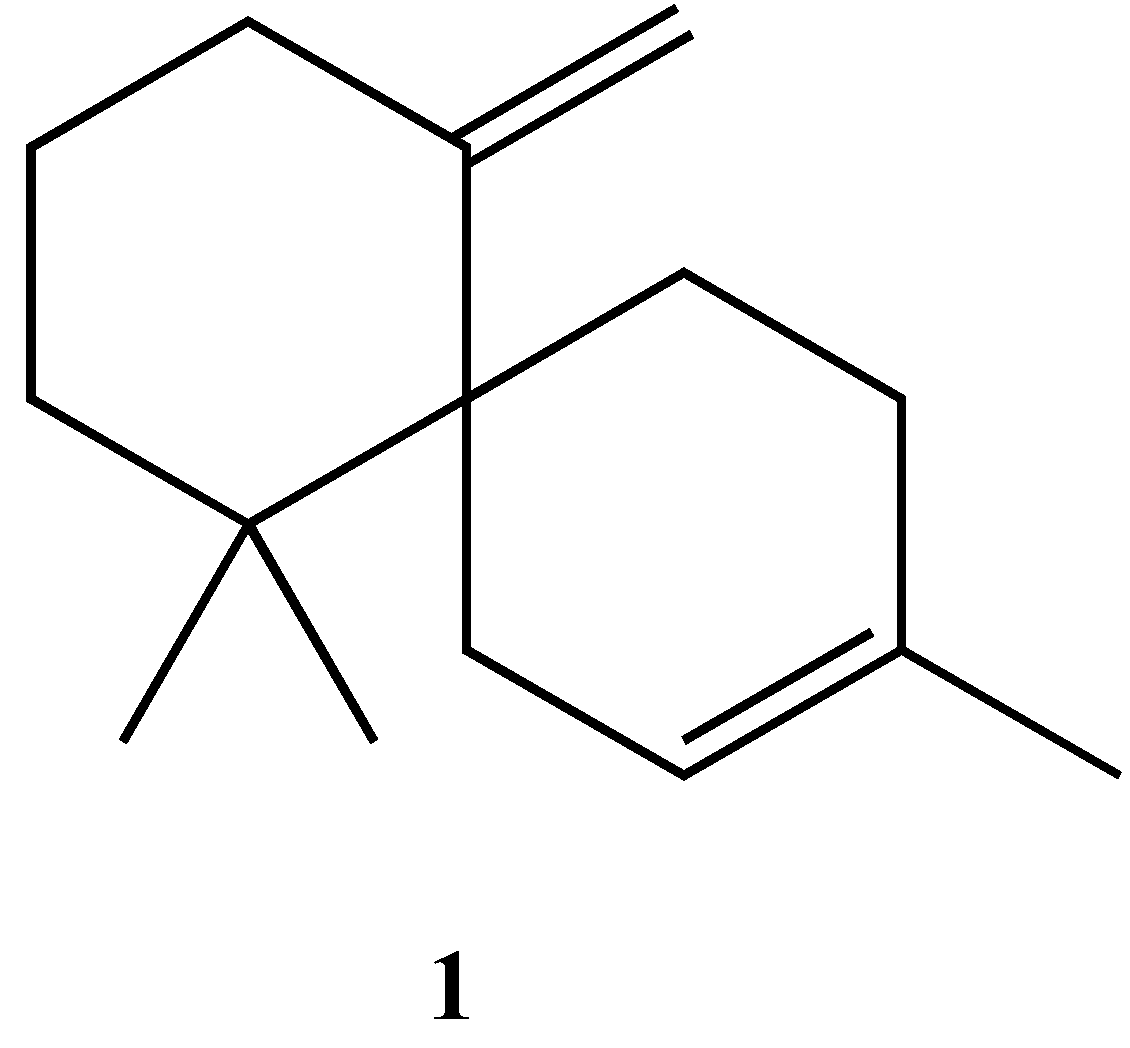
Synthesis of Racemic β-Chamigrene, a Spiro[5.5]undecane Sesquiterpene


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

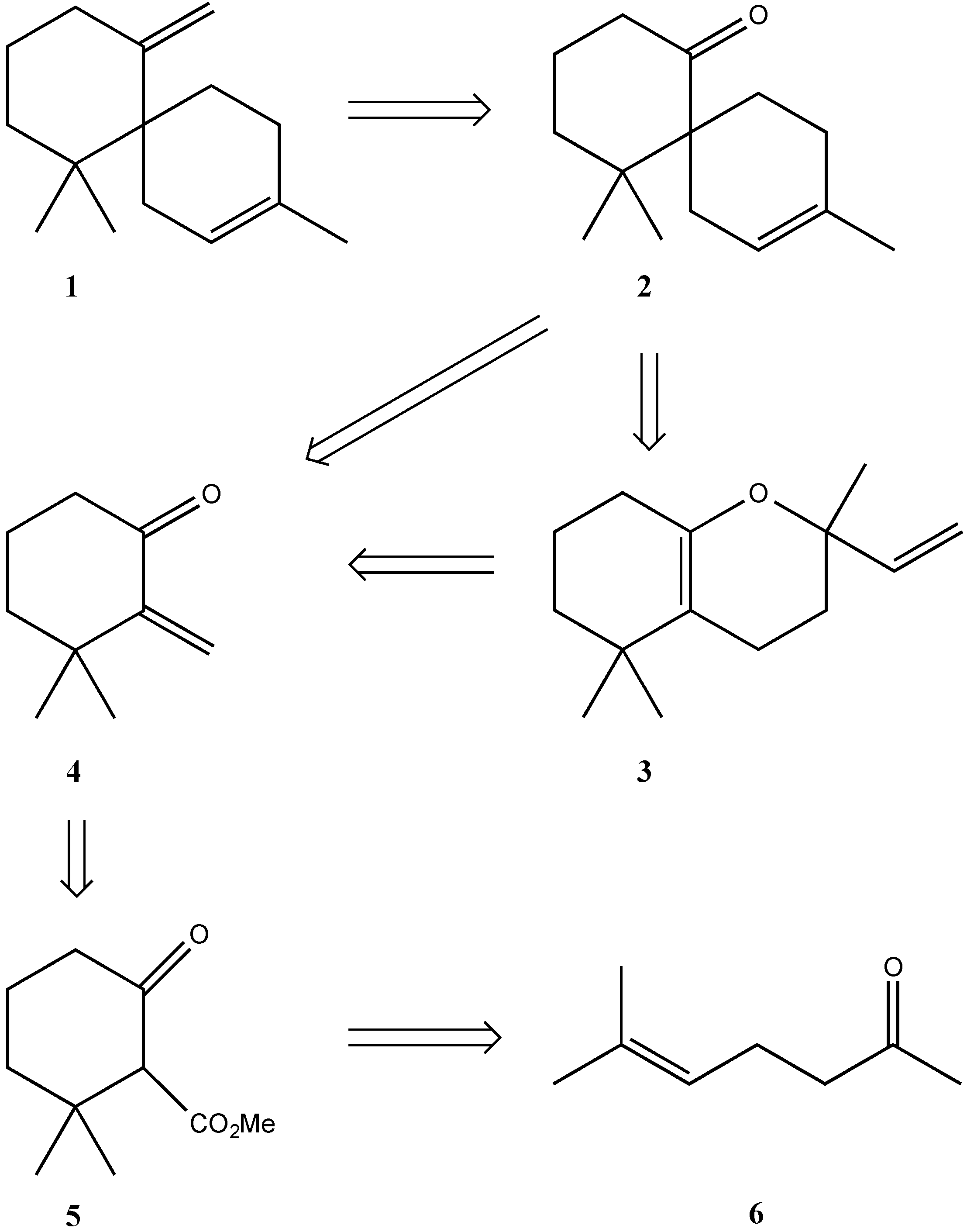
2. Results and Discussion


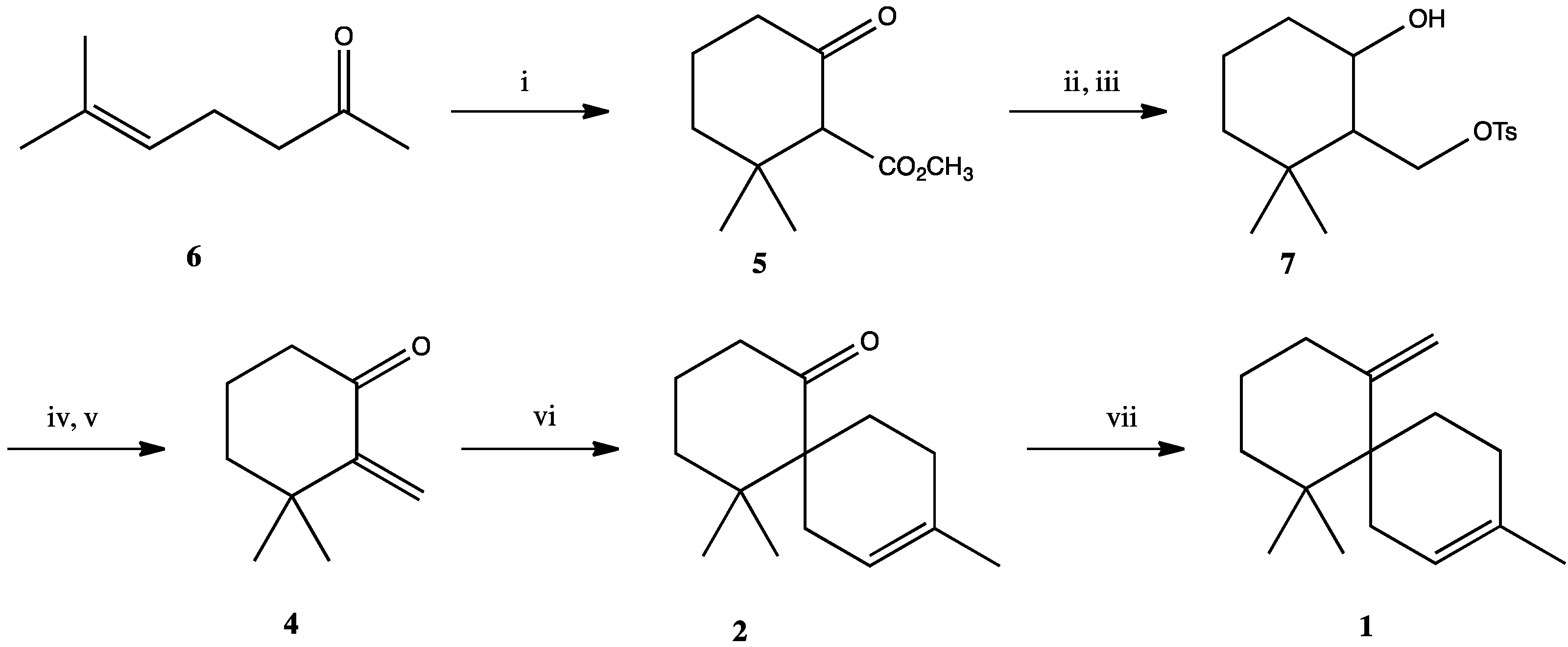
3. Experimental Section
3.1. 2-Carbomethoxy-3,3-Dimethylcyclohexanon (5)
3.2. 3,3-Dimethyl-2-Tosyloxymethylcyclohexanol (7)
3.3. 3,3-Dimethyl-2-Methylenecyclohexanone (4)
3.4. 5,5,9-Trimethylspiro[5.5]undec-8-en-1-one (2)
3.5. 5,5,9-Trimethylspiro[5.5]undec-8-en-1-one (2) (With Microwave Heating)
3.6. 3,7,7-Trimethyl-11-Methylenespiro[5.5]undec-2-ene (1)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Srikrishna, A.; Ramesh Babu, R. Total synthesis of (±)-β-chamigrene and (±)-laurencenone C via Ireland ester Claisen rearrangement and an intramolecular type II carbonyl ene reaction sequence. Tetrahedron 2008, 64, 10501–10506. [Google Scholar] [CrossRef]
- Ito, S.; Endo, K.; Yoshida, M.; Kodama, M. Chemical constituents in the essential oil of the leaves of Chamaecyparis taiwanensis. Yakugaku Zasshi 1967, 87, 434–439. [Google Scholar] [PubMed]
