMS (DART) calcd. for C17H15Cl4O3 [M+H]+: 408.9747; found 408.9772.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