- Otha, Y.; Hirose, Y. New sesquiterpenoids from Schizandra chinensis. Tetrahedron Lett. 1968, 9, 2483–2485. [Google Scholar] [CrossRef]
- Martin, J.D.; Perez, C.; Ravelo, J.L. Enantioselective ring construction: Synthesis of halogenated marine natural spiro[5.5]undecane sesquiterpenes. J. Am. Chem. Soc. 1986, 108, 7801–7811. [Google Scholar] [CrossRef] [PubMed]
- Dias, T.; Brito, I.; Moujir, L.; Paiz, N.; Darias, J.; Cueto, M.J. Cytotoxic sesquiterpenes from Aplysia dactylomela. J. Nat. Prod. 2005, 68, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Lepine-Frenette, C.; Spero, D. Intramolecular cyclopropanation-ring fragmentation leading to spirocyclic ring construction: A stereoselective synthesis of β-chamigrene. J. Org. Chem. 1991, 56, 4494–4498. [Google Scholar] [CrossRef]
- White, J.D.; Skeean, R.W.; Trammell, G.L. Lewis acid and photochemically mediated cyclization of olefinic β-keto esters. J. Org. Chem. 1985, 50, 1939–1948. [Google Scholar] [CrossRef]
- Ireland, R.E.; Dow, W.C.; Godfrey, J.D.; Thaisrivongs, S. Total synthesis of (±)-aphidicolin and (±)-β-chamigrene. J. Org. Chem. 1984, 49, 1001–1013. [Google Scholar] [CrossRef]
- Tanaka, A.; Uda, H.; Yoshikoshi, A. The total synthesis of (±)-chamigrene. Chem. Commun. 1967, 188–189. [Google Scholar]
- Peterson, D.J. Carbonyl olefination reaction using silyl-substituted organometallic compounds. J. Org. Chem. 1968, 33, 780–784. [Google Scholar] [CrossRef]
- Quartieri, F.; Mesiano, L.E.; Borghi, D.; Desperati, V.; Gennari, C.; Papeo, G. Total Synthesis of (+)-7,11-Helianane and (+)-5-Chloro-7,11-helianane through Stereoselective Aromatic Claisen Rearrangement. Eur. J. Org. Chem. 2011, 33, 6794–6801. [Google Scholar] [CrossRef]
- Payack, J.F.; Hughes, D.L.; Cai, D.; Cottrell, I.F.; Verhoeven, T.R. Dimethyltitanocene. Org. Syn. 2002, 79, 19–22. [Google Scholar] [CrossRef]
- Yan, T.; Tsai, C.; Chien, C.; Cho, C.; Huang, P. Dichloromethane activation. Direct methylenation of ketones and aldehydes with CH2Cl2 promoted by Mg/TiCl4/THF. Org. Lett. 2004, 6, 4961–4963. [Google Scholar] [CrossRef] [PubMed]
- Stenstrøm, Y. Facile activation of zinc. Preparation of cyclobutanones via dichloroketene and cyclopropanes using the Simmons-Smith reaction. Synth. Commun. 1992, 22, 2801–2810. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the title compound, β-chamigrene (1), is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonsen, S.; Skattebøl, L.; Stenstrøm, Y. Synthesis of Racemic β-Chamigrene, a Spiro[5.5]undecane Sesquiterpene. Molecules 2014, 19, 20664-20670. https://doi.org/10.3390/molecules191220664
Antonsen S, Skattebøl L, Stenstrøm Y. Synthesis of Racemic β-Chamigrene, a Spiro[5.5]undecane Sesquiterpene. Molecules. 2014; 19(12):20664-20670. https://doi.org/10.3390/molecules191220664
Chicago/Turabian StyleAntonsen, Simen, Lars Skattebøl, and Yngve Stenstrøm. 2014. "Synthesis of Racemic β-Chamigrene, a Spiro[5.5]undecane Sesquiterpene" Molecules 19, no. 12: 20664-20670. https://doi.org/10.3390/molecules191220664
APA StyleAntonsen, S., Skattebøl, L., & Stenstrøm, Y. (2014). Synthesis of Racemic β-Chamigrene, a Spiro[5.5]undecane Sesquiterpene. Molecules, 19(12), 20664-20670. https://doi.org/10.3390/molecules191220664